
An NMR study on a pseudo-intramolecular transacylation reaction of an α-aryl-β-keto ester†
Abstract
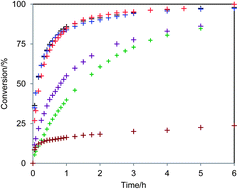
The pseudo-intramolecular transacylation reaction efficiently proceeds like an intramolecular reaction, even though it is actually an intermolecular reaction. We have obtained valuable insights by monitoring the reaction by 1H NMR spectroscopy.
 Please wait while we load your content...
Please wait while we load your content...

