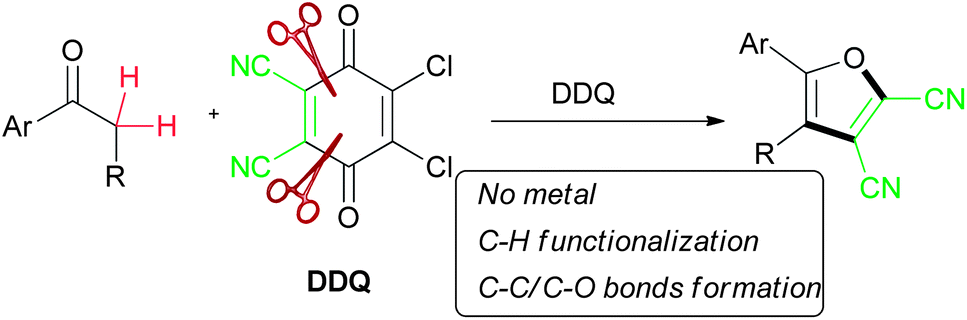
One-step metal-free construction of fluorescent 5-aryl-2,3-dicyanofurans from simple aryl ketones with DDQ†
Jiang-Sheng
Li
*a,
Fei-Fei
Cai
a,
Zhi-Wei
Li
*a,
Wei-Dong
Liu
b,
Jim
Simpson
c,
Yuan
Xue
a,
Huai-Lin
Pang
b,
Peng-Mian
Huang
a,
Zhong
Cao
a and
Dao-Lin
Li
a
aHunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation, School of Chemistry and Biological Engineering, Changsha University of Science & Technology, Changsha 410114, P. R. China. E-mail: jslichem@gmail.com; lizwhn@126.com; Fax: +86 731 85258733
bNational Engineering Research Center for Agrochemicals, Hunan Research Institute of Chemical Industry, Changsha 410007, P. R. China
cDepartment of Chemistry, University of Otago, P. O. Box 56, Dunedin 9054, New Zealand
First published on 1st November 2013
Abstract
A variety of fluorescent 5-aryl-2,3-dicyanofurans were prepared by the treatment of electron-rich aryl ketones with DDQ. The presence of an alkoxy substituent on the benzene ring in the substrates was essential for an efficient cyclization to occur. This novel approach allows the metal-free construction of furan rings from simple aryl ketones.
Direct utility of C–H functionalization to create C–C and C–heteroatom bonds is of great significance and also a challenge to synthetic chemists. Direct C–H functionalization represents a highly attractive strategy for an ideal chemical synthesis. Compared to traditional chemical reactions, such coupling reactions based on C–H activation1 have an inherent advantage in that they do not require substrates with highly functionalized groups, such as organohalides or organometallic species. However, these coupling reactions1g–i usually involve the presence of terminal alkyne Csp–H bonds, arene/alkene Csp2–H bonds and Csp3–H bonds adjacent to heteroatoms (N, O and S) or π-systems. Ketones with α-H atoms undergo such oxidative coupling but usually act as nucleophiles.2
α-Functionalization of carbonyl compounds represents one of the most versatile and valuable procedures in organic synthesis. With such a strategy, various important synthetic building blocks such as 1,4-dicarbonyl compounds,3 1-arylated or alkynylated ketones/esters4 could be readily prepared. Although some progress has been made, such functionalization is currently limited to readily available α-halocarbonyl compounds. The direct oxidative α-C–H functionalization of carbonyl compounds usually refers to such transformations as halogenation and oxygenation. The application of oxidative functionalization of simple ketones to construct heterocycles is still rare.5
Furans are important fundamental structures with many interesting optoelectronic and biological properties.6 Further-more, they occur widely in a variety of natural products, pharmaceuticals and organic solar cell dyes, and also serve as versatile building blocks in organic synthesis. Generally, further functionalization of the furan ring itself is an efficient approach to the synthesis of furans by means of Stille or Heck reactions.7 Alternatively, metal-catalyzed intramolecular annulation of alkyne-containing substrates or their equivalents has also been developed.8 However, the requirement to pre-functionalize the starting materials in these two strategies limited their application. Besides, oxidative intermolecular cyclization using basic starting materials, mediated by the use of metal catalysts, provided another facile approach to furans.9 It is no doubt that such direct oxidative formation of C–C/C–O bonds in one step via C–H functionalization would be an attractive approach to synthesize the furan scaffold.
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) is a well-known oxidant in organic chemistry. In recent years, it has been successfully used in cross dehydrogenative coupling (CDC) for C–X (X = C, N, O, S, etc.) bond formation based on C–H activation.2g,10 During our continued investigation to DDQ-mediated CDC reactions,11 we accidentally discovered that DDQ can also serve as a maleonitrile building block12 when simple electron-rich aryl ketones were used as reactant partner, which meanwhile led to an efficient approach toward preparing 5-aryl-2,3-dicyanofurans from simple aryl ketones. To the best of our knowledge, only very few examples regarding 2,3-dicyanofurans have been documented.12,13
Herein, we report our efforts on the construction of polysubstituted furan derivatives from simple aryl ketones via C–H functionalization/C–C and C–O bond formation by using DDQ without the requirement of a metal catalyst (Scheme 1).
 | ||
| Scheme 1 A novel approach for the construction of furan derivatives. | ||
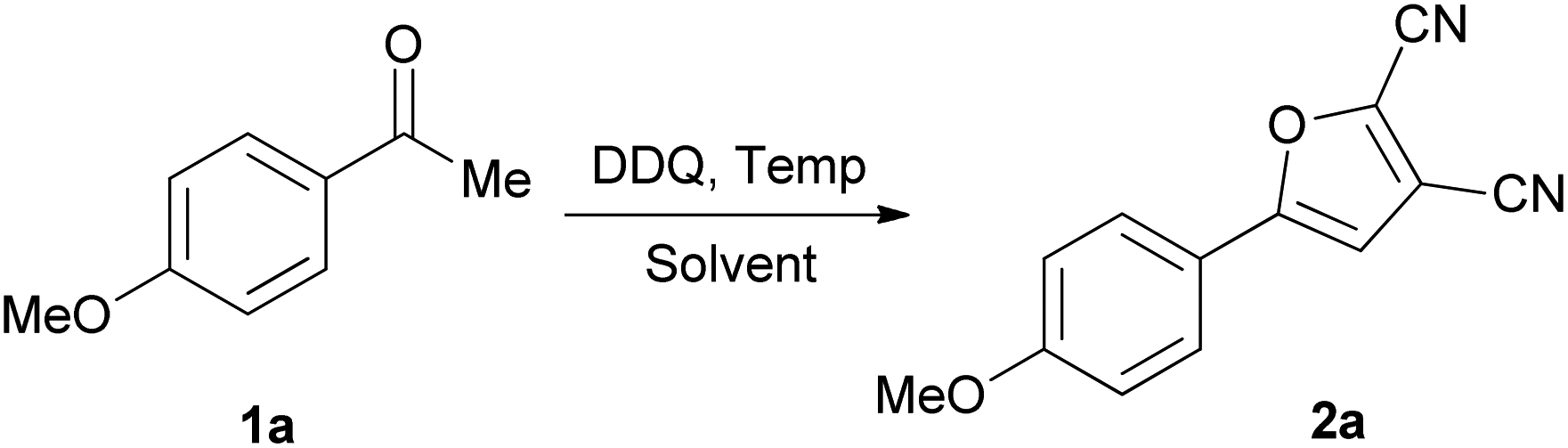
With this discovery in mind, we set out to optimize the reaction parameters based on a model reaction of 1-(4-methoxyphenyl)ethanone (1a) with DDQ in 1,2-dichloroethane (DCE) in a sealed tube.
Selected results are compiled in Table 1. Firstly, the variation of the amount of DDQ was investigated. When the ratio of DDQ/1a was 1.2, 2.0 and 2.5, the conversion of 1a to 5-(4-methoxyphenyl)furan-2,3-dicarbonitrile (2a) went gradually to completion (Table 1, entries 1–3). When 3.0 equivalents of DDQ were used, the reaction went to completion and the optimum yield (52%) of 2a was obtained (Table 1, entry 4). Continued increasing the DDQ amount (4.0 equiv.) did not afford better yield. Next, several other solvents such as benzene, HOAc, MeCN, MeNO2, EtOAc and dichloromethane (DCM) were examined (Table 1, entries 5–10) using the optimum 3/1 ratio of DDQ/1a. Compared to DCE, MeCN, MeNO2 and DCM gave moderate yields while with HOAc and EtOAc the yields were poor. Furthermore, reacting 1a with neat DDQ furnished a 36% yield of 2a. The effects of reaction temperature and time on the yield of 2a were also investigated. When the reaction was carried out at 80 or 120 °C, 2a was produced in a lower yield (Table 1, entries 12 and 13). No reaction was observed at ambient temperature. When the reaction time was shortened to 10 h or increased to 15 h, the yield of 2a still remained essentially the same. In an attempt to further improve the reaction yield, metal catalysts such as CuBr, CuBr2, CuCl2 and Cu(OAc)2 were used. Much to our disappointment, they were ineffective and even led to a decrease in the yield (Table 1, entries 16–19). Thus, the optimal conditions for such transformation were established to be as follows: a DDQ/ketone molar ratio of 3.0, in DCE as solvent, at a temperature of 100 °C for 13 h under N2 without any metal catalyst. Under such optimal reaction conditions, the yield of 2a was 52%.
|
|
|||||
|---|---|---|---|---|---|
| Entry | DDQ/1ab | Solvent | Temp (°C) | Time (h) | Yieldc (%) |
| a Reaction conditions: 1a (0.5 mmol), solvent (2 mL), N2 in sealed tube. b The molar ratio. c Isolated yield. d CuBr (5 mol %). e CuBr2 (5 mol %). f CuCl2 (5 mol %). g Cu(OAc)2 (5 mol %). | |||||
| 1 | 1.2 | DCE | 100 | 13 | 20 |
| 2 | 2.0 | DCE | 100 | 13 | 31 |
| 3 | 2.5 | DCE | 100 | 13 | 37 |
| 4 | 3.0 | DCE | 100 | 13 | 52 |
| 5 | 3.0 | benzene | 100 | 13 | 36 |
| 6 | 3.0 | HOAc | 100 | 13 | 14 |
| 7 | 3.0 | MeCN | 100 | 13 | 43 |
| 8 | 3.0 | MeNO2 | 100 | 13 | 47 |
| 9 | 3.0 | EtOAc | 100 | 13 | 21 |
| 10 | 3.0 | DCM | 100 | 13 | 44 |
| 11 | 3.0 | Neat | 100 | 13 | 36 |
| 12 | 3.0 | DCE | 80 | 13 | 43 |
| 13 | 3.0 | DCE | 120 | 13 | 40 |
| 14 | 3.0 | DCE | 100 | 10 | 50 |
| 15 | 3.0 | DCE | 100 | 15 | 52 |
| 16d | 3.0 | DCE | 100 | 13 | 32 |
| 17e | 3.0 | DCE | 100 | 13 | 32 |
| 18f | 3.0 | DCE | 100 | 13 | 22 |
| 19g | 3.0 | DCE | 100 | 13 | 13 |
Next, we sought to probe the scope and limitations of the substrate on these DDQ-mediated annulation reactions. The reaction was attempted using acetophenone, 1-(p-tolyl)ethanone and 1-(4-chlorophenyl)ethanone in place of 1a. However, it was found that they were unreactive and none of the desired furan derivatives were obtained. These results implied that the presence of the electron-donating methoxy substituent on the benzene ring of the substrate was crucial for such oxidative intermolecular cyclization to occur.
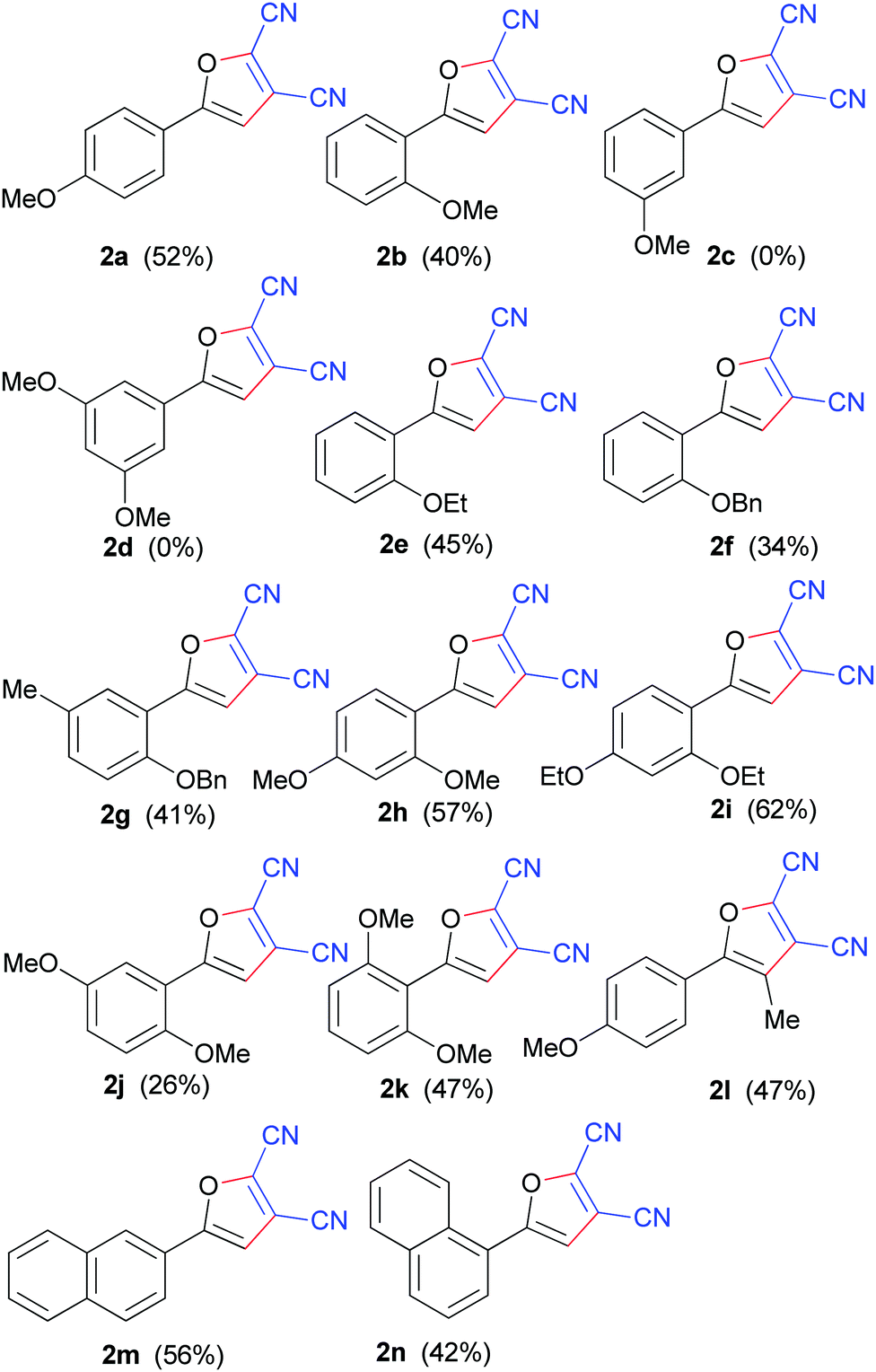
These findings pointed to the importance of an electron-rich substrate so we investigated changing the position of the methoxy group on the benzene ring and tested the ortho- and meta-analogues of 1a. Delightedly, substrate 1b, with the methoxy substitutent in the ortho-position, was found to be effective and gave the desired product 2b in moderate yield. However, we were disappointed to find that the meta-methoxy group 1c yielded none of the desired product under the same conditions, even using a longer reaction time and a higher reaction temperature. Furthermore, 1-(3,5-dimethoxyphenyl)-ethanone (1d), with two methoxy groups located at the meta-positions of the benzene ring, also gave no desired furan product. On the basis of the above experimental facts, therefore, we tentatively conclude that an ortho- and/or a para-alkoxy group is essential for such oxidative annulation. Gratifyingly, replacement of a methoxy group with an ethoxy group afforded similar yield (Table 2, 2bvs.2e). However, replacing a methoxy group with a benzyloxyl one resulted in a relatively lower yield (Table 2, 2f). This was attributed to competitive DDQ-mediated debenzylation under such oxidative conditions,14 which could be supported by the fact that 1-(2-hydroxyphenyl)ethanone was detected. Introduction of a methyl group into the benzene ring of substrate 1f could promote this transformation and enhance the yield of 2g (Table 2, 2fvs.2g). Furthermore, two alkoxy substituents located at different positions on the benzene ring were investigated (Table 2, 2h–k). All the substrates tested gave satisfactory yields. Substrates 1h, i, k, with both alkoxy groups at ortho-/para- or ortho-/ortho-positions, gave relatively better yields. In particular, substrate 1i, 1-(2,4-diethoxyphenyl)ethanone, gave rise to 2i, 5-(2,4-diethoxyphenyl)furan-2,3-dicarbonitrile, in 62% yield. It is notable that substrate 1j with two ortho-/meta-methoxy groups afforded 2j in a less than satisfactory yield of 26%. To directly functionalize furan ring at 4-position, 1-(4-methoxyphenyl)-propan-1-one (1l) was used for this oxidative reaction under the established conditions. As is all known, substrate 1l is vulnerable to oxidative elimination, providing α, β-unsaturated ketone using DDQ. Interestingly, substrate 1l reacted smoothly to form a tetrasubstituted furan 2l in 47% yield. The oxidative reaction went equally well with substrates 1m–n, in which the naphthyl ring could be considered as an electron-rich benzene ring,15 with a ortho- or para-methoxy group. In addition, attempts were made to use 1-(2-hydroxyphenyl)ethanone, 1-(2-hydroxy-4-methoxyphenyl)ethanone, and 2-acetylphenyl acetate as substrates for this protocol. As expected, it was found that no desired furan products were obtained and the ketones were largely unreacted.
To further identify product structure, the molecular structure of furan 2f was determined by X-ray diffraction as shown in Fig. 1 together with the atom numbering scheme using Mercury software.16 The central C8⋯C13 benzene ring is inclined to the furan ring at 4.32(17)° such that the phenyl-furan-dicarbonitrile unit is close to planar.
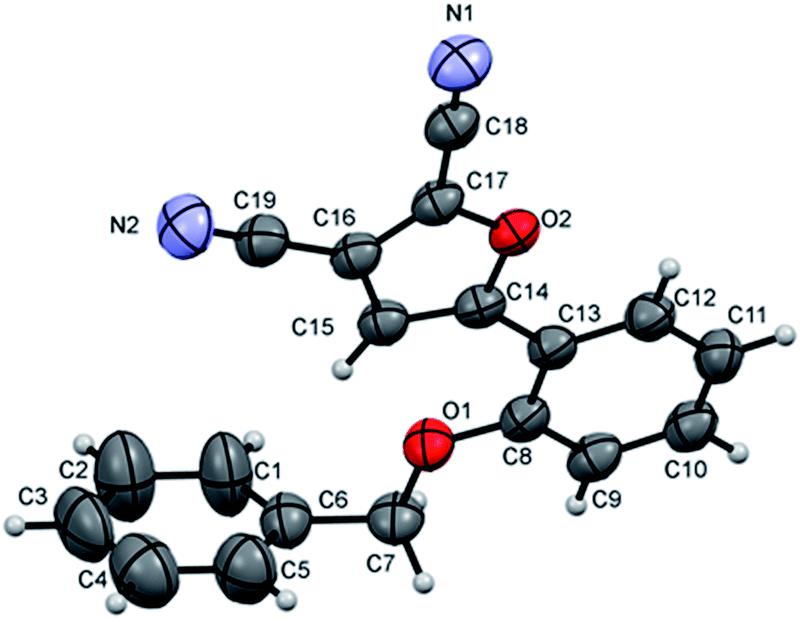 | ||
| Fig. 1 X-ray structure of compound 2f. | ||
To probe whether radical species are produced in this oxidative cyclization, the radical scavenger TEMPO was utilized in the cyclization of 1a (Scheme 2). As expected, the formation of the desired product 2a is completely inhibited by the addition of 6 equivalents of TEMPO. This result indicates that this reaction may involve radical intermediates.
 | ||
| Scheme 2 Radical-trapping experiment. | ||
Based on the above experimental results and our earlier work,12a a tentative mechanism for this protocol is proposed, as depicted in Scheme 3. Firstly, 1a reacts with DDQ to form an adduct (geminate radical ion pair) via single electron transfer (SET), followed by radical combination and subsequent proton transfer to give rise to the intermediate I. Secondly, I undergoes intramolecular addition followed by proton transfer, affording the intermediate II. Thirdly, with the aid of a second molecule of DDQ, II converts from the diol form to a dione form and a subsequent retro-Diels Alder reaction occurs to yield the product 2a, together with the intermediate IV. Hydrolysis of IV followed by intramolecular cyclization produces 2,3-dichloro-5-hydroxyfuran-2(5H)-one V. Finally, V is subjected to dehydrogenation at the presence of a third molecule of DDQ, affording the side-product 2,3-dichloromaleic anhydride. This interpretation of the reaction is further supported by the observation that washing the reaction mixture with a dilute solution of aqueous NaHCO3 could simplify the work-up.
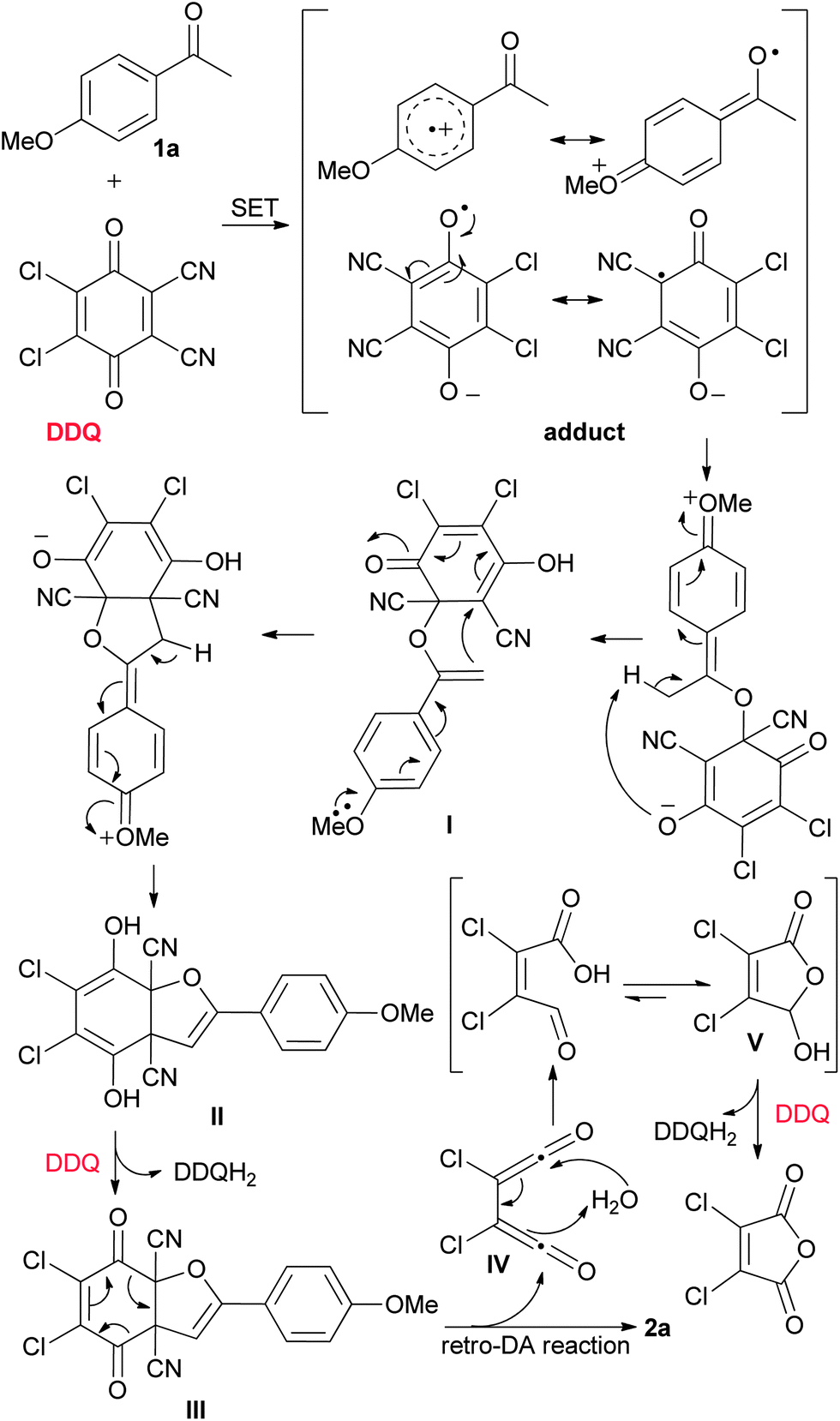 | ||
| Scheme 3 Proposed mechanism for the formation of 2,3-dicyanofurans solely using 3 equivalents of DDQ. | ||
It's well known that fluorescent substances are of vital importance in a variety of fields.17 Facile and efficient construction of fluorescent core frameworks using new synthetic methodologies is starting to attract great interest.18 To our delight, all the obtained 5-aryl-2,3-dicyanofurans were found to possess significantly strong photonic fluorescence both in solution and in the solid state (see Fig. S1 in the ESI†).
Such fluorescent properties are due to the molecular coplanarity as revealed in X-ray crystallographic analysis, facilitating a “push–pull” π-electron system. Novel fluorescent probe molecules and solar cell dyes will be achieved using this reaction protocol combined with the metal-catalyzed coupling technique.
In conclusion, we have developed a metal-free and efficient approach to the synthesis of fluorescent 5-aryl-2,3-dicyano-furans using DDQ in moderate to good yields. This novel protocol allows the construction of a furan core from simple aryl ketones in one step. Further investigations into the reaction mechanism and applications of this protocol in the production of fluorescent probes and solar cell dyes are still on-going.
Acknowledgements
Supports of this work by the National Natural Science Foundation of China (21202010 & 21275022), and the Hunan Provincial Natural Science Foundation of China (12JJ4021) are gratefully acknowledged.Notes and references
- (a) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (b) S. Y. Zhang, F. M. Zhang and Y. Q. Tu, Chem. Soc. Rev., 2011, 40, 1937 RSC; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (d) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (e) J.-Q. Yu and Z. Shi, C–H Activation, Springer, New York, 2010 Search PubMed; (f) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540 RSC; (g) W. J. Yoo and C. J. Li, Top. Curr. Chem., 2010, 292, 281 CrossRef; (h) C. J. Scheuermann, Chem.–Asian. J., 2010, 5, 436 CrossRef CAS PubMed; (i) C. J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed.
- (a) G. Zhang, Y. Ma, S. Wang, W. Kong and R. Wang, Chem. Sci., 2013, 4, 2645 RSC; (b) M. Zhao, F. Wang and X. Li, Org. Lett., 2012, 14, 1412 CrossRef CAS PubMed; (c) F. Yang, J. Li, J. Xie and Z.-Z. Huang, Org. Lett., 2010, 12, 5214 CrossRef CAS PubMed; (d) W. J. Yoo, C. A. Correia, Y. Zhang and C. J. Li, Synlett, 2009, 138 CAS; (e) Y. M. Shen, M. Li, S. Z. Wang, T. G. Zhan, Z. Tan and C. C. Guo, Chem. Commun., 2009, 953 RSC; (f) A. Sud, D. Sureshkumar and M. Klussmann, Chem. Commun., 2009, 3169 RSC; (g) Y. Zhang and C. J. Li, J. Am. Chem. Soc., 2006, 128, 4242 CrossRef CAS PubMed.
- (a) Y. Ito, T. Konoike, T. Harada and T. Saegusa, J. Am. Chem. Soc., 1977, 99, 1487 CrossRef CAS; (b) R. H. Frazier and R. L. Harlow, J. Org. Chem., 1980, 45, 5408 CrossRef CAS; (c) C. Liu, Y. Deng, J. Wang, Y. Yang, S. Tang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 7337 CrossRef CAS PubMed.
- (a) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234 CrossRef CAS PubMed; (b) T. Hama, X. Liu, D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 11176 CrossRef CAS PubMed; (c) C. He, S. Guo, L. Huang and A. Lei, J. Am. Chem. Soc., 2010, 132, 8273 CrossRef CAS PubMed; (d) W. Shi, C. Liu, Z. Yu and A. Lei, Chem. Commun., 2007, 2342 RSC.
- Y.-P. Zhu, M. Lian, F.-C. Jia, M.-C. Liu, J.-J. Yuan, Q.-H. Gao and A.-X. Wu, Chem. Commun., 2012, 48, 9086 RSC.
- (a) A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Handbook of Heterocyclic chemistry, Elsevier, Amsterdam, 3rd edn, 2010 Search PubMed; (b) A. R. Katritzky, Comprehensive Heterocyclic Chemistry III, Elsevier, 1st edn, Amsterdam, 2008 Search PubMed.
- L. Li and S. Ma, Chin. J. Org. Chem., 2000, 20, 701 CAS.
- (a) W. J. Moran and A. Rodriguez, Org. Prep. Proced. Int., 2012, 44, 103 CrossRef CAS; (b) Y. Lu, F. Song, X. Jia and Y. Liu, Prog. Chem., 2010, 22, 58 CAS.
- (a) C. He, S. Guo, J. Ke, J. Hao, H. Xu, H. Chen and A. Lei, J. Am. Chem. Soc., 2012, 134, 5766 CrossRef CAS PubMed; (b) M. Zheng, L. Huang, W. Wu and H. Jiang, Org. Lett., 2013, 15, 1838 CrossRef CAS PubMed; (c) H. Cao, H. Zhan, J. Cen, J. Lin, Y. Lin, Q. Zhu, M. Fu and H. Jiang, Org. Lett., 2013, 15, 1080 CrossRef CAS PubMed.
- (a) H. F. He, K. Wang, B. Xing, G. Sheng, T. Ma and W. Bao, Synlett, 2013, 24, 211 CrossRef CAS PubMed; (b) H. Yi, Q. Liu, J. Liu, Z. Zeng, Y. Yang and A. Lei, ChemSusChem, 2012, 5, 2143 CrossRef CAS PubMed; (c) H. Wang, X. Li, F. Wu and B. Wan, Tetrahedron Lett., 2012, 53, 681 CrossRef CAS PubMed; (d) D. Ramesh, U. Ramulu, K. Mukkanti and Y. Venkateswarlu, Tetrahedron Lett., 2012, 53, 2904 CrossRef CAS PubMed; (e) Y. Cui and P. E. Floreancig, Org. Lett., 2012, 14, 1720 CrossRef CAS PubMed; (f) G. J. Brizgys, H. H. Jung and P. E. Floreancig, Chem. Sci., 2012, 3, 438 RSC; (g) J. Xie and Z.-Z. Huang, Angew. Chem., Int. Ed., 2010, 49, 10181 CrossRef CAS PubMed; (h) C. Guo, J. Song, S.-W. Luo and L.-Z. Gong, Angew. Chem., Int. Ed., 2010, 49, 5558 CrossRef CAS PubMed; (i) D. Cheng and W. Bao, Adv. Synth. Catal., 2008, 350, 1263 CrossRef CAS; (j) Y. Zhang and C. J. Li, Angew. Chem., 2006, 118, 1983 CrossRef.
- J. S. Li, F. F. Cai, Z. W. Li, Y. Xue, C. Cheng, W. D. Liu and Z. Cao, Chin. J. Chem., 2012, 30, 1699 CrossRef CAS.
- (a) J. S. Li, Y. Xue, Z. W. Li, W. D. Liu, C. H. Lu and P. X. Zhao, Synlett, 2013, 24, 2003 CrossRef CAS PubMed; (b) A. G. M. Barrett, F. Blaney, A. D. Campbell, D. Hamprecht, T. Meyer, A. J. P. White, D. Witty and D. J. Williams, J. Org. Chem., 2002, 67, 2735 CrossRef CAS PubMed; (c) W. Deng, Z. Wang, Z. Zhang and H. Li, Faming Zhuanli Shenqing Gongkai Shuomingshu, 2012, CN102516210 Search PubMed.
- L. S. Boulos and I. T. Hennawy, Phosphorus, Sulfur, and Silicon, 1993, 84, 173 CrossRef CAS.
- (a) S. Chandrasekhar, G. Sumithra and J. S. Yadav, Tetrahedron Lett., 1996, 37, 1645 CrossRef CAS; (b) B. P. Ying, B. G. Trogden, D. T. Kohlman, S. X. Liang and Y. C. Xu, Org. Lett., 2004, 6, 1523 CrossRef CAS PubMed.
- Z. Liang, W. Hou, Y. Du, Y. Zhang, Y. Pan, D. Mao and K. Zhao, Org. Lett., 2009, 11, 4978 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. V. Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- (a) J. R. Lakowicz, Probe Design and Chemical Sensing, in Topics in Fluorescence Spectroscopy, ed. J. R. Lakowicz, Kluwer, New York, 2002, vol. 4 Search PubMed; (b) I. Johnson and M. T. Spence, The Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies, Live Technologies Corporation, Carlsbad, CA, 11th edn, 2010 Search PubMed; (c) B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH, Weinheim, 2012 Search PubMed.
- (a) M. Maruyama, M. König, D. M. Guldi, E. Nakamura and Y. Matsuo, Angew. Chem., Int. Ed., 2013, 52, 3015 CrossRef CAS PubMed; (b) M. Arisawa, Y. Fujii, H. Kato, H. Fukuda, T. Matsumoto, M. Ito, H. Abe, Y. Ito and S. Shuto, Angew. Chem., Int. Ed., 2013, 52, 1003 CrossRef CAS PubMed; (c) A. V. Anzalone, T. Y. Wang, Z. Chen and V. M. Cornish, Angew. Chem., Int. Ed., 2013, 52, 650 CrossRef CAS PubMed; (d) E. Benedetti, L. S. Kocsis and K. M. Brummond, J. Am. Chem. Soc., 2012, 134, 12418 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra45462b |
| This journal is © The Royal Society of Chemistry 2014 |