
A novel trigeminal zinc porphyrin and corresponding porphyrin monomers for dye-sensitized solar cells†
Abstract
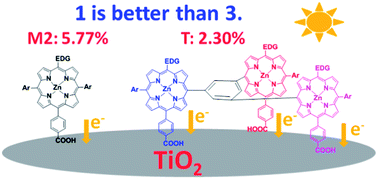
A novel trigeminal zinc porphyrin sensitizer (T) and two zinc porphyrin monomers (M1 and M2) were successfully designed and synthesized. The spectral, electrochemical, and photovoltaic properties of the porphyrin dyes were investigated. Compared with M1, the molecule of M2 has an additional aliphatic n-hexyloxyl chain at the meso-position of the porphyrin framework, and such a structure is favorable for the formation of a compact hydrophobic layer at the TiO2 surface and the retardation of the diffusion of I3− ions into the nanoporous TiO2 electrode, resulting in more effective suppression of the charge recombination process and a higher Voc. Meanwhile, M2 has larger IPCE values than those of M1, leading to the higher Jsc value. Thus, the DSSC devices based on M2 demonstrated a relatively high power conversion efficiency of 5.77%, with the Jsc, Voc and ff values of 13.93 mA cm−2, 732 mV, and 0.566, respectively. Even though dye T has the highest molar absorption coefficients and multiple binding moieties, the corresponding power conversion efficiency is 2.30%, which is lower than those for M1 and M2. These observations may be ascribed to the low efficiency of the electron injection process caused by the isolation of the LUMOs from the anchoring carboxyl groups in addition to the lowest adsorption amount.
 Please wait while we load your content...
Please wait while we load your content...

