New insight into the anisotropic effects in solution-state NMR spectroscopy
Marija
Baranac-Stojanović
*
Faculty of Chemistry, University of Belgrade, Studentski trg 16, P.O.Box 158, 11000 Belgrade, Serbia. E-mail: mbaranac@chem.bg.ac.rs
First published on 5th November 2013
Abstract
Nuclear magnetic resonance (NMR) spectroscopy is an important technique for structure determination. Within it, anisotropic effects of different functional groups and ring systems, depicted as familiar “anisotropy cones”, are broadly used to deduce the stereochemistry, for chemical shift assignments and to explain shielding or deshielding of nuclei spatially close, or directly attached to the corresponding functional group, or ring. Progress in computational methods has enabled the quantification of anisotropic effects, an insight into their origin and to the source of (de)shielding of proximal nucleus. Some widely accepted traditional explanations, presented in NMR spectroscopy textbooks, have been questioned. The purpose of this review is to collect and discuss the research, mainly based on theoretical calculations, that provided new insight into the anisotropic effects, their origin and factors responsible for (de)shielding of proximal protons.
 Marija Baranac-Stojanović | Marija Baranac-Stojanović studied chemistry at the Faculty of Chemistry, University of Belgrade (Serbia), where she received her diploma in 1995. She obtained her MSc and PhD degrees from the same university, working on the chemistry of heterocyclic compounds. She carried out her postdoctoral research in theoretical chemistry at the University of Potsdam (Germany). She is currently an associate professor at the Faculty of Chemistry, University of Belgrade. |
Introduction
Nuclear magnetic resonance (NMR) spectroscopy is a powerful and inevitable tool for structure determination. It is based on the fact that nuclei residing in different environments in a molecule resonate at different frequencies (ν), since they do not sense the same magnetic field.1–3 The frequency at which resonance occurs is defined as: ν =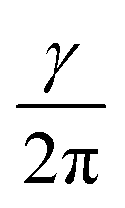 Beff. Here, γ represents the gyromagnetic ratio which is a constant for each isotope of each element. Beff is the effective magnetic field acting at the nucleus. It is determined as the sum of the applied external magnetic field (B0) and magnetic field induced by the surrounding electrons (Bind): Beff = B0 + Bind. The induced magnetic field is related to the applied field by the shielding constant (σ): Bind = −σB0. Hence, the resonance frequency depends on σ as following: ν =
Beff. Here, γ represents the gyromagnetic ratio which is a constant for each isotope of each element. Beff is the effective magnetic field acting at the nucleus. It is determined as the sum of the applied external magnetic field (B0) and magnetic field induced by the surrounding electrons (Bind): Beff = B0 + Bind. The induced magnetic field is related to the applied field by the shielding constant (σ): Bind = −σB0. Hence, the resonance frequency depends on σ as following: ν = 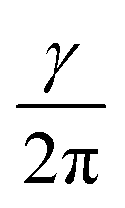 B0(1 − σ). If σ > 0, the Beff is reduced which is termed as diamagnetic or shielding effect. If σ < 0, the Beff is increased, i.e. a nucleus is deshielded, which is known as paramagnetic effect. Chemical shift (δ) is obtained as a difference between the resonance frequencies of a sample and a standard substance (in Hz) divided by the operating frequency of the spectrometer (in MHz). It is thus in units Hz/MHz, or parts per million (ppm), and is proportional to the difference in shielding of the reference (σref) and the nucleus (σ): δ ∼ σref − σ. A reference substance often used in 1H and 13C NMR spectroscopy is tetramethylsilane (TMS). Main factors influencing the shielding constant in proton NMR spectroscopy include: inductive and resonance effects of substituents, hybridization, hydrogen bonds, steric and electric field effects, solvent effects and contribution from the magnetic anisotropy of neighbouring groups, i.e. anisotropic effects.
B0(1 − σ). If σ > 0, the Beff is reduced which is termed as diamagnetic or shielding effect. If σ < 0, the Beff is increased, i.e. a nucleus is deshielded, which is known as paramagnetic effect. Chemical shift (δ) is obtained as a difference between the resonance frequencies of a sample and a standard substance (in Hz) divided by the operating frequency of the spectrometer (in MHz). It is thus in units Hz/MHz, or parts per million (ppm), and is proportional to the difference in shielding of the reference (σref) and the nucleus (σ): δ ∼ σref − σ. A reference substance often used in 1H and 13C NMR spectroscopy is tetramethylsilane (TMS). Main factors influencing the shielding constant in proton NMR spectroscopy include: inductive and resonance effects of substituents, hybridization, hydrogen bonds, steric and electric field effects, solvent effects and contribution from the magnetic anisotropy of neighbouring groups, i.e. anisotropic effects.
Due to different susceptibilities along the three directions in space, chemical bonds are in general magnetically anisotropic. As a consequence, the shielding of a proximal nucleus (σaniz) depends on its spatial position relative to the magnetically anisotropic group. In 1957, McConnell4 modeled the anisotropic effect as the influence of a magnetic dipole (X–Y, Fig. 1) on a point in space at which a proton resides.
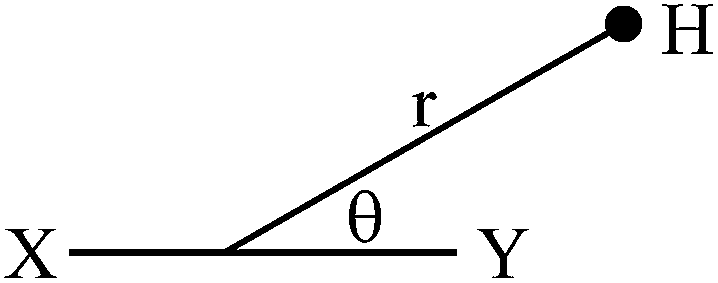 | ||
| Fig. 1 McConnell's point dipole model. | ||
He considered it to be a function of both the distance (r) to the point dipole center and the angle (θ) between the line joining this center to the proton and the direction of the induced magnetic moment. The derived equation can be written as:

Calculation of anisotropic effects
Calculation of anisotropic effects is based on the nucleus independent chemical shift (NICS) concept, introduced by Schleyer and co-workers.6,7 The calculated NICS value represents the magnitude of (de)shielding of a point in the vicinity of the molecule. Two-dimensional and three-dimensional grids of NICS points around molecules are useful to get an insight into diatropic (magnetically shielded) and paratropic (magnetically deshielded) regions of the molecules, their magnitude, and to visualize anisotropic effects.7–28 NICS isosurfaces have been named isochemical shielding surfaces (ICSSs) by Klod and Kleinpeter.16 The ICSSs have been applied to quantify and analyze anisotropic effects,16–22 for stereochemistry determinations,16–21 and to separate the anisotropic effect of functional groups from other influences on proton chemical shifts.23–26 Merino et al.14 and Islas et al.15 calculated and graphically represented the “induced magnetic field” of a molecule as the response to an external magnetic field applied in a specific direction, and found such representations useful to get an information about electron delocalization. The induced magnetic field was calculated according to the equation: Bind = −σBext, where Bext represents external magnetic field, assumed to amount 1 T. Hence, Bind was given in units 1 μT, which is equivalent to 1 ppm of the shielding tensor.14,15 It should be noted that the negative of the NICS values computed at ring centers, 1 Å or 2 Å above the ring center are now widely used to estimate a degree of (anti)aromaticity.6,7Further development of NICS concept resulted in “dissected” NICS techniques which allow one to estimate an orbital contribution to the NICS value and thus to anisotropic effects. Two types of “dissected” NICS analyses exist. The localized molecular orbital dissection (LMO-NICS) separates the total shielding into contributions from bonds, lone pairs and core electrons,8,29 while the CMO-NICS provides contributions from each canonical molecular orbital.30–32 These two methods are complementary. The total NICS are pretty much the same, but their decompositions are different. The CMO-NICS differentiates among the individual canonical orbitals, but it cannot separate the σ framework effects into contributions from C–C and C–H bonds. The LMO-NICS gives the contributions from both C–C (σ) and C–H (σ) bonds, as well as from the localized π bonds. The separation of the total NICS or nuclear shielding into orbital contributions can be done by using the individual gauge for localized orbitals (IGLO) method,33–37 the localized orbital/local origin (LORG) method,38–40 the continuous transformation of the origin of the current density (CTOCD) methods,41 or by using the gauge-including atomic orbital (GIAO) method42,43 in connection with the natural chemical shielding (NCS) analysis44 within the natural bond orbital (NBO) method.45,46
Magnetic shielding47,48 is a second-rank dimensionless tensor fully specified by its diagonal elements, σ11, σ22 and σ33, along its principal axes (the principal axis system, PAS, is the coordinate system for which the tensor is diagonal). Individual tensor components can be characterized by solid-state NMR measurements, where orientation of a molecule is fixed. However, when an NMR spectrum is recorded in solution, rapid molecular tumbling averages magnetic shielding tensor components to the isotropic value, σiso = (σ11 + σ22 + σ33)/3 = (σxx + σyy + σzz)/3, where σxx, σyy and σzz refer to the tensor components in molecular Cartesian axis system. Thus, the calculation of σiso gives us the same information that we obtain from NMR recording in solution. Computation of tensor components provides information about magnitude of (de)shielding/induced magnetic field arising from a specific orientation of the molecule with respect to the direction of the applied magnetic field. For example, if a planar ring lies in the xy plane of the coordinate system, the σzz component (also termed out-of-plane component of the shielding tensor) would correspond to the perpendicular orientation of the molecule relative to the magnetic field, i.e. when magnetic field is applied along the z direction. The σxx and σyy components then correspond to the two parallel orientations, that is, when the field is applied along the x and y axes, respectively.
It is important to realize that NICS values reflect only anisotropic effects. Total through-space shielding experienced by a proximal proton also depends on steric and electric field effects. Martin and co-workers49–51 have shown that in order to assess the total through-space effect it is necessary to include a covalently bonded hydrogen, such as diatomic hydrogen or methane, in the vicinity of the molecule in question. Owing to the presence of the polarizable bond, such covalently bonded hydrogen is sensitive to steric and electric field effects, for which NICS is insensitive. A difference in the isotropic shielding value for proton in an isolated molecule, H2 or CH4, and that for the supramolecule corresponds to the total through-space effect.49–51 The shielding increments obtained in this way are usually mapped as a function of their position in Cartesian coordinates, thus providing a three-dimensional shielding increment surface around a molecule.52–54
New insight into the anisotropic effects and their influence on 1H NMR chemical shifts
Benzene
In the early days of NMR spectroscopy it was recognized that arene protons resonate at higher frequency relative to those in alkenes.55 This difference was interpreted by Pople in terms of the ring current model (RCM),56 which was based on the Pauling–Lonsdale–London π model57–60 used to explain anisotropy of diamagnetic susceptibility of aromatic hydrocarbons that was found to be greater in a direction normal to the molecular plane than in the direction parallel to it.58 According to the RCM, an external magnetic field (B0) acting at right angles to the ring plane induces a ring current in the mobile π electrons. This ring current generates a counter field which reinforces the external field in area around the ring, but opposes it above, below and inside the ring (Fig. 2).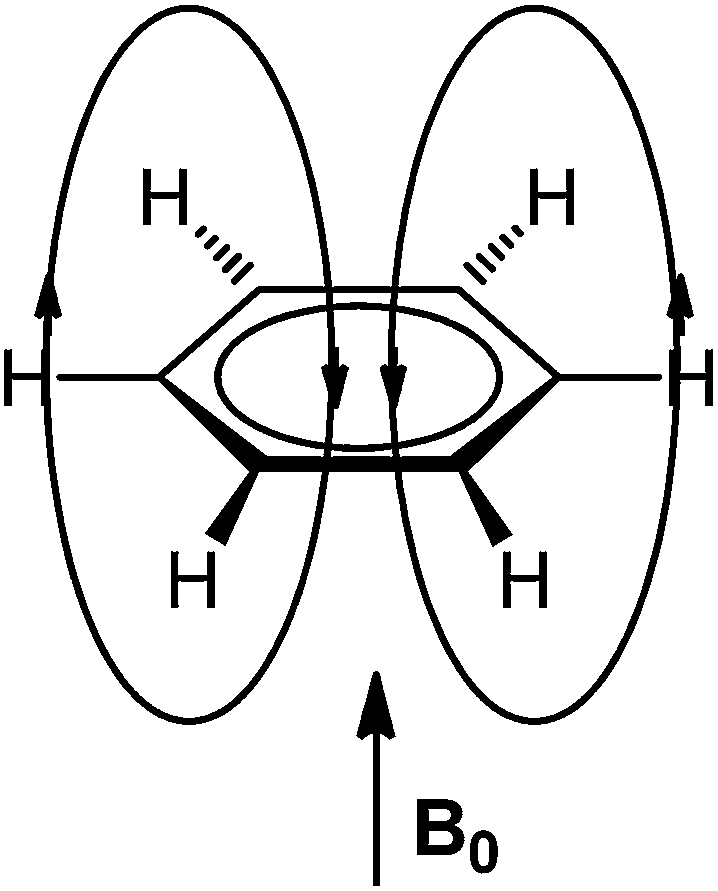 | ||
| Fig. 2 Induced magnetic field of benzene by an external field acting perpendicularly to the ring plane. | ||
This leads to regions of increased and reduced shielding around an arene molecule, with protons located in the deshielding area (Fig. 3).
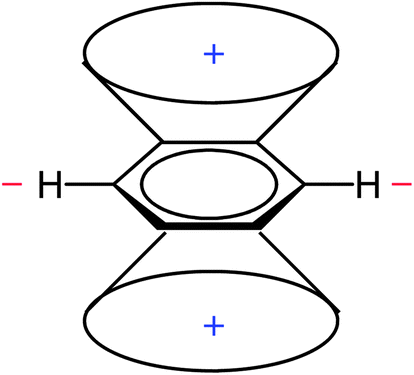 | ||
| Fig. 3 Shielding (denoted by “+”) and deshielding areas (denoted by “−”) around benzene. | ||
The RCM (Fig. 2) and the resulting anisotropy cone (Fig. 3) are presented in almost all NMR spectroscopy textbooks and serve as an explanation of arene proton chemical shifts, which are shifted downfield relative to those of olefins.
The RCM has been supported and criticized.61 The severest criticism was Musher's suggestion that the anisotropic magnetic susceptibility and chemical shift of aromatic hydrocarbons can be correctly represented as a sum of local contributions from both π and σ electrons.62,63 Some support to Musher's “antitheory” has been given by Lazzeretti et al.64,65 and Ferraro et al.66 By computing the magnetic field induced electron current density64 and magnetic shielding density,65,66 the authors concluded that deshielding effect of π electrons on proton chemical shift in benzene is mainly of local character and that the core and σ electrons also play a role in determining the magnitude of total proton shielding. However, their later study has revealed how an apparently localized shielding density map can be a consequence of the delocalized electron flow.67 Current density maps presented by Steiner and Fowler68 have shown that an external magnetic field applied perpendicularly to the benzene ring induces a diatropic ring current in π electrons, characteristic for aromatic molecules, and that this ring current arises from the four HOMO electrons.69–71 An assessment of relative weights of σ and π electron currents in benzene, induced by the perpendicular magnetic field, has shown that the majority of delocalized currents is transported by π electrons, while σ electrons sustain mainly localized currents.72
An analysis of the orbital contributions to the proton shielding tensor of benzene has shown that the π electron ring current is not the sole deshielding effect73 and also not the dominant factor that influences benzene proton chemical shifts.74 Thus, employing the IGLO method for proton shielding tensor dissection, Fleischer et al.74 found that proton chemical shift is mainly determined by local effects (the contribution from the C–H bond in which the proton is involved), and to a much lesser extent by global ones, such as ring current. This finding was, again, some support to Musher's description of magnetic properties of benzene in terms of localized quantities.62,63 The first three-dimensional visualization of benzene anisotropic effects as ICSSs, done by Klod and Kleinpeter in 2001,16 surprisingly revealed that protons are situated in the shielding, rather than the deshielding region (Fig. 4).
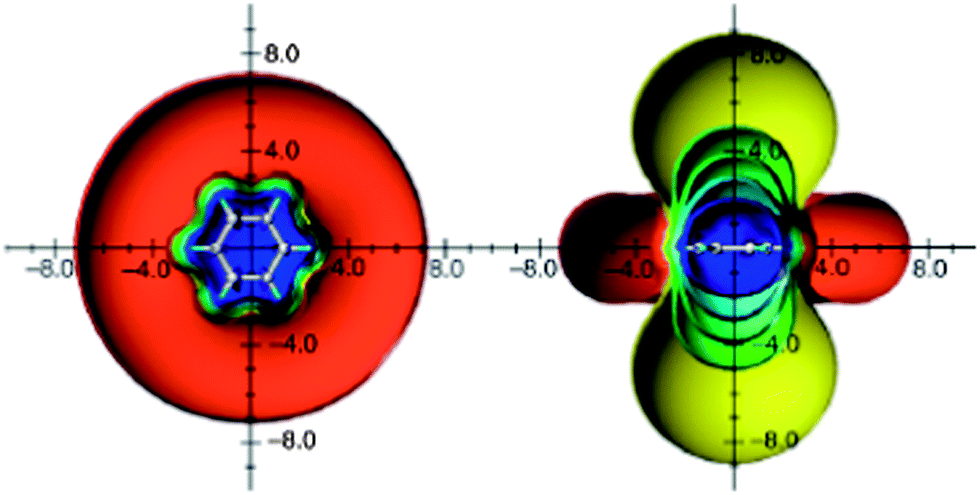 | ||
| Fig. 4 Calculated anisotropic effect of benzene (shielding surfaces at 0.1 ppm in yellow, at 0.5 ppm in green, at 1 ppm in green-blue, at 2 ppm in cyan, and 5 ppm in blue, respectively; deshielding surface at 0.1 ppm in red). View from perpendicular to the molecule and in the plane of the molecule. Reproduced from ref. 16. | ||
At about the same time, Schleyer et al.8 presented a two-dimensional grid of isotropic NICS points around the benzene molecule, which concurred with the results of Klod and Kleinpeter. These graphical representations showed that the deshielding zone in benzene starts approximately 3 Å from the ring center, so that it does not encompass hydrogen atoms. By using the IGLO method, Wannere and Schleyer9 studied orbital contributions to the isotropic values of proton shielding tensor of a series of aromatic, antiaromatic and nonaromatic molecules. In agreement with the previous study,74 they found that the main contribution to the benzene proton shielding comes from the intrinsic C–H bond (25.8 ppm). The most important finding was that π electrons actually shield protons by 1.9 ppm, while deshielding originates from the C–C framework (−3.6 ppm) and to a lesser extent from the rest of the C–H bonds (−0.1 ppm). This finding, certainly, triggered a question: if π electrons shield benzene protons, why then these protons resonate at higher frequency than those attached at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond? The answer given by the authors9 was that benzene protons are shielded less (1.9 ppm) than vinylic protons in ethene (2.6 ppm) and cyclohexene (2.9 ppm). Hence, the difference between benzene and vinyl proton chemical shifts can be accounted for by the less π electron shielding combined with the paratropic contributions from the σ bonds.9 The findings from this study were used as an argument against the RCM model: “The conventional explanation for the unusual downfield chemical shift of arene hydrogens needs a fundamental revision: arene hydrogens are NOT deshielded by ring current effects”.9 However, the authors erroneously ascribed the average (isotropic) π electron effect to the ring current. It is important to make a distinction between the ring current and the average effect, which was clearly done by Viglione et al.11 and Lazzeretti.75 A ring current can be induced only when a magnetic field acts at right angles to the ring plane. Thus, the only component of the magnetic shielding tensor which is biased by ring currents is the out-of-plane component (σzz in the case of benzene). By visualizing the π electron contribution to the out-of-plane component of the tensor at points uniformly distributed around benzene molecule, Viglione et al.11 obtained the classical “anisotropy cone” for benzene (Fig. 5). The same was observed later by Islas et al.15 and Heine et al.76 In fact, the π electron ring current deshields benzene protons by approximately −3 ppm, which means approximately −1 ppm deshielding to the average chemical shift value.11,75 This deshielding is, however, covered by the π electron shielding contributions (positive σ values) coming from the two parallel orientations of the molecule with respect to the magnetic field direction (the average of the two in-plane components is: (σxx + σyy)/2 ≈ 3.6 ppm) resulting in the average π shielding of approximately 1.4 ppm (ref. 11 and 77) which is close to the result obtained by Wannere and Schleyer.9
C double bond? The answer given by the authors9 was that benzene protons are shielded less (1.9 ppm) than vinylic protons in ethene (2.6 ppm) and cyclohexene (2.9 ppm). Hence, the difference between benzene and vinyl proton chemical shifts can be accounted for by the less π electron shielding combined with the paratropic contributions from the σ bonds.9 The findings from this study were used as an argument against the RCM model: “The conventional explanation for the unusual downfield chemical shift of arene hydrogens needs a fundamental revision: arene hydrogens are NOT deshielded by ring current effects”.9 However, the authors erroneously ascribed the average (isotropic) π electron effect to the ring current. It is important to make a distinction between the ring current and the average effect, which was clearly done by Viglione et al.11 and Lazzeretti.75 A ring current can be induced only when a magnetic field acts at right angles to the ring plane. Thus, the only component of the magnetic shielding tensor which is biased by ring currents is the out-of-plane component (σzz in the case of benzene). By visualizing the π electron contribution to the out-of-plane component of the tensor at points uniformly distributed around benzene molecule, Viglione et al.11 obtained the classical “anisotropy cone” for benzene (Fig. 5). The same was observed later by Islas et al.15 and Heine et al.76 In fact, the π electron ring current deshields benzene protons by approximately −3 ppm, which means approximately −1 ppm deshielding to the average chemical shift value.11,75 This deshielding is, however, covered by the π electron shielding contributions (positive σ values) coming from the two parallel orientations of the molecule with respect to the magnetic field direction (the average of the two in-plane components is: (σxx + σyy)/2 ≈ 3.6 ppm) resulting in the average π shielding of approximately 1.4 ppm (ref. 11 and 77) which is close to the result obtained by Wannere and Schleyer.9
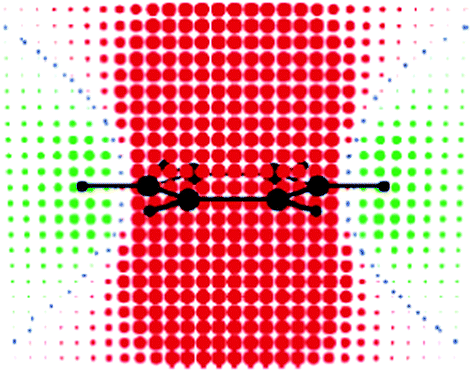 | ||
| Fig. 5 Benzene: π-electron contribution to σ‖ at points uniformly distributed in space (0.5 b apart in each direction) on a symmetry plane containing the C6 axis of symmetry and a pair of C–H bonds. Red and green circular spots represent shielding (positive) and deshielding (negative) zones, respectively. The radius of each spot is proportional to the absolute value of the contribution. Small blue spots indicate points in space where the contribution is vanishing. Reprinted with permission from ref. 11. Copyright 2004, American Chemical Society. | ||
A conclusion to be reached at, by now, is that while benzene protons do lie in the deshielding zone of the π electron ring current, as conventionally interpreted, this effect is small and overcome by π shielding contributions from other orientations of the molecule relative to magnetic field and by the σ framework effects, the most prominent being the intrinsic C–H bond shielding. Overall, benzene protons are shielded by all electrons, that is lie in the isotropic shielding zone.78 Even though it is small, the ring current effect (approximately −1 ppm to the average chemical shift) can account for the chemical shift difference between benzene (δ = 7.3 ppm) and fully localized cyclohexatriene (calculated δ = 6.2 ppm).79
Now, what makes the deshielding zone around benzene (Fig. 4)? An evaluation of the orbital contributions (both LMO and CMO) to the isotropic NICS values at points located along the two in-plane Cartesian axes (x and y), with a step width of 1 Å, was done by Baranac-Stojanović et al.80 employing the NCS analysis to the GIAO calculated shielding values. This study showed that the π electron shielding effect reaches up to the end of the deshielding zone, which actually arises from the σ electron effects, overcoming the π electron shielding. The same analysis applied to the z axis, coinciding with the C6 symmetry axis, revealed that both σ and π electrons contribute to the shielding zone above/below the benzene ring. The π electron contribution is significant up to 2–3 Å, then it weakens and is overcome by the σ electron effects. In fact, the π electron shielding ends up at approximately 5 Å. Farther away, a weak deshielding was observed.80 These results are in sharp contrast with the conventional interpretations of benzene anisotropic effects, but do not dispute the ring current model. As already pointed out, a ring current is induced only by perpendicular magnetic field. Several computational studies have shown that it does deshield the in-plane area around benzene.11,15,22,76 However, in a real NMR experiment, molecular tumbling averages the magnetic shielding tensor components and isotropic shielding is the quantity which we observe. It should also be noted that even in the perpendicular orientation of the molecule with respect to the magnetic field, the benzene ring current is not the only deshielding source. The σ framework effects deshield the in-plane area around benzene to even higher degree than the ring current.22
Borazine
Borazine (B3N3H6) is an inorganic counterpart of benzene, planar, with equal B–N bond lengths and six π electrons (nitrogen lone pairs). Aromaticity of borazine was a long debated issue, resulted in its description as nonaromatic or weakly aromatic.15,29,72,79,81–88 This is a consequence of the large electronegativity difference between boron (2.0) and nitrogen (3.0) which places the six π electrons formally on nitrogen atoms. Whereas anisotropic effects of carbon-containing groups and rings are discussed in every NMR spectroscopy textbook,1–3 there is a lack of data on purely inorganic rings. Hence, anisotropic effects of borazine were visualized as ICSSs (Fig. 6) and discussed only recently.22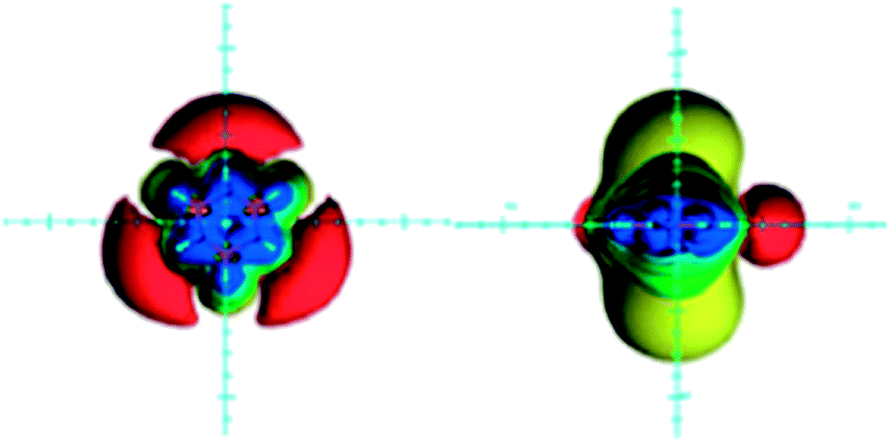 | ||
| Fig. 6 Visualization of the anisotropic effects of borazine, in-plane view on the left and side view on the right. ICSSs: blue represents 5 ppm shielding, cyan 2 ppm shielding, green-blue 1 ppm shielding, green 0.5 ppm shielding, yellow 0.1 ppm shielding, orange −0.5 ppm deshielding, red −0.1 ppm deshielding. Reproduced from ref. 22. Copyright 2012, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
There are two main differences between anisotropic effects of benzene and borazine (compare Fig. 4 and 6): (1) deshielding belt existing around the benzene ring is replaced with the three separated deshielding surfaces located near the nitrogen atoms of borazine, (2) the extent of the in-plane deshielding and the shielding above/below the borazine ring is reduced compared to benzene. Although, the three isolated in-plane deshielding areas may point to the deshielding effect coming from the local circulations of π electrons in an external magnetic field, this is not the case. The NCS analysis of the GIAO calculated isotropic shielding values at points located along the two in-plane axes (x and y), separated by 1 Å, revealed that the in-plane region is shielded by π electrons and deshielded by σ electrons, which obviously dominates in the regions around nitrogen atoms.22 This finding compares with the results for benzene.80 At the boron side, however, the B–N bonds deshielding effect is covered by the B–H bond shielding, which is most prominent when the B–H bond is parallel with the magnetic field direction. As a consequence, the deshielding belt is disrupted.22
The region above/below the borazine ring is shielded by π electrons only up to 1–2 Å, while deshielding is observed farther away. Thus, the net shielding starting from that distance is caused by the σ electron effects.22 The shorter range of borazine anisotropic effects, compared to benzene, mainly stems from the transition from the global ring current in benzene to mostly localized π electron circulations in borazine, induced by the perpendicular magnetic field.81,84–86,88 This reduces the π electron contributions to the out-of-plane component of the shielding tensor (deshielding in-plane around borazine, shielding above/below the ring).22 The same was observed by Islas et al.15,87 By computing the induced magnetic field of borazine π electrons as a response to the perpendicular magnetic field, the authors found it similar in shape to that of benzene, but smaller in magnitude.15,87 Consequently, the π electron contributions to the isotropic shielding values are dominated more by the two in-plane components of the tensor, the effect of which (shielding in-plane around borazine, deshielding above/below the ring from 1–2 Å and away) is opposite to that of the out-of-plane component. Since the average π electron effects oppose those of the σ electrons (deshielding in-plane around borazine, shielding above/below the ring), the magnitude of the total anisotropic effects of borazine is smaller with respect to benzene.22 These anisotropic effects, like those of benzene, are actually dominated by the (de)shielding contributions from the σ framework.22,80 The calculated NICS(π) isolines also showed that the effects of currents induced by magnetic fields parallel to the ring are not negligible, and that they have an impact on the total shielding cone.87
Borazine hydrogen atoms are less deshielded by π electrons than benzene protons when a magnetic field is applied perpendicularly to the ring plane (σπzz (CH) = −2.91 ppm, σπzz (BH) = −0.98 ppm, σπzz (NH) = −0.61 ppm). This, again, reflects the local nature of π electron circulations around nitrogen atoms.22
Cyclobutadiene
Although cyclobutadiene is widely accepted as a typical antiaromatic compound, its antiaromaticity has been a topic of discussion.89–91 It has been shown recently that the high energy of cyclobutadiene originates from angle strain, torsional strain, π–π and σ–σ repulsion, rather than from the antiaromatic destabilization.91 Nevertheless, cyclobutadiene is magnetically antiaromatic,92 displaying shielding in the plane surrounding the ring and deshielding above/below and inside the ring (Fig. 7).14,15,22 An NCS analysis of magnetic shielding values at points located along the three Cartesian axes revealed that, in this case, the π electron shielding contributions follow the prediction: the in-plane region is shielded by π electrons, while zones above/below the ring are deshielded. These π electron effects overcome the opposing σ electron in-plane deshielding and above/below the plane shielding contributions.80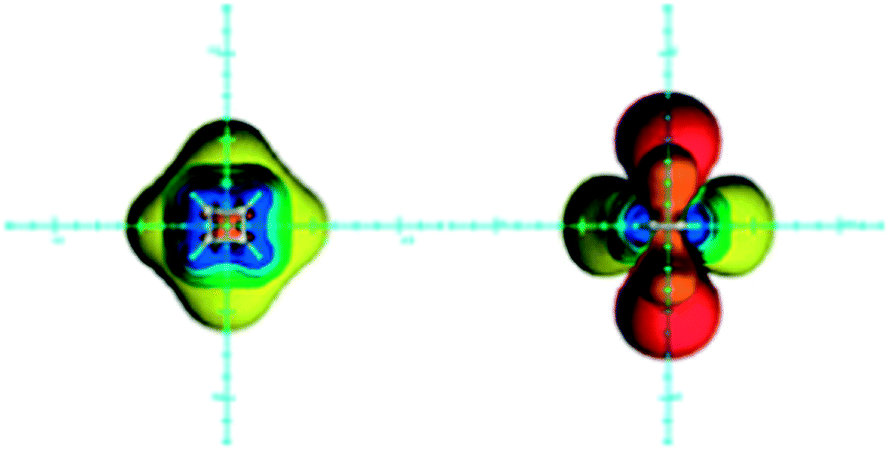 | ||
| Fig. 7 Visualization of the anisotropic effects of cyclobutadiene, in-plane view on the left and side view on the right, taken from the side of the double bond. ICSSs: blue represents 5 ppm shielding, cyan 2 ppm shielding, green-blue 1 ppm shielding, green 0.5 ppm shielding, yellow 0.1 ppm shielding, orange −0.5 ppm deshielding, red −0.1 ppm deshielding. Reproduced from ref. 22. Copyright 2012, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Visualization of the induced magnetic field of π electrons as a response to an external field applied in the perpendicular direction also showed a (de)shielding pattern typical for an antiaromatic system.76 However, it is interesting to note that the influence of the paramagnetic ring current of cyclobutadiene to the average anisotropic effects in plane surrounding the ring is surprisingly small. At the side of the C–C single bonds, it compares in magnitude to the π electron shielding contributions coming from the two parallel orientations of the molecule with respect to the magnetic field direction. At the side of the CC double bonds it is highly overcome by the π contributions arising from one of the parallel orientations in which the magnetic field direction coincides with the C2 symmetry axis bisecting the CC bonds.22
A puzzling observation related to cyclobutadiene 1H NMR chemical shift was that even though it is magnetically antiaromatic, its proton chemical shift is olefin-like (5.7 ppm) rather than shifted to the lower frequency. This anomaly has been rationalized by employing the block-localized wave function for the computation of NMR chemical shifts. Thus, compared to the 1H NMR chemical shift of an ideal non-antiaromatic cyclobutadiene (7.5 ppm), its real proton chemical shift is found at lower frequency.79
A net deshielding above/below the molecular plane of cyclobutadiene was explained to arise from two deshielding effects: the additive deshielding effects of the two π bonds and paratropic ring current.116
Ethene and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C double bond
C double bond
According to the classical interpretations and “anisotropy cone” depictions,1–3 the π electron circulations induced by a magnetic field perpendicular to the plane containing the sp2-hybrid orbitals deshields the in-plane region around the CC double bond and shields the area above/below the bond plane (Fig. 8). It is believed that hydrogen atoms attached at the CC double bond reside in this deshielding zone.
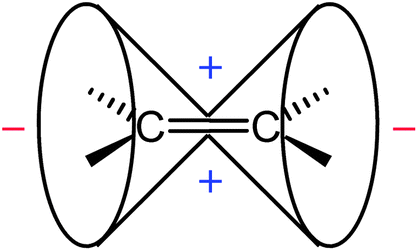 | ||
| Fig. 8 Anisotropy cone of ethene, as represented in NMR spectroscopy textbooks (“−” indicates deshielding, “+” denotes shielding). | ||
Modern quantum-chemical calculations proved the existence of the shielding zone above/below the CC double bond of ethene and deshielding area in the plane around the bond. Fig. 9 portrays anisotropic effects of ethene, visualized as ICSSs.16,25 The magnitude of the deshielding along the bond is obviously too small (less than −0.1 ppm) and is not presented.
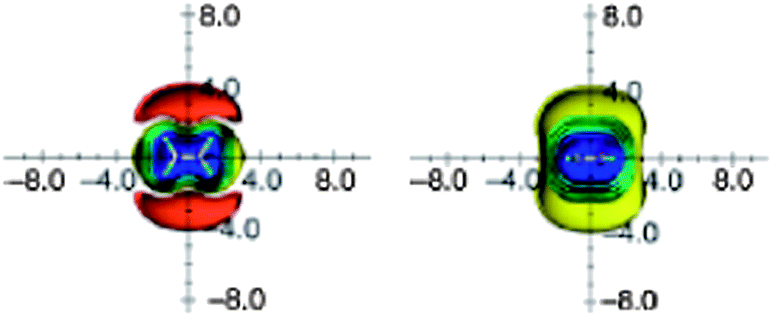 | ||
| Fig. 9 Calculated anisotropy effect of the double bond: ethylene (shielding surfaces at 0.1 ppm yellow, at 0.5 ppm in green, at 1 ppm in green-blue, at 2 ppm in cyan, at 5 ppm in blue, respectively; deshielding surface at 0.1 ppm in red); view from perpendicular to the molecule (left) and in the plane of the molecule (right). Reproduced from ref. 16. | ||
Opposite to what is believed, ethene protons lie in the shielding, isotropic zone (Fig. 9). The main shielding contribution comes from the same C–H bond in which the proton is involved.9,74 The study of Fleischer et al.74 and later of Wannere and Schleyer9 showed that, on average, π electrons of the double bond in ethene and cyclohexene shield the vinylic protons by σπiso = 2.5–2.6 ppm (ref. 9 and 74) and σπiso = 2.9 ppm,9 respectively. Even though the familiar anisotropy cone for the CC double bond is easily recognizable in the visualization of the π electron response to the perpendicular magnetic field,11,76Fig. 10, the influence of such π electron currents on proton chemical shift in ethene is negligible (−0.02–0.17 ppm),11,74,76 since protons are positioned almost on the anisotropy cone. The contribution of π electrons to the vinylic hydrogen chemical shift is thus just shielding,9,11,74 originating from the two parallel orientations of the molecule relative to the magnetic field direction.11,74
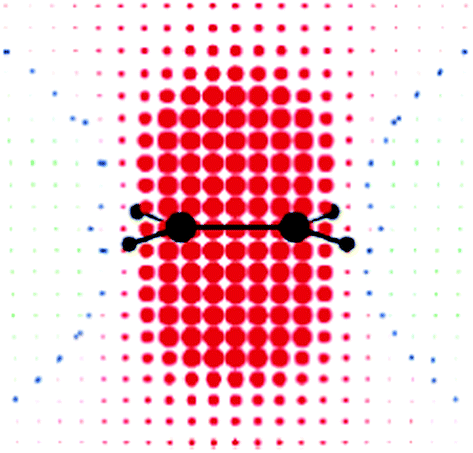 | ||
| Fig. 10 Ethene: π-electron contribution to σ‖ at points uniformly distributed in space (0.5 b apart in each direction) on a symmetry plane orthogonal to the molecular plane, containing the C2 axis of symmetry and the C–C bond. Red and green circular spots represent shielding (positive) and deshielding (negative) zones, respectively. The radius of each spot is proportional to the absolute value of the contribution. Small blue spots indicate points in space where the contribution is vanishing. Reprinted with permission from ref. 11. Copyright 2004, American Chemical Society. | ||
Hence, contrary to the classical explanations, ethene hydrogen atoms are not deshielded by π electrons, but rather by the C–C σ bond and the C–H bonds other than the one in which the proton is involved, σiso (C–C) ≈ −2 ppm,9,74 and σiso (C–H) = −0.8 ppm.9 Though, overall, they are shielded by all electrons.
Furthermore, the deshielding zone around ethene, which certainly exists16,25,80 (Fig. 9), does not originate from the π electron effects, which are actually shielding, but from the σ electron deshielding contributions.80 The π electron contribution to the total shielding above/below the plane of the CC double bond is significant up to approximately 1 Å. Farther away, their contribution is deshielding. Thus, the total shielding zone above/below the plane of the double bond results from the σ electron shielding effects. Consequently, σ electrons, not π electrons, are responsible for the net anisotropy of the CC double bond.80
Norbornene (1, Fig. 11) found its place in NMR spectroscopy textbooks as a nice example for the magnetic anisotropy of the CC double bond: syn and endo protons fall into the shielding zone of the classical anisotropy cone (Fig. 8), thereby their δ values are shifted to lower frequency with respect to those of the anti and exo protons.2 In fact, this anisotropy cone, whatever its real origin is, predicts that any nucleus situated above the double bond is shielded, and that which is in the plane of the double bond is deshielded.
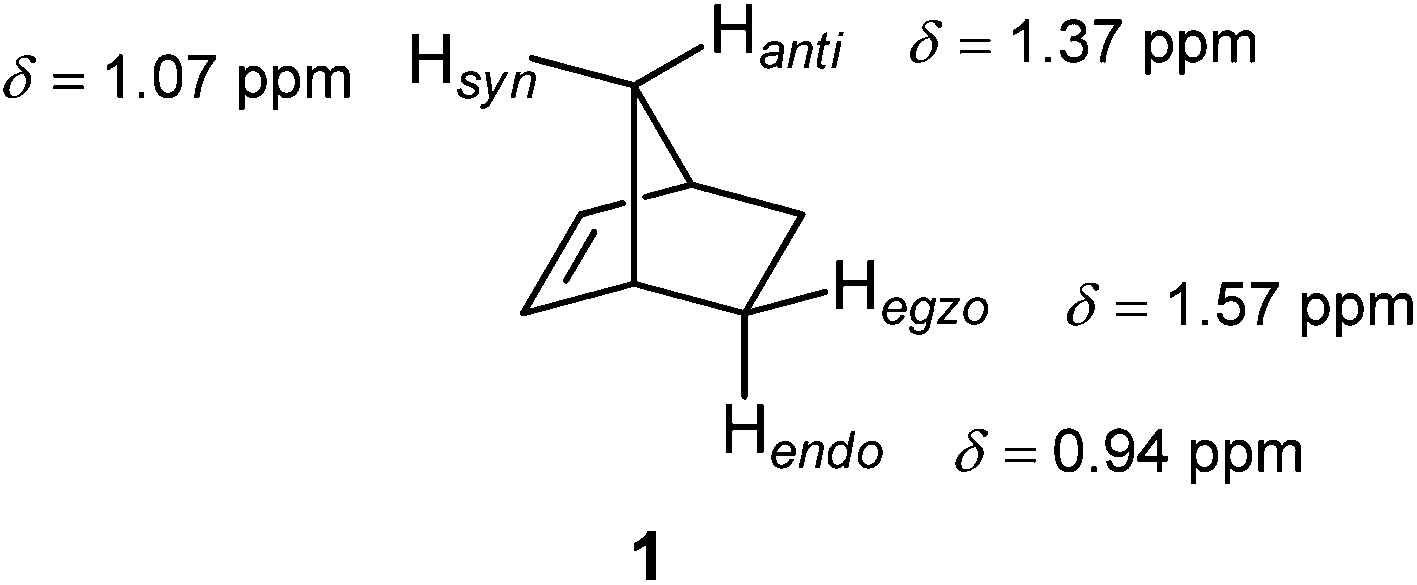 | ||
| Fig. 11 Proton chemical shifts in norbornene traditionally used to illustrate the magnetic anisotropy of the CC double bond.2 | ||
Tori et al.93 assigned the Hsyn of norbornene to higher field compared to Hanti, and this difference rationalized by the magnetic anisotropy of the CC double bond. However, in the second paper, Tori and co-workers94 reversed their initial assignment. Subsequent deuteration studies unequivocally assigned the Hsyn to higher frequency.95,96 Franzus et al.95 suggested that small variations in molecular geometry in norbornene derivatives can cause the Hsyn to fall either into shielding or deshielding zone, since Hsyn is shielded in some compounds, but deshielded in others. These latter assignments of the bridge methylene proton resonances in norbornene were further confirmed by high-field NMR measurements and computer simulations.97 A computational DFT-GIAO (density functional theory-gauge including atomic orbitals) study of Alkorta and Elguero98 showed that a methane hydrogen atoms positioned in the plane of the double bond at a distance of 3.7 Å at its side and along the bond experienced deshielding of −0.06 ppm and −0.11 ppm, respectively. However, a methane proton placed 2.5 Å above the CC double bond of ethene was deshielded, too, by −1.27 ppm.98 The origin of this deshielding, which strongly opposes the traditional anisotropy cone predictions, was suggested by Martin et al.53 to arise from steric interactions, not from anisotropic effects. While anisotropy cone predicts only the effect of the magnetic anisotropy, Martin and Brown99 presented a new model for the net through-space effects of the CC double bond (Fig. 12). It is clearly seen from the model that a proton located above the double bond at a distance which is less than ∼3 Å, depending on its actual position, is deshielded. An “unusual” deshielding of the bridge methylene protons in norbornadiene, observed by Tori et al.,93 might serve as an experimental proof for this steric effect which obviously dominate the anisotropic effect at short distances. At larger distances, proton chemical shifts are influenced only by the bond anisotropy which means the shielding effect.100 A proton situated in the plane of the CC double bond will always be deshielded.100
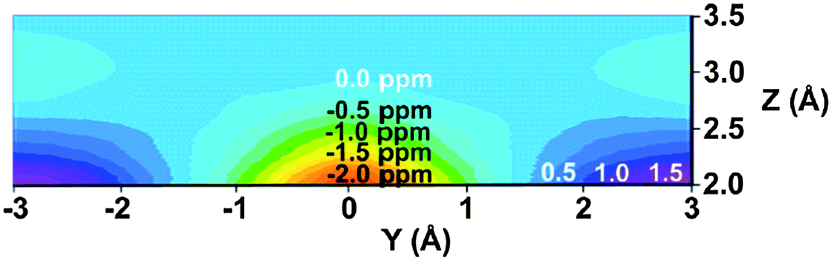 | ||
| Fig. 12 (De)shielding isosurface above the plane of ethene (Z direction) along the CC bond (Y) axis predicted by GIAO-SCF calculations (predicted shielding in ppm indicated on the graph; positive (white) numbers represent shielding, negative (black) numbers represent deshielding). Reproduced from ref. 99. Copyright 2000, authors. Published by MDPI. | ||
Compound 2 provided a test for the through-space effects of the CC double bond. Thus, Hsyn is located almost vertically above the olefinic bond, at a distance of approximately 2 Å. Whereas the magnetic anisotropy does shield this proton,25 its strongly deshielded position arises from the steric strain.25,100
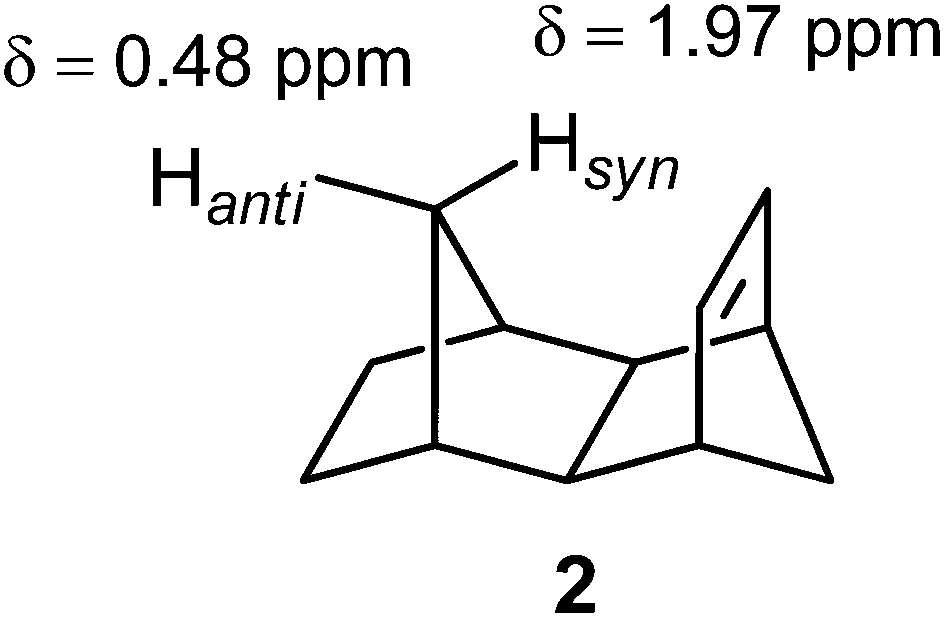
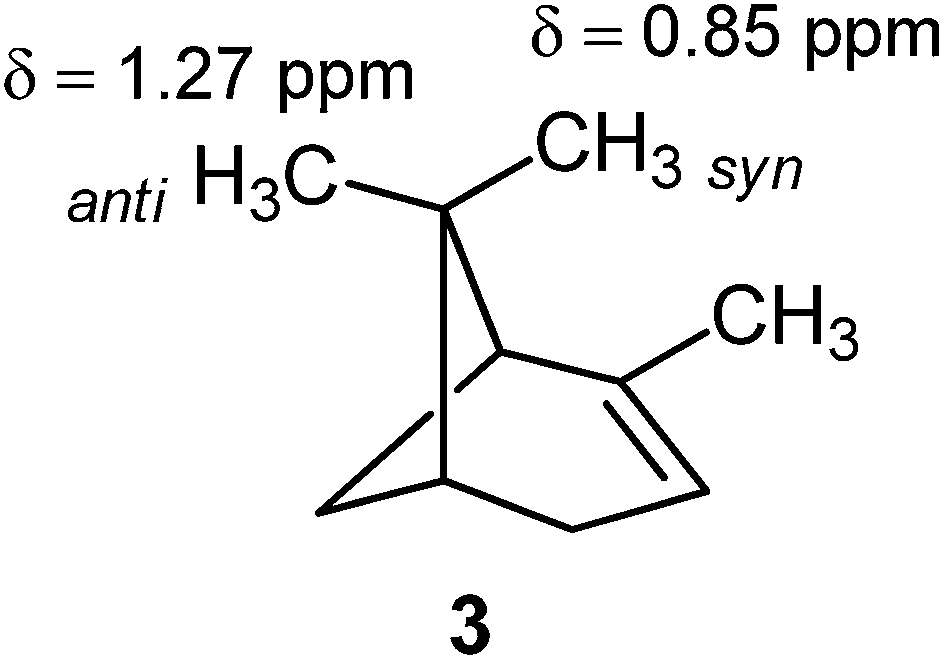
However, this is not always the case. For example, the syn methyl group in α-pinene (3) is shielded with respect to the anti methyl group.1,100
Ethyne and the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C triple bond
C triple bond
The unusually low chemical shift of protons bound to an sp-hybridized carbon of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond (2–3 ppm vs. 5–6 ppm for vinylic protons) is explained by the magnetic anisotropy of the bond. Thus, a magnetic field applied parallel to the triple bond induces the π electron currents flowing around the bond. These currents, in turn, create a secondary field reinforcing the external magnetic field around the triple bond, but opposing it along the bond. The resulting, familiar anisotropy cone, showing (de)shielding regions around the CC triple bond, is presented in Fig. 13.
C triple bond (2–3 ppm vs. 5–6 ppm for vinylic protons) is explained by the magnetic anisotropy of the bond. Thus, a magnetic field applied parallel to the triple bond induces the π electron currents flowing around the bond. These currents, in turn, create a secondary field reinforcing the external magnetic field around the triple bond, but opposing it along the bond. The resulting, familiar anisotropy cone, showing (de)shielding regions around the CC triple bond, is presented in Fig. 13.
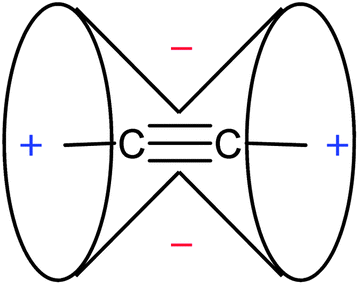 | ||
| Fig. 13 Anisotropy cone for the CC triple bond (“−” indicates deshielding, “+” denotes shielding). | ||
Theoretical calculations supported this explanation.11,76,98 Visualizations of the induced magnetic field of π electrons, as well as those of σ and all-electrons (σ + π), as a response to the external field applied parallel to the bond, all matched the classical anisotropy cone.76 Ethyne hydrogen atoms were found inside the shielding zone of both π and σ electrons. While π electrons shield the proton by 13.565 ppm in this orientation, the shielding influence of core and σ electrons is twice as much, 27.133 ppm.11 Computational study of Alkorta and Elguero98 showed that a methane proton placed perpendicularly to the CC triple bond at a distance of 2.5 Å is deshielded by −1.54 ppm, while methane proton positioned 3.7 Å along the bond is shielded by 0.44 ppm, which is again in agreement with the anisotropy cone predictions.
However, molecular tumbling in solution lowers the magnitude of these effects. Ethyne anisotropic effects, calculated on the basis of the isotropic (average) shielding values and visualized as ICSSs, are presented in Fig. 14. The most striking feature of these pictures is very weak and spatially restricted deshielding area around the triple bond.16
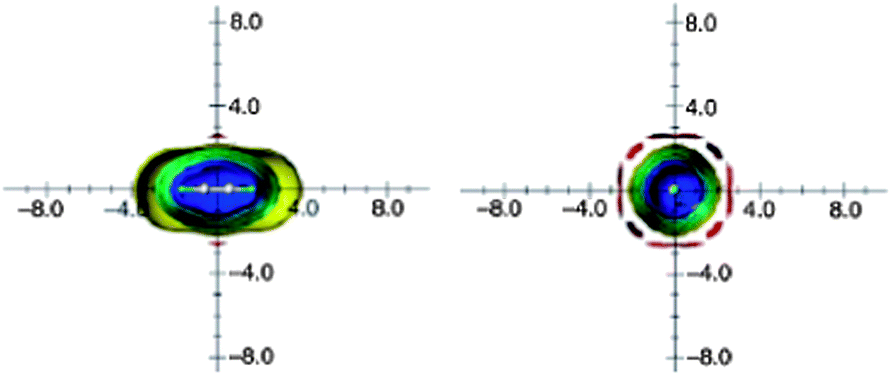 | ||
| Fig. 14 Calculated anisotropy effect of the triple bond: acetylene (shielding surfaces at 0.1 ppm in yellow, at 0.5 ppm in green, at 1 ppm in green-blue, at 2 ppm in cyan, and 5 ppm in blue, respectively; deshielding surface at 0.1 ppm in red). View from perpendicular to the bond axis (left) and along the bond axis (right). Reproduced from ref. 16. | ||
The deshielding of H(5) in 4-ethynylphenantrene (5) by 1.6 ppm with respect to H(4) in phenanthrene (4), observed by Mallory and Baker,101 has long been considered as a prime example of the anisotropic effects of the CC triple bond. It was believed that the H(5) in 5 rested in the deshielding region of the conventional anisotropy cone (Fig. 13).
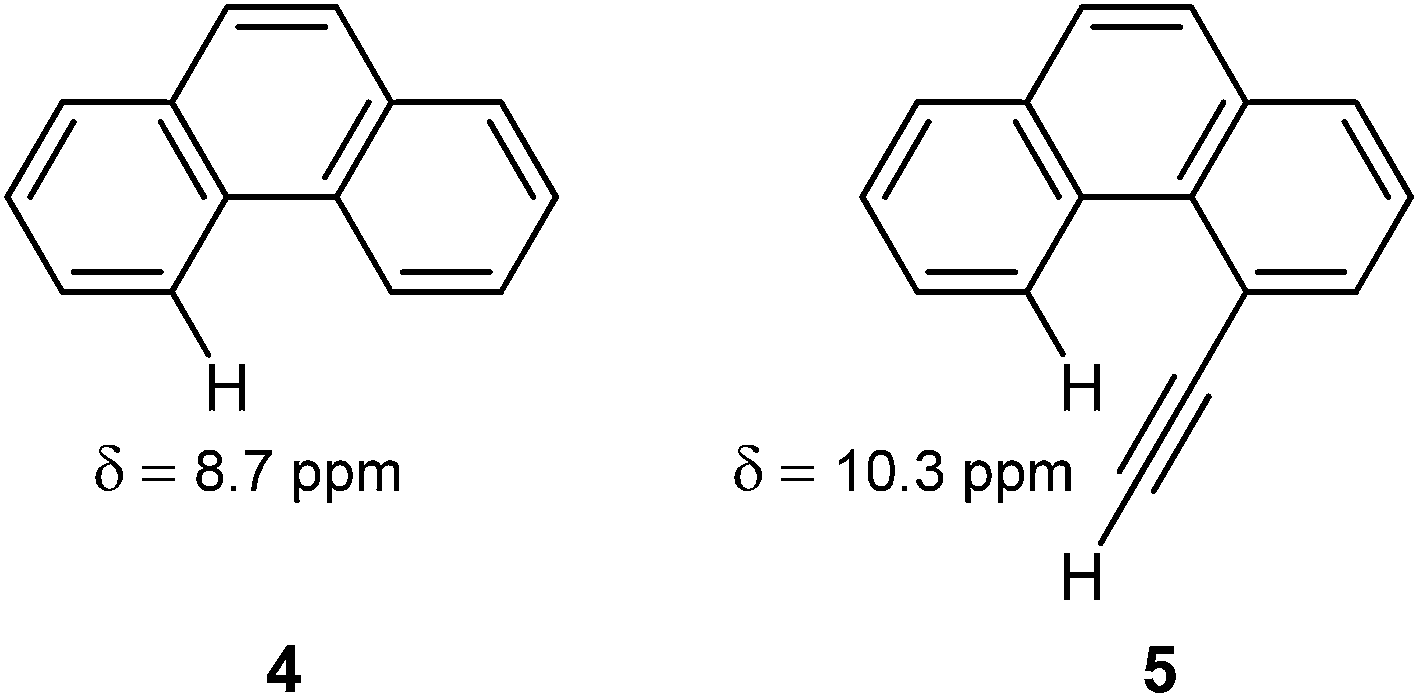
However, the very weak deshielding area around the triple bond, observed by Klod and Kleinpeter (Fig. 14),16 questioned the explanations based on this anisotropy cone. They suggested steric compression, and not the anisotropic effect, as the real reason for the downfield shift of H(5) in 5.16 By using the CHARGE7 model, which discriminates between substituent electronic and long-range effects (electric fields, anisotropic and steric effects), Abraham and Reid102 found that proton chemical shifts of different acetylenic compounds are influenced by both anisotropic and steric effects of the acetylene group. In particular, they predicted that steric effects dominate in the case of 4-ethynylphenantrene.102 The following study of Kleinpeter and Klod,23 by NCS analysis applied to the GIAO calculated shieldings, estimated that magnetic anisotropy of the triple bond affects the chemical shift of H(5) in 5 by only 0.06 ppm, the rest of the deshielding originates from the steric interaction.
The small averaged anisotropy of ethyne also led Wannere and Schleyer9 to a conclusion that there is no indication of the shielding-deshielding cone. They found that shielding of hydrogen atoms per π bond is the same for ethene (2.6 ppm) and ethyne (2.5 ppm), that is the π electron effect is additive rather than enhanced in ethyne.9 In fact, as discussed before, there is a shielding cone, but only for parallel orientation of the triple bond with respect to the magnetic field direction (as originally assumed). This shielding cone is, however, highly reduced due to the molecular tumbling in solution.
Ethane and the C–C single bond
The anisotropy cone for the C–C single bond, as presented in NMR spectroscopy textbooks, is shown in Fig. 15. It predicts shielding at the side of the bond and deshielding along its end. | ||
| Fig. 15 Anisotropy cone for the C–C single bond (“−” indicates deshielding, “+” denotes shielding). | ||
A shift to a higher frequency in the series methane (CH4, δ = 0.23 ppm), ethane (CH3CH3, δ = 0.86 ppm), propane (CH3CH2CH3, δ = 1.33 ppm) and isobutane ((CH3)3CH, δ = 1.56 ppm) is traditionally attributed to deshielding effect of one, two and three C–C bonds, respectively.
This classical “anisotropy cone” picture was first reversed in the computational study of Alkorta and Elguero.98 They found that a methane proton residing 2.5 Å at the side of the C–C bond in ethane is deshielded by −0.6 ppm, while the one positioned collinearly with the bond, at a distance of 3.7 Å from the bond is shielded by 0.04 ppm. The authors also commented on possible influences of the six C–H bonds on the observed (de)shielding values.98 Baranac-Stojanović and Stojanović13 excluded the C–H bond effects by employing the NCS analysis on the GIAO calculated NICS values at points around ethane, thus revealing the anisotropy of the C–C bond only (Fig. 16).13 Visualizations are given in the plane containing the HCCH bonds (left) and the one perpendicular to it (right).
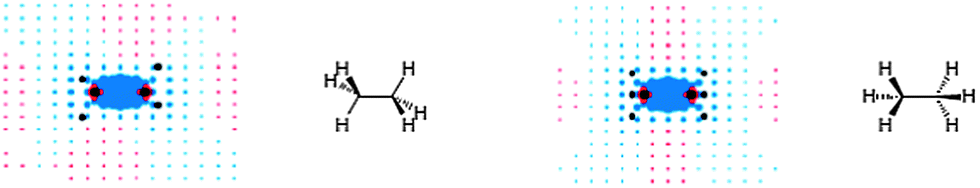 | ||
| Fig. 16 Visualization of anisotropic effects of the C–C single bond in ethane: view in the plane containing the HCCH bonds (left) and in the plane perpendicular to it (right). Blue and red points denote shielding and deshielding effects, respectively. The radius of points is proportional to the absolute value of the contribution (the points merge into one another near the bond). Positions of carbon and hydrogen nuclei are marked by black points. Reproduced from ref. 13. Copyright 2013, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
These pictures are apparently opposite to the classical anisotropy cone (Fig. 15). Although, deshielding is also seen along the bond, which is in accord with the classical model, its magnitude is very low and does not exceed −0.05 ppm. In fact, magnetic anisotropy of the C–C single bond is small and becomes insignificant ∼2.5–3 Å from the bond center and away in all directions.13 This agrees with conclusions of Abraham and co-workers103 who found that the C–C single bond anisotropy does not in general contribute significantly to proton chemical shifts. By visualizing the anisotropic effects as ICSSs, Kleinpeter and co-workers17 found only shielding effect along the C–C bond in ethane. However, ICSSs did not show the deshielding zone near the side of the bond, as is seen in Fig. 16. The asymmetry of the shielding pattern in the HCCH plane (Fig. 16, left) was ascribed either to an orbital distorsion in the HCCH planes occurring in an external magnetic field, or to a formation of common electron currents between the C–C and C–H bonds assuming magnetic field perpendicular to the bond, in addition to local electron circulations within the bonds.13 This anisotropic effect is also environment dependent and differs for the staggered and eclipsed conformation of ethane in the HCCH planes, as well as for cyclohexane,13 as will be discussed later.
It turned out that the C–C single bond does not deshield methyl, methylene and methyne hydrogen atoms as is conventionally explained in NMR spectroscopy textbooks. They are shielded by the C–C bond(s), but less than by the C–H bonds attached at the same carbon.13 Previous IGLO calculations of the proton shielding tensor of ethane yielded similar shielding contributions of different localized orbitals9,35 as obtained in ref. 13, but they were not further discussed in light of the anisotropy of the C–C bond.
Magnetic anisotropy of the C–C single bond is generally accepted to be the source of the chemical shift difference between axial and equatorial protons of a rigid cyclohexane ring, which may range from 0.1 to 0.7 ppm. Thus, axial hydrogen atoms are thought to lie within the shielding region of the classical anisotropy cone (Fig. 15) of the C(2)–C(3) and C(5)–C(6) bonds, whereas equatorial protons are situated in its deshielding zone (Fig. 17).
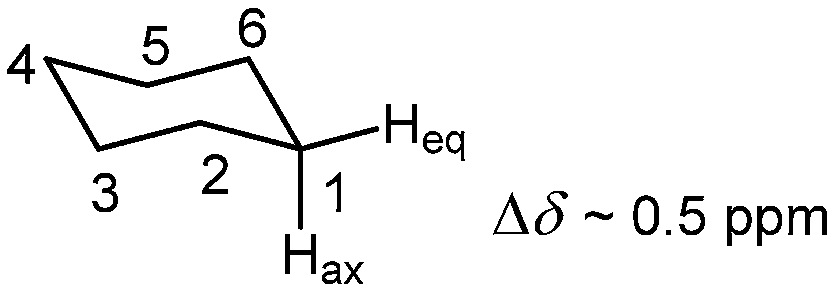 | ||
| Fig. 17 Chemical shift difference between axial and equatorial protons of cyclohexane. The difference may range from Δδ = 0.1–0.7 ppm. | ||
Abraham et al.,104 however, found that other factors, too, are responsible for this difference in chemical shifts. While equatorial proton is deshielded by the C–C bond anisotropic effects, the axial proton is shielded by the two synaxial hydrogen atoms by approximately the same amount as the sum of the magnetic anisotropy of the Cβ–Cγ and Cγ–Cδ bonds, with a smaller electric field effect.104 Later refinement of their CHARGE scheme103 predicted this Δδ difference still as a multifunctional, but caused mostly by steric and electric field effects. The electric field component was actually found to be similar for the two protons, but the steric shielding effect coming from the two axial C(3)–H and C(5)–H bonds was greater at the axial position. The Cδ provided the only steric deshielding term, again larger at the axial proton.103 Klod et al.,17 too, argued against the classical explanation, but found hyperconjugation as a dominant factor determining the Hax/Heq chemical shift difference. By visualizing the cyclohexane C–C bond and the CC framework response to an applied magnetic field, Baranac-Stojanović and Stojanović13 concluded that the anisotropy of a single C–C bond is significantly altered by the formation of common electron currents between the C–C bonds when the magnetic field direction coincides with the C3 symmetry axis. In this orientation, the CC framework shields the Hax by σ = 4.20 ppm and deshields the Heq by σ = −0.45 ppm, thus contributing 4.65/3 = 1.55 ppm to the chemical shift difference, which is greater than actually observed. In the other two orientations of the cyclohexane ring relative to a magnetic field direction the two adjacent C(1)–C(2) and C(1)–C(6) bonds increase the difference to Δδ = 2.19 ppm. The contribution of the C(2)–C(3) and C(5)–C(6) bonds to the chemical shift difference of Hax and Heq in these orientations is pretty small, 0.06 ppm. The influences of the C–H bonds in which the Hax and Heq are involved increase the difference further to Δδ = 2.42 ppm. Now, deshielding contributions from the other C–H bonds and core electrons diminish this value to the one experimentally observed.13 Thus, it is not the anisotropy of the C(2)–C(3) and C(5)–C(6) bonds that is responsible for the Hax/Heq chemical shift difference, but magnetic contributions from all bonds.
Cyclopropane
The chemical shift of cyclopropane hydrogen atoms is unusually low, δ = 0.22 ppm versus δ = 1.44–1.45 ppm for cyclopentane and larger cycloalkanes. This is conventionally accounted for in two ways. According to the first explanation, it is the anisotropy of the C–C bond, which is just opposite to CH2 group, that shields the cyclopropane protons. However, the study of Baranac-Stojanović and Stojanović13 on magnetic anisotropy of the C–C single bond revealed that a proton located at its side would be deshielded, or shielded by at most 0.02 ppm if eclipsed conformation is taken into account. Hence, this first explanation does not seem appropriate. The second explanation is based on an aromatic-like ring current involving the six electrons in the three C–C bonds,105,106 the so-called σ aromaticity. This σ electron ring current is believed to shield the cyclopropane protons. The concept of σ aromaticity in cyclopropane was proposed by Dewar107–109 and is still debated issue. According to magnetic criterion for aromaticity, NICS, cyclopropane was characterized as aromatic,10 while energetic criteria described it as nonaromatic.110 Even computations of the magnetically induced current density did not agree with each other: while Fowler et al.111 and Fliegl et al.112 found a strong diatropic ring current in cyclopropane induced by a perpendicular magnetic field, no evidence for strong diatropism and, hence, σ aromaticity was observed by Lazzeretti et al.113 and Pelloni et al.114 However, the revised three-dimensional model of current density induced by the perpendicular magnetic field revealed a strong delocalized flow circulating beyond the cyclopropane skeleton and extending above/below the ring more than the plane of the hydrogen atoms.115 The strength of the diatropic σ ring current in cyclopropane was estimated to be comparable to that of the π ring current of benzene.72Is it the CC σ electron ring current (σ aromaticity) that makes cyclopropane hydrogen atoms so highly shielded? Pelloni et al.114 found that protons are immersed in a strong localized diatropic current about the C–H bonds, assuming that magnetic filed is applied perpendicularly to the ring. This was recognized as the significant shielding contribution. The revised ring current model of cyclopropane115 showed equally important shielding contribution from the already mentioned delocalized current, particularly from the current flowing on planes close to hydrogen nuclei. The electron flow on the plane of the carbon atoms, involving cyclic CC σ electron delocalization and referred to as σ aromaticity, provided just minor shielding contributions.115 An NCS analysis of the GIAO calculated proton shielding values indicated that both C–C and C–H bonds contribute to the unusual chemical shift of cyclopropane.12 The major shielding contribution certainly comes from the intrinsic C–H bond, as already observed.113–115 However, its mean value, σiso = 25.6 ppm, is lower than for some other (a)cyclic hydrocarbons (for example, σisoC–H = 26.54 ppm for cyclohexane).12 Hence, other shielding mechanisms must be operative. They are most prominent when magnetic field acts perpendicularly to the ring, when the CC framework and the C–H bonds other than the one in which proton is involved shield the hydrogen nucleus by 5.84 ppm and 2.9 ppm, respectively, contributing 5.84/3 ≈ 1.9 and 2.9/3 ≈ 1 ppm to the average shielding.12 The shielding contributions of the CC framework arise from a combined effect of local electron circulations within the cyclopropane bent bonds and delocalized ring current, as deduced from the visualization of its shielding pattern as a response to the action of the perpendicular magnetic field.12 The shielding value of 5.84 ppm for cyclopropane, compared to the corresponding much lower values for planar cyclopentane (0.26 ppm) and cyclohexane (1.21 ppm) is also indicative of the existence of some CC ring currents,12 not observed in previous studies.113–115 Importantly, the shielding pattern of the C–H bonds as a response to the field applied perpendicularly to the ring, implied the formation of a delocalized σ C–H diatropic current,12 probably referred to just as a delocalized current in ref. 115. It is also worth noting that when a field direction is parallel to the C2 symmetry axis of cyclopropane, the C–C bonds provide an equally strong shielding (5.88 ppm) to the two protons attached at the carbon lying on the axis. It is mainly caused by the two adjacent bonds (5.12 ppm) and contributes 5.88/3 × 3 ≈ 0.7 ppm to the proton chemical shift.12
By mapping the total through-space effects of cyclopropane, Martin et al.116 observed that a nucleus located above/below the ring was shielded, which they ascribed to the diatropic effect of the ring current arising from σ aromaticity. At positions near the cyclopropane hydrogen atoms a deshielding was experienced, due to steric interactions.116 The results of Abraham et al.117 showed that anisotropic effects were significant for near protons above/below the ring, but their magnitude rapidly decreased with distance. For nuclei at the edges of the ring, anisotropic effects were of less significance.117
Cyclobutane
Contrary to cyclopropane, 1H NMR chemical shift of cyclobutane protons is unusually high, δ = 1.98 ppm compared to δ = 1.44–1.45 ppm for larger cycloalkanes. In recent literature, this high value is attributed to a deshielding coming from the σ antiaromatic CC framework,10,118 involving eight electrons in the four C–C bonds. The σ antiaromaticity in cyclobutane, proposed by Dewar,108 was supported in the work of Moran et al.10 Computation of a two-dimensional grid of total, isotropic NICS values at points 0.5 Å apart from each other on the plane bisecting the planar cyclobutane showed the (de)shielding pattern typical of an antiaromatic system (Fig. 18; deshielding above/below and inside the ring, shielding around it; (de)shielding regions are reversed for an aromatic molecule: shielding above/below and inside the ring, deshielding around it).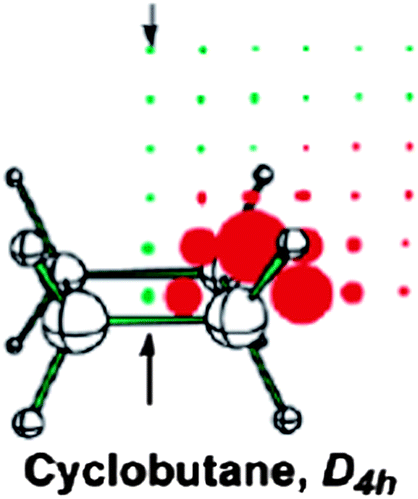 | ||
| Fig. 18 Planar cyclobutane NICS(total) grids. Red and green points denote positive and negative NICS values, respectively, and the arrows point to the ring centers and above. Reprinted with permission from ref. 10. Copyright 2003, American Chemical Society. | ||
Merino et al.14 and Islas et al.,15 however, computed and represented the induced magnetic field of the molecule in an external field acting at right angles to the planar ring and concluded that cyclobutane should be described as nonaromatic (Fig. 19). While the aromatic benzene and the antiaromatic cyclobutadiene show long-range diatropic and paratropic responses, respectively, the responses of cyclobutane (planar) and cyclohexane (chair conformation) are short-range, weakly paratropic inside the rings and surrounded by a diatropic region.14,15 Recall that a diatropic/paratropic ring current characteristic for an aromatic/antiaromatic system can be induced only by the magnetic field applied perpendicularly to the ring plane.
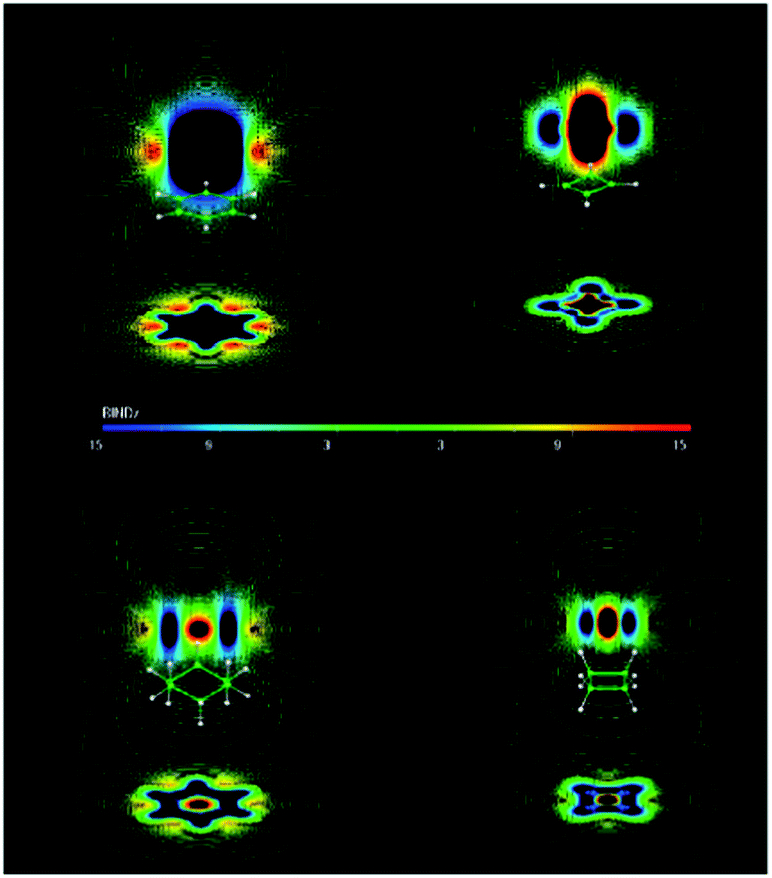 | ||
| Fig. 19 B ind z of benzene, cyclobutadiene, cyclohexane, and cyclobutane. Red zones show paratropic (deshielding) regions around the molecule. Blue ones show diatropic (shielding) zones. The induced magnetic field is given in ppm, which is equivalent to μT. Bind computations were performed at the PW91/IGLO-III level. Reprinted with permission from ref. 15. Copyright 2012, American Chemical Society. | ||
Finally, Baranac-Stojanović and Stojanović12 visualized the response of just the CC framework of planar, D4h, cyclobutane (a transition state) and nonplanar, D2d, cyclobutane (an energy minimum) to a perpendicular magnetic field (Fig. 20) and found it quite similar to responses of other planar CC frameworks, such as those of cyclopentane and cyclohexane. Although, the intensity of the paratropic region inside the cyclobutane molecule is stronger, giving its CC framework slightly paratropic character. The same authors observed that the total anisotropic effects of cyclobutane, typical of an antiaromatic system, actually arise from the two parallel orientations of the molecule relative to the magnetic field, not from the antiaromatic character of the CC framework.12
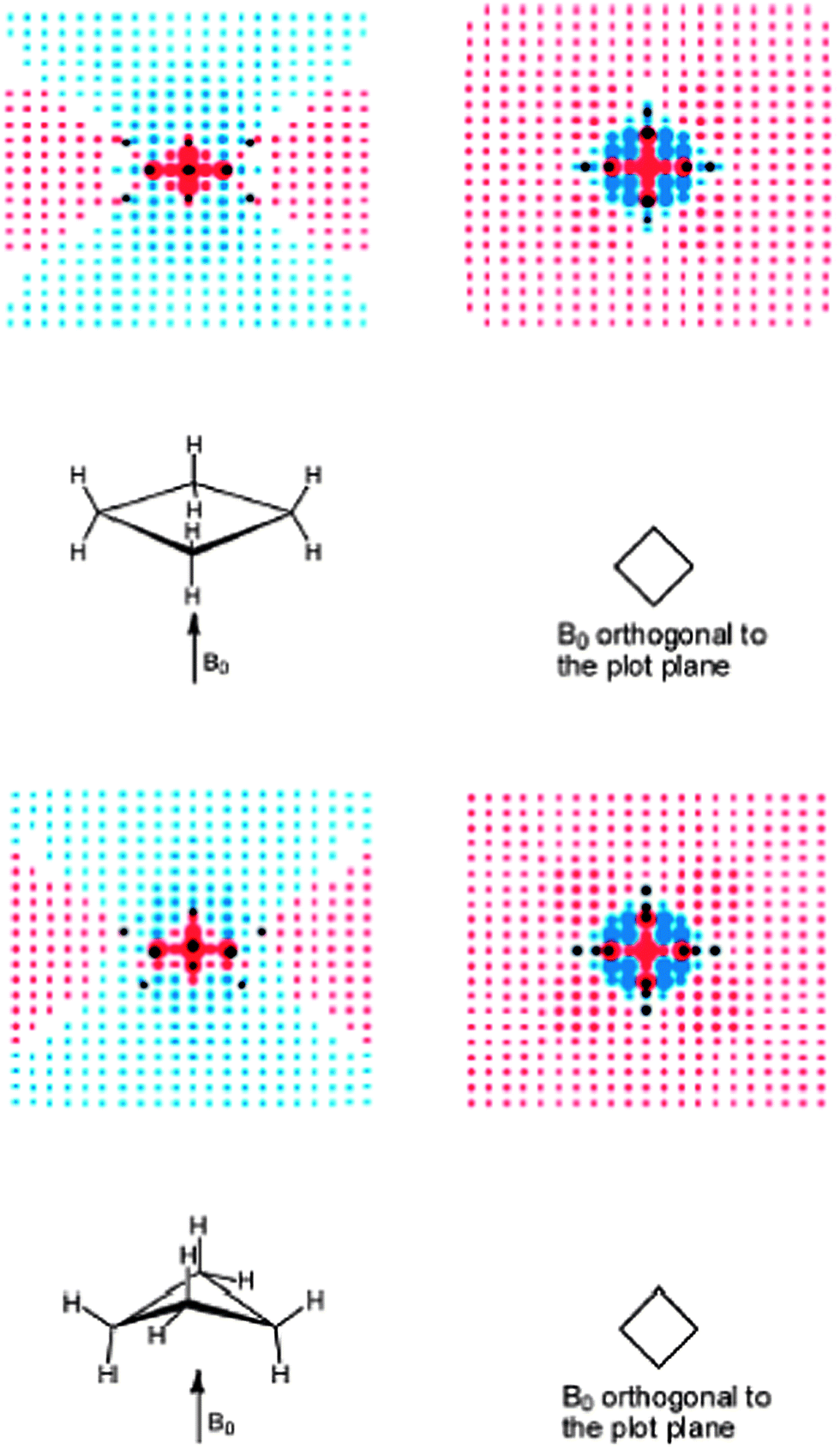 | ||
| Fig. 20 Visualization of (de)shielding contributions of cyclobutane D4h (upper part) and D2d (lower part) CC framework for a magnetic field applied perpendicularly to the ring plane: the first view is in the σv plane, passing through the CH2 groups and the second view is in the plane orthogonal to the first (plane of carbon nuclei for D4h). Blue and red points denote shielding and deshielding effects, respectively. The radius of points is proportional to the absolute value of the contribution. Positions of carbon and hydrogen nuclei are marked by black points. Reprinted with permission from ref. 12. Copyright 2013, American Chemical Society. | ||
Computations of the magnetically induced current density in cyclobutane by Fowler et al.111 and the energy associated with the extra (de)stabilization arising from the cyclic electron delocalization, estimated by Wu et al.,110 are also in accord with the very weak paratropicity of cyclobutane.
Therefore, unlike the belief that the “antiaromatic” CC framework deshields cyclobutane hydrogen nuclei, it shields them by 1.84 ppm, on average.12 In fact, there is no any specific deshielding effect that could account for the experimentally observed high 1H NMR chemical shift. Cyclobutane protons are just less shielded by the C–C and C–H bonds than protons in other cyclic hydrocarbons.12
The total through-space effects of cyclobutane on a chemical shift of a nucleus positioned above the ring are obviously deshielding, and originate from both the anisotropic effect and van der Waals deshielding near the hydrogen atoms.116
Conclusions
In this review, the most important literature providing novel insight into the anisotropic effects and the source of (de)shielding of proximal protons has been collected and discussed. Some classical “anisotropy cone” predictions are reversed, as that for the C–C single bond, or shown to be highly diminished in magnitude, like that for the CC triple bond.
It appears that the explanations of the origin of anisotropic effects should not be based on just one orientation of the molecule with respect to the magnetic field direction – magnetic fields induced in other positions may give significant contributions, too. In the case of unsaturated groups and rings, conventional interpretations take into account π electron effects. However, the influence of σ electrons should not be neglected – it may be even stronger than that of the π electrons. For example, net anisotropic effects of benzene, borazine and ethene are actually dominated by the σ electron effects.
1H NMR chemical shifts of protons spatially close to a functional group are affected not only by magnetic anisotropy of the corresponding group, but by steric and electric field effects, as well. This should be considered when explaining the chemical shift values or making the chemical shift assignments.
Acknowledgements
Financial support from the Ministry of Education, Science and Technological Development of the Republic of Serbia, Grant no. 172020, is acknowledged.References
- E. Breitmaier, in Structure Elucidation by NMR in Organic Chemistry, A Practical Guide, John Wiley and Sons Ltd., West Sussex, England, 2002 Search PubMed.
- J. B. Lambert and E. P. Mazzola, in Nuclear Magnetic Resonance Spectroscopy: An Introduction to Principles, Applications, and Experimental Methods, Pearson Education, Inc., Upper Saddle River, New Jersey, 2004 Search PubMed.
- H. Friebolin, in Basic One- and Two-Dimensional NMR Spectroscopy, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005 Search PubMed.
- H. M. McConnell, J. Chem. Phys., 1957, 27, 226–229 CrossRef CAS.
- K. Peter and C. Vollhardt, in Organic Chemistry, W. H. Freeman and Company, New York, 1987 Search PubMed.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- P. v. R. Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R. v. E. Hommes, Org. Lett., 2001, 3, 2465–2468 CrossRef CAS PubMed.
- C. S. Wannere and P. v. R. Schleyer, Org. Lett., 2003, 5, 605–608 CrossRef CAS PubMed.
- D. Moran, M. Manoharan, T. Heine and P. v. R. Schleyer, Org. Lett., 2003, 5, 23–26 CrossRef CAS PubMed.
- R. S. Viglione, R. Zanasi and P. Lazzeretti, Org. Lett., 2004, 6, 2265–2267 CrossRef CAS PubMed.
- M. Baranac-Stojanović and M. Stojanović, J. Org. Chem., 2013, 78, 1504–1507 CrossRef PubMed.
- M. Baranac-Stojanović and M. Stojanović, Chem.–Eur. J., 2013, 19, 4249–4254 CrossRef PubMed.
- G. Merino, T. Heine and G. Seifert, Chem.–Eur. J., 2004, 10, 4367–4371 CrossRef CAS PubMed.
- R. Islas, T. Heine and G. Merino, Acc. Chem. Res., 2012, 45, 215–228 CrossRef CAS PubMed and references cited therein.
- S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001, 1893–1898 CAS.
- S. Klod, A. Koch and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2002, 1506–1509 RSC.
- E. Kleinpeter, A. Schulenburg, I. Zug and H. Hartmann, J. Org. Chem., 2005, 70, 6592–6602 CrossRef CAS PubMed.
- E. Kleinpeter and A. Koch, Tetrahedron, 2011, 67, 5740–5743 CrossRef CAS PubMed.
- E. Kleinpeter, A. Lämmermann and H. Kühn, Org. Biomol. Chem., 2011, 9, 1098–1111 CAS.
- R. Csütörtöki, I. Szatmári, A. Koch, M. Heydenreich, E. Kleinpeter and F. Fülöp, Tetrahedron, 2012, 68, 4600–4608 CrossRef PubMed.
- M. Baranac-Stojanović, A. Koch and E. Kleinpeter, ChemPhysChem, 2012, 13, 3803–3811 CrossRef PubMed.
- E. Kleinpeter and S. Klod, J. Am. Chem. Soc., 2004, 126, 2231–2236 CrossRef CAS PubMed.
- E. Kleinpeter and A. Schulenburg, J. Org. Chem., 2006, 71, 3869–3875 CrossRef CAS PubMed.
- E. Kleinpeter, A. Koch and P. R. Seidl, J. Phys. Chem. A, 2008, 112, 4989–4995 CrossRef CAS PubMed.
- E. Kleinpeter, I. Szatmári, L. Lázár, A. Koch, M. Heydenreich and F. Fülöp, Tetrahedron, 2009, 65, 8021–8027 CrossRef CAS PubMed.
- G. Sánchez-Sans, I. Alkorta, C. Trujillo and J. Elguero, Tetrahedron, 2012, 68, 6548–6556 CrossRef PubMed.
- G. Sánchez-Sans, C. Trujillo, I. Rozas and J. Elguero, Tetrahedron, 2013, 69, 7333–7344 CrossRef PubMed.
- P. v. R. Schleyer, H. Jiao, N. J. R. v. E. Hommes, V. G. Malkin and O. L. Malkin, J. Am. Chem. Soc., 1997, 119, 12669–12670 CrossRef CAS.
- T. Heine, P. v. R. Schleyer, C. Corminboeuf, G. Seifert, R. Reviakine and J. Weber, J. Phys. Chem. A, 2003, 107, 6470–6475 CrossRef CAS.
- C. Corminboeuf, T. Heine and J. Weber, Phys. Chem. Chem. Phys., 2003, 5, 246–251 RSC.
- C. Corminboeuf, T. Heine, G. Seifert, P. v. R. Schleyer and J. Weber, Phys. Chem. Chem. Phys., 2004, 6, 273–276 RSC.
- W. Kutzelnigg, Isr. J. Chem., 1980, 19, 193–200 CAS.
- M. Schindler and W. Kutzelnigg, J. Chem. Phys., 1982, 76, 1919–1933 CrossRef CAS.
- M. Schindler and W. Kutzelnigg, J. Am. Chem. Soc., 1983, 105, 1360–1370 CrossRef CAS.
- M. Schindler and W. Kutzelnigg, Mol. Phys., 1983, 48, 781–798 CrossRef CAS.
- W. Kutzelnigg, J. Mol. Struct.: THEOCHEM, 1989, 202, 11–61 CrossRef.
- A. E. Hansen and T. D. Bouman, J. Chem. Phys., 1985, 82, 5035–5047 CrossRef CAS.
- A. E. Hansen and T. D. Bouman, J. Chem. Phys., 1989, 91, 3552–3560 CrossRef CAS.
- M. Bilde and A. E. Hansen, Mol. Phys., 1997, 92, 237–250 CAS.
- P. Lazzeretti, Theor. Chem. Acc., 2012, 131, 1222 CrossRef.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- J. A. Bohmann, F. Weinhold and T. C. Farrar, J. Chem. Phys., 1997, 107, 1173–1184 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, WIREs Comput. Mol. Sci., 2012, 2, 1–42 CrossRef CAS.
- F. Weinhold and C. R. Landis, in Discovering Chemistry with Natural Bond Orbitals, John Wiley & Sons, Inc., Hoboken, New Jersey, 2012 Search PubMed.
- T. Heine, C. Corminboeuf and G. Seifert, Chem. Rev., 2005, 105, 3889–3910 CrossRef CAS PubMed.
- J. C. Facceli, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 58, 176–201 CrossRef PubMed.
- N. H. Martin, N. W. Allen III, E. K. Minga, S. T. Ingrassia and J. D. Brown, J. Am. Chem. Soc., 1998, 120, 11510–11511 CrossRef CAS.
- N. H. Martin, D. M. Loveless and D. C. Wade, J. Mol. Graphics Modell., 2004, 23, 285–290 CrossRef CAS PubMed.
- N. H. Martin, J. D. Brown, K. H. Nance, H. F. Schaefer III, P. v. R. Schleyer, Z.-X. Wang and H. L. Woodcock, Org. Lett., 2001, 3, 3823–3826 CrossRef CAS PubMed.
- N. H. Martin, N. W. Allen III, K. D. Moore and L. Vo, J. Mol. Struct.: THEOCHEM, 1998, 454, 161–166 CrossRef CAS.
- N. H. Martin, N. W. Allen III, E. K. Minga, S. T. Ingrassia and J. D. Brown, Struct. Chem., 1998, 9, 403–410 CrossRef CAS.
- N. H. Martin, N. W. Allen III and J. C. Moore, J. Mol. Graphics Modell., 2000, 18, 242–246 CrossRef CAS.
- L. H. Meyer, A. Saika and H. S. Gutowsky, J. Am. Chem. Soc., 1953, 75, 4567–4573 CrossRef CAS.
- J. A. Pople, J. Chem. Phys., 1956, 24, 1111 CrossRef CAS.
- L. Pauling, J. Chem. Phys., 1936, 4, 673–677 CrossRef CAS.
- K. Lonsdale, Proc. R. Soc. London, Ser. A, 1937, 159, 149–161 CrossRef CAS.
- F. London, J. Phys. Radium, 1937, 8, 397–409 CrossRef CAS.
- F. London, J. Chem. Phys., 1937, 5, 837–838 CrossRef CAS.
- P. Lazzeretti, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 36, 1–88 CrossRef CAS.
- J. I. Musher, J. Chem. Phys., 1965, 43, 4081–4083 CrossRef CAS.
- J. I. Musher, J. Chem. Phys., 1967, 46, 1219–1221 CrossRef CAS.
- P. Lazzeretti, E. Rossi and R. Zanasi, J. Chem. Phys., 1982, 77, 3129–3139 CrossRef CAS.
- P. Lazzeretti and R. Zanasi, Chem. Phys. Lett., 1983, 100, 67–69 CrossRef CAS.
- M. B. Ferraro, P. Lazzeretti, R. G. Viglione and R. Zanasi, Chem. Phys. Lett., 2004, 390, 268–271 CrossRef CAS PubMed.
- A. Soncini, P. W. Fowler, P. Lazzeretti and R. Zanasi, Chem. Phys. Lett., 2005, 401, 164–169 CrossRef CAS PubMed.
- E. Steiner and P. W. Fowler, Int. J. Quantum Chem., 1996, 60, 609–616 CrossRef CAS.
- E. Steiner and P. W. Fowler, Chem. Commun., 2001, 2220–2221 RSC.
- E. Steiner and P. W. Fowler, J. Phys. Chem. A, 2001, 105, 9553–9562 CrossRef CAS.
- P. W. Fowler, E. Steiner, R. W. A. Havenith and L. W. Jenneskens, Magn. Reson. Chem., 2004, 42, S68–S78 CrossRef CAS PubMed.
- G. Monaco, R. Zanasi, S. Pelloni and P. Lazzeretti, J. Chem. Theory Comput., 2010, 6, 3343–3351 CrossRef CAS.
- P. Lazzeretti, M. Malagoli and R. Zanasi, J. Mol. Struct.: THEOCHEM, 1991, 234, 127–145 CrossRef.
- U. Fleischer, W. Kutzelnigg, P. Lazzeretti and V. Mühlenkamp, J. Am. Chem. Soc., 1994, 116, 5298–5306 CrossRef CAS.
- P. Lazzeretti, Phys. Chem. Chem. Phys., 2004, 6, 217–223 RSC.
- T. Heine, R. Islas and G. Merino, J. Comput. Chem., 2007, 28, 302–309 CrossRef CAS PubMed.
- E. Steiner and P. W. Fowler, Phys. Chem. Chem. Phys., 2004, 6, 261–272 RSC.
- C. S. Wannere, C. Corminboeuf, W. D. Allen, H. F. Schaefer III and P. v. R. Schleyer, Org. Lett., 2005, 7, 1457–1460 CrossRef CAS PubMed.
- S. N. Steinmann, D. F. Jana, J. I.-C. Wu, P. v. R. Schleyer, Y. Mo and C. Corminboeuf, Angew. Chem., Int. Ed., 2009, 48, 9828–9833 CrossRef CAS PubMed.
- M. Baranac-Stojanović, A. Koch and E. Kleinpeter, Chem.–Eur. J., 2012, 18, 370–376 CrossRef PubMed.
- P. W. Fowler and E. Steiner, J. Phys. Chem. A, 1997, 101, 1409–1413 CrossRef CAS.
- E. D. Jemmis and B. Kiran, Inorg. Chem., 1998, 37, 2110–2116 CrossRef CAS PubMed.
- B. Kiran, A. K. Phukan and E. D. Jemmis, Inorg. Chem., 2001, 40, 3615–3618 CrossRef CAS.
- E. Steiner, P. W. Fowler and R. W. A. Havenith, J. Phys. Chem. A, 2002, 106, 7048–7056 CrossRef CAS.
- J. J. Engelberts, R. W. A. Havenith, J. H. v. Lenthe, L. W. Jenneskens and P. W. Fowler, Inorg. Chem., 2005, 44, 5266–5272 CrossRef CAS PubMed.
- A. Soncini, C. Domene, J. J. Engelberts, P. W. Fowler, A. Rassat, J. H. v. Lenthe, R. W. A. Havenith and L. W. Jenneskens, Chem.–Eur. J., 2005, 11, 1257–1266 CrossRef CAS PubMed.
- R. Islas, E. Chamorro, J. Robles, T. Heine, J. C. Santos and G. Merino, Struct. Chem., 2007, 18, 833–839 CrossRef CAS and references therein.
- R. Carion, V. Liégeois, B. Champagne, D. Bonifazi, S. Pelloni and P. Lazzeretti, J. Phys. Chem. Lett., 2010, 1, 1563–1568 CrossRef CAS.
- K. B. Wiberg, Chem. Rev., 2001, 101, 1317–1331 CrossRef CAS PubMed.
- T. Bally, Angew. Chem., Int. Ed., 2006, 45, 6616–6619 CrossRef CAS PubMed.
- J. I.-C. Wu, Y. Mo, F. A. Evangelista and P. v. R. Schleyer, Chem. Commun., 2012, 48, 8437–8439 RSC.
- S. Pelloni, A. Ligabue and P. Lazzeretti, Org. Lett., 2004, 6, 4451–4454 CrossRef CAS PubMed.
- K. Tori, Y. Hata, R. Muneyuki, Y. Takano, T. Tsuji and H. Tanida, Can. J. Chem., 1964, 42, 926–933 CrossRef CAS.
- K. Tori, K. Aono, Y. Hata, R. Muneyuki, T. Tsuji and H. Tanida, Tetrahedron Lett., 1966, 9–17 CrossRef CAS.
- B. Franzus, W. C. Baird Jr, N. F. Chamberlain, T. Hines and E. I. Snyder, J. Am. Chem. Soc., 1968, 90, 3721–3724 CrossRef CAS.
- A. P. Marchand and J. E. Rose, J. Am. Chem. Soc., 1968, 90, 3724–3731 CrossRef CAS.
- R. J. Abraham and J. Fischer, Magn. Reson. Chem., 1985, 23, 856–861 CrossRef CAS.
- I. Alkorta and J. Elguero, New J. Chem., 1998, 22, 381–385 RSC.
- N. H. Martin and J. D. Brown, Int. J. Mol. Sci., 2000, 1, 84–91 CrossRef CAS.
- R. J. Abraham, M. Canton and L. Griffiths, Magn. Reson. Chem., 2001, 39, 421–431 CrossRef CAS.
- F. B. Mallory and M. B. Baker, J. Org. Chem., 1984, 49, 1323–1326 CrossRef CAS.
- R. J. Abraham and M. Reid, J. Chem. Soc., Perkin Trans. 2, 2001, 1195–1204 RSC.
- R. J. Abraham, M. A. Warne and L. Griffiths, Magn. Reson. Chem., 1998, 36, S179–S188 CrossRef CAS.
- R. J. Abraham, M. A. Warne and L. Griffiths, J. Chem. Soc., Perkin Trans. 2, 1997, 31–39 RSC.
- K. B. Wiberg and B. J. Nist, J. Am. Chem. Soc., 1961, 83, 1226–1230 CrossRef CAS.
- D. J. Patel, M. E. H. Howden and J. D. Roberts, J. Am. Chem. Soc., 1963, 85, 3218–3223 CrossRef CAS.
- M. J. S. Dewar, Bull. Soc. Chim. Belg., 1979, 88, 957–967 CrossRef CAS.
- M. J. S. Dewar and M. L. McKee, Pure Appl. Chem., 1980, 52, 1431–1441 CrossRef CAS.
- M. J. S. Dewar, J. Am. Chem. Soc., 1984, 106, 669–682 CrossRef CAS.
- W. Wu, B. Ma, J. I.-C. Wu, P. v. R. Schleyer and Y. Mo, Chem.–Eur. J., 2009, 15, 9730–9736 CrossRef CAS PubMed.
- P. W. Fowler, J. Baker and M. Lillington, Theor. Chem. Acc., 2007, 118, 123–127 CrossRef CAS.
- H. Fliegl, D. Sundholm, S. Taubert, J. Jusélius and W. Klopper, J. Phys. Chem. A, 2009, 113, 8668–8676 CrossRef CAS PubMed.
- P. Lazzeretti, E. Rossi and R. Zanasi, J. Am. Chem. Soc., 1983, 105, 12–15 CrossRef CAS.
- S. Pelloni, P. Lazzeretti and R. Zanasi, J. Phys. Chem. A, 2007, 111, 8163–8169 CrossRef CAS PubMed.
- R. Carion, B. Champagne, G. Monaco, R. Zanasi, S. Pelloni and P. Lazzeretti, J. Chem. Theory Comput., 2010, 6, 2002–2018 CrossRef CAS.
- N. H. Martin, D. M. Loveless, K. L. Main and D. C. Wade, J. Mol. Graphics Modell., 2006, 25, 389–395 CrossRef CAS PubMed.
- R. J. Abraham, P. Leonard and C. F. Tormena, Magn. Reson. Chem., 2012, 50, 305–313 CrossRef CAS PubMed.
- K. B. Wiberg, in The Chemistry of Cyclobutanes, John Wiley and Sons Ltd., New York, 2005 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |