Highly crystallized Fe2O3 nanocrystals on graphene: a lithium ion battery anode material with enhanced cycling
Hongwei
Zhang
,
Liang
Zhou
* and
Chengzhong
Yu
*
Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: l.zhou1@uq.edu.au; c.yu@uq.edu.au; Fax: +61 7-334-63973; Tel: +61 7-334-63283
First published on 31st October 2013
Abstract
A facile one-step hydrothermal method has been developed to synthesise Fe2O3-graphene nanocomposites using L-arginine as the hydrolysis-controlling agent. In the synthesised composites, highly crystallized Fe2O3 nanocrystals with an average size of ∼50 nm are firmly anchored on the graphene sheets. When applied as the anode material for lithium ion batteries, the resulting Fe2O3-graphene nanocomposites show excellent cycling stability and good rate performance. The composites deliver a capacity of 946 and 634 mA h g−1 at a current density of 200 and 2000 mA g−1, respectively. After cycling at 200 mA g−1 for 450 cycles, a capacity of 1049 mA h g−1 can still be maintained.
1. Introduction
Lithium ion batteries (LIBs) have been widely used in portable electronic devices such as smartphones, tablets, and laptop computers. They are also being seriously considered as the power sources of choice for upcoming hybrid, plug-in hybrid, and all electric vehicles.1,2 Graphite is the most widely employed anode materials in commercialised LIBs. However, the limited capacity (theoretical capacity: 372 mA h g−1) as well as the safety hazards arising from an operating voltage close to that of Li/Li+ avoid its large-scale applications. To meet the increasing demands of newly emerging applications, advanced LIBs with better performance in terms of energy density, power density, calendar life, and safety are required.Transition metal oxides have been considered as promising anode materials for LIBs due to their high capacity.3 Among them, Fe2O3 has attracted much attention due to its high theoretical specific capacity (1007 mA h g−1), low cost, abundance, and environmental friendliness.4–7 However, the practical application of Fe2O3 in LIBs is still hindered by the huge volume variation during charge–discharge as well as the low electrical conductivity. While the former leads to notorious problems such as active material pulverization and electrode disintegration, the later results in poor rate performance. It has been reported that the introduction of a carbon matrix, such as graphene, in the active material to form a Fe2O3-carbon nanocomposite can significantly enhance the electrochemical performance.8–10 The carbon matrix can not only buffer the huge volume expansion but also significantly enhance the electrical conductivity. Besides the carbon matrix, the structural parameters of Fe2O3, such as morphology,11–13 particle size14,15 and crystallinity,13 also have significant effects on the electrochemical performances. Although there are several reports on the synthesis of Fe2O3-graphene nanocomposites and their electrochemical performance, most of them focus on controlling the Fe2O3 shape and composite architecture.8–10,16–20 The role of crystallinity of Fe2O3 on the electrochemical performance of Fe2O3-graphene nanocomposites has attracted little attention. Moreover, hydrazine, a toxic chemical, is usually utilised to reduce the graphene oxide to graphene.8,17,20
Herein, we report a facile one-step hydrothermal synthesis of highly crystallized Fe2O3-graphene nanocomposites using L-arginine as the hydrolysis-controlling agent. When employed as the anode material for LIBs, the obtained Fe2O3-graphene nanocomposites, designated as Fe2O3/G(H), exhibit significantly improved cycling stability than bare Fe2O3 nanocrystals and poorly crystallized Fe2O3-graphene composites (Fe2O3/G(P)).
2. Experimental
Synthesis of Fe2O3/G(H) and Fe2O3/G(P)
Graphene oxide (GO) suspension (2 mg ml−1) was prepared according to a previous report.21 For the synthesis of highly crystallized Fe2O3-graphene nanocomposites (Fe2O3/G(H)), 0.8 g of Fe(NO3)3·9H2O and 0.4 g of L-arginine was dissolved in 20 ml of water and then mixed with 20 ml of GO suspension. The mixture was ultrasonicated for 1 hour before hydrothermal treatment at 180 °C for 12 hours. The resulting precipitation was centrifuged, washed repeatedly with deionized water, and dried at 50 °C for 12 hours. Poorly crystallized Fe2O3-graphene nanocomposites (Fe2O3/G(P)) was prepared by replacing L-arginine with urea, while keeping other synthetic conditions unchanged. Bare Fe2O3 nanoparticles were prepared by the same procedure as Fe2O3/G(H) except no graphene oxide was added.Materials characterizations
The morphology of the samples was investigated by field emission scanning electron microscope (FESEM, JEOL 7001F) operated at 15 kV and transmission electron microscope (TEM, Tecnai F20) at 200 kV. Thermo gravimetric analysis (TGA) was carried out on a TGA/DSC1 STARe System under air flow (25–600 °C, 5 °C min−1). X-ray diffraction (XRD) patterns were collected on a Rigaku Miniflex X-ray Diffractometer with Co Kα radiation (λ = 0.179 nm). X-ray photoelectron spectra (XPS) were collected on a Kratos Axis ULTRA X-ray photoelectron spectrometer using a monochromatic Al Kα (1486.6 eV) X-ray source and a 165 mm hemispherical electron energy analyzer.Electrochemical measurements
The electrochemical measurements were carried out on a MTI 8 Channels Battery Analyser at room temperature. Lithium metal chips were used as both the counter electrode and reference electrode. The working electrode is consisted of active material, conductive acetylene black, and polyvinylidene fluoride (PVDF) binder in a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10. The electrolyte is composed of 1 M LiPF6 in a mixture of ethylene carbonate, dimethyl carbonate, and diethyl carbonate (1:1:1 in volume). The electrode was punched into small disks with a diameter of 14 mm. The active material was around 200 μm in thickness, 3 mg in mass, and the loading density was calculated to be around 2 mg cm−2 on copper foil. CR 2032 coin-type cells were fabricated in an Ar-filled glovebox with moisture and oxygen concentrations below 0.1 ppm and sealed with a MSK-110 Compact Hydraulic Crimping Machine.
:10:10. The electrolyte is composed of 1 M LiPF6 in a mixture of ethylene carbonate, dimethyl carbonate, and diethyl carbonate (1:1:1 in volume). The electrode was punched into small disks with a diameter of 14 mm. The active material was around 200 μm in thickness, 3 mg in mass, and the loading density was calculated to be around 2 mg cm−2 on copper foil. CR 2032 coin-type cells were fabricated in an Ar-filled glovebox with moisture and oxygen concentrations below 0.1 ppm and sealed with a MSK-110 Compact Hydraulic Crimping Machine.
3. Results and discussion
The synthetic procedure for Fe2O3/G(H) is schematically illustrated in Fig. 1. Firstly, iron nitrate, L-arginine and graphene oxide were dispersed in water to form a homogeneous suspension. During the subsequent hydrothermal treatment, hydroxyl ions were released from L-arginine through hydrolysis, and Fe(OH)3 nuclei began to form. Due to the existence of oxygen-containing functional groups, such as carboxyl, hydroxyl and epoxyl groups, the newly formed Fe(OH)3 nanoparticles were anchored on the graphene oxide sheets. With extended hydrothermal treatment time, the Fe(OH)3 nanoparticles were converted into Fe2O3 through dehydrogenation and the graphene oxide was reduced to graphene.22 After hydrothermal treated at 180 °C for 12 hours, Fe2O3/G(H) was obtained.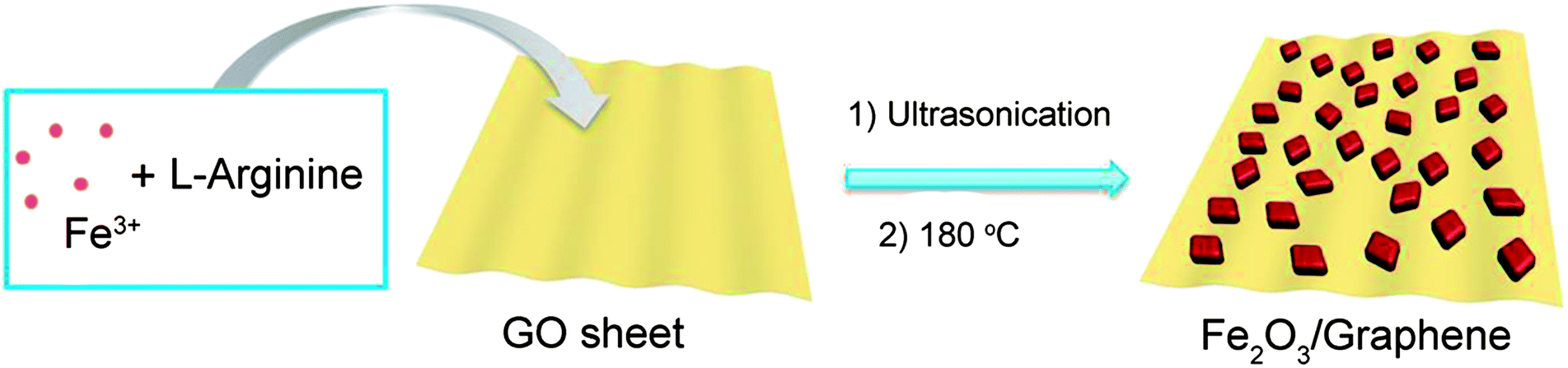 | ||
| Fig. 1 Schematic illustration for the formation of Fe2O3/G(H). | ||
The crystalline structures of the as-synthesised samples were studied by XRD (Fig. 2). For both Fe2O3/G(H) and Fe2O3/G(P), all the diffraction peaks can be exclusively assigned to rhombohedral α-Fe2O3 (JCPDF no. 33-0664), also known as hematite. No apparent diffraction peaks from graphene or graphene oxide can be detected, indicating the ultrathin characteristic of the graphene sheets in the products. Compared to Fe2O3/G(P), Fe2O3/G(H) shows a much stronger intensity in XRD, indicating its better crystallinity. The high crystallinity of Fe2O3/G(H) may be attribute to the weak basic conditions due to the slow release of OH− from L-arginine under hydrothermal treatment.23 By applying Scherrer equation to the strongest (104) diffraction peak, the grain size of Fe2O3 in Fe2O3/G(H) and Fe2O3/G(P) is determined to be 27.17 and 21.45 nm, respectively.
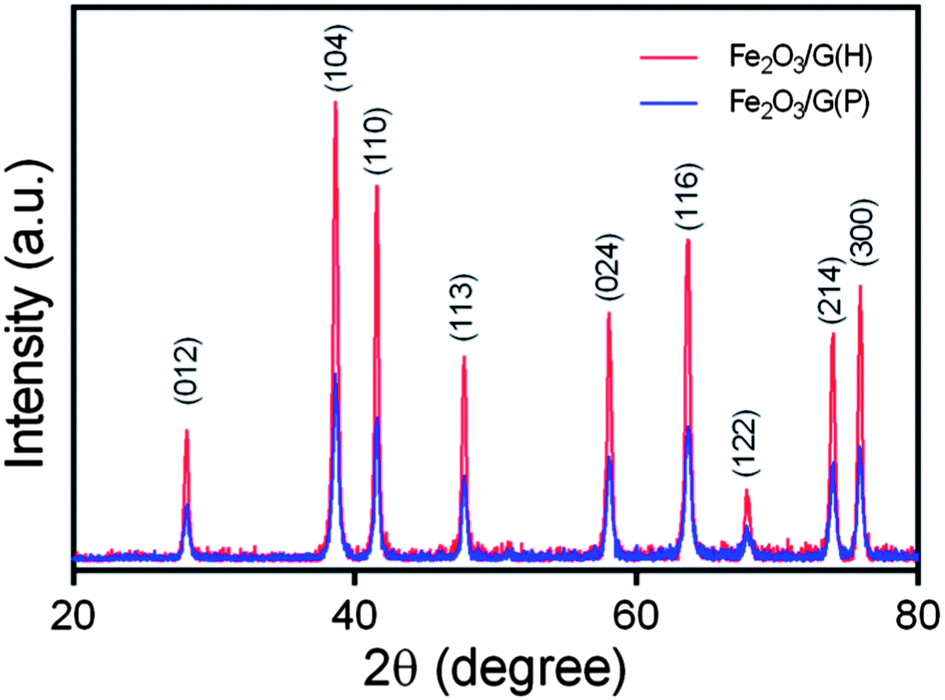 | ||
| Fig. 2 XRD patterns of Fe2O3/G(H) and Fe2O3/G(P). | ||
The morphology and structure of the samples were investigated by SEM and TEM. Low-magnification SEM and TEM images reveal that polyhedral Fe2O3 nanocrystals with sizes of ∼50 nm homogenously decorated on the surface of two dimensional graphene sheets (Fig. 3a and b). Fig. 3c shows the selected area electron diffraction (SAED) pattern of Fe2O3/G(H), where a serious of concentric rings can be found. The diffraction rings from inside to outside can be assigned to the (012), (104), (110), (113), (024) and (116) diffractions of rhombohedral α-Fe2O3, respectively. The high resolution TEM (HRTEM) image of Fe2O3/G(H) is shown in Fig. 3d. From Fig. 3d, well-resolved lattice fringes with spacings of 0.37 nm and 0.27 nm can be clearly distinguished, corresponding to the (012) and (104) interplane spacings of α-Fe2O3, respectively. Fig. 3e and f present the SEM and TEM images of Fe2O3/G(P), which is prepared by using urea as the hydrolysis controlling agent. A morphology quite similar to that of Fe2O3/G(H) can be observed.
 | ||
| Fig. 3 SEM image (a), TEM image (b) SAED pattern (c) and HRTEM image (d) of Fe2O3/G(H); SEM (e) and TEM (f) images of Fe2O3/G(P). | ||
To determine the chemical composition of the Fe2O3-graphene composites, TGA analysis was carried out in air from 50 °C to 600 °C with a heating rate of 5 °C min−1. The results are shown in Fig. 4. The slight weight loss below 200 °C can be attributed to the loss of residual moisture in the samples. The major weight loss between 250 °C and 450 °C can be ascribed to the combustion of graphene in air. From TGA, the graphene content in the nanocomposites is determined to be 17.1% for Fe2O3/G(H) and 16.9% for Fe2O3/G(P).
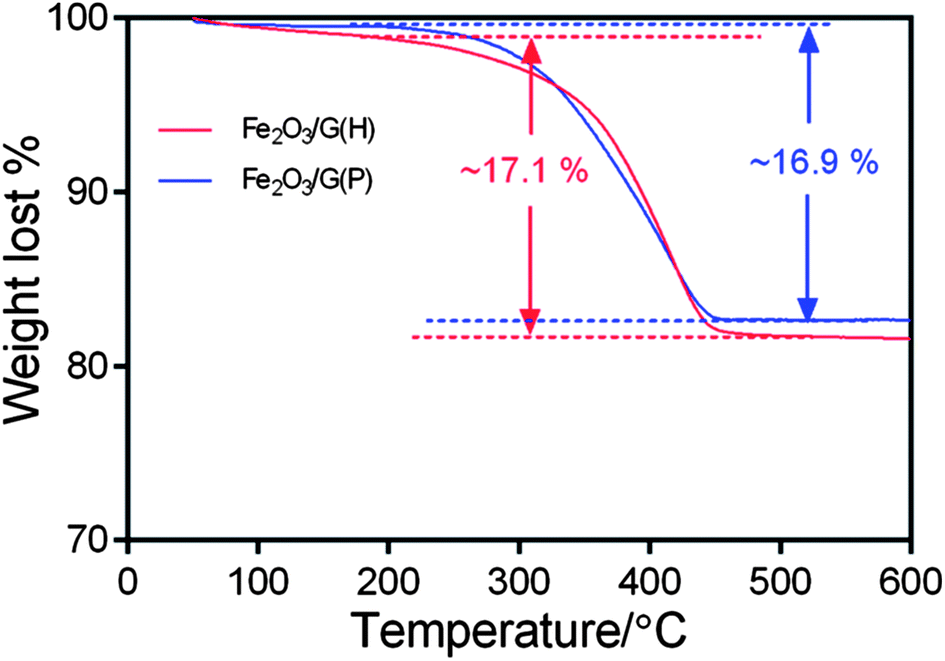 | ||
| Fig. 4 TGA curves of Fe2O3/G(H) and Fe2O3/G(P). | ||
The conversion of graphene oxide into graphene was confirmed by XPS analysis, and the results are shown in Fig. 5. The high resolution C1s XPS spectrum of graphene oxide (Fig. 5a) can be fitted into four components, corresponding to carbon atoms in four different chemical environments: non-oxygenated carbon (C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, 284.8 eV), C in C–O bonds (287.2 eV), carbonyl carbon (CO, 288.9 eV) and carboxylate carbon(O–CO, 289.6 eV).24 Although the C1s spectra of Fe2O3-G(P) (Fig. 5b) and Fe2O3-G(H) (Fig. 5c) also exhibit the same oxygen functionalities, their peak intensities are much weaker than those in graphene oxide. The significant intensity enhancement of non-oxygenated carbon and the decrease of oxygen-containing functionalities demonstrate the efficient reduction of graphene oxide during hydrothermal treatment.
C, 284.8 eV), C in C–O bonds (287.2 eV), carbonyl carbon (CO, 288.9 eV) and carboxylate carbon(O–CO, 289.6 eV).24 Although the C1s spectra of Fe2O3-G(P) (Fig. 5b) and Fe2O3-G(H) (Fig. 5c) also exhibit the same oxygen functionalities, their peak intensities are much weaker than those in graphene oxide. The significant intensity enhancement of non-oxygenated carbon and the decrease of oxygen-containing functionalities demonstrate the efficient reduction of graphene oxide during hydrothermal treatment.
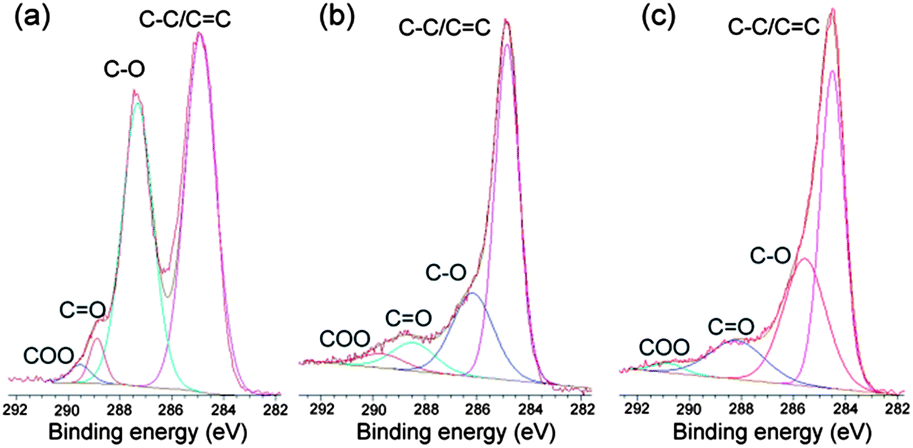 | ||
| Fig. 5 XPS of graphene oxide (a), Fe2O3/G(P) (b) and Fe2O3/G(H) (c). | ||
The electrochemical performances of the samples were then evaluated by galvanostatic charge–discharge in 2032 coin-type cells using lithium chips as the counter electrode. Fig. 6a shows the representative charge–discharge profiles of Fe2O3/G(H) at a current density of 200 mA g−1 between 0.05 and 3 V. In the first discharge profile, three voltage plateaus can be clearly identified. The minor plateau at 1.6 V can be ascribed to the insertion of lithium ion into Fe2O3 crystal structure.6,25 The plateau at 1.1 V can be attributed to the reduction of Fe3+ to Fe2+; while the plateau at 0.8 V can be attributed to the reduction of Fe2+ to Fe0.6,25 The first discharge process delivers an initial discharge capacity of 1268 mA h g−1, the subsequent charge step delivers a charge capacity of 856 mA h g−1, resulting a Coulombic efficiency of 67.5%. The irreversible capacity loss (32.5%) observed in the first cycle may be attributed to the irreversible decomposition of electrolyte and the subsequent formation of solid electrolyte interface (SEI) layer on the surface of the electrode.26 Worth mentioning is that the Fe2O3/G(H) nanocomposites contain a relatively high graphene content, 17.1% by weight. Considering the fact that graphene is also electrochemically active,27 a significant fraction of the reversible capacity should be resulted from graphene.
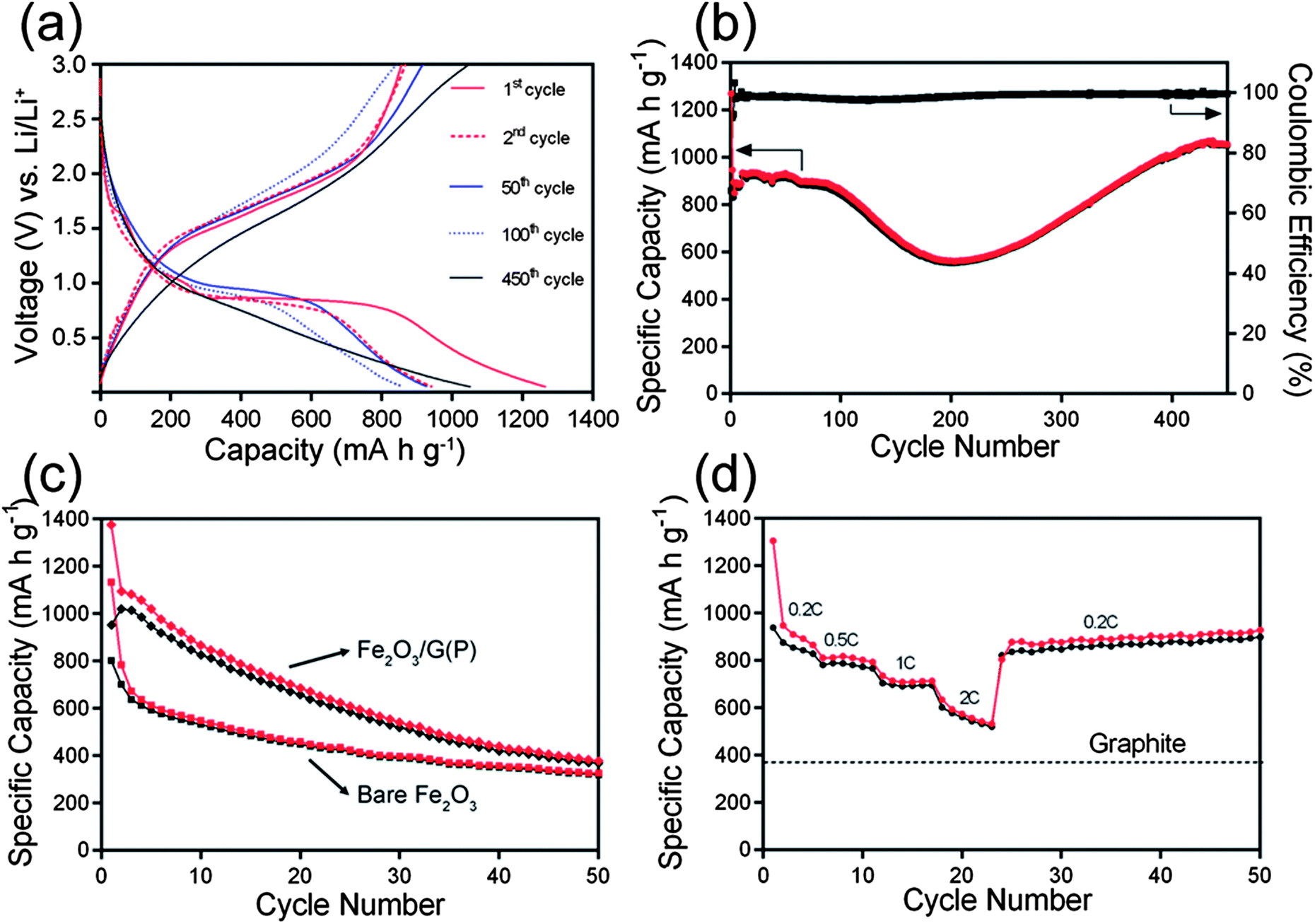 | ||
| Fig. 6 Charge–discharge profiles (a) and cycling performance (b) of Fe2O3/G(H); cycling performance (c) of Fe2O3/G(P) and bare Fe2O3; and rate performance (d) of Fe2O3/G(H). (1 C = 1000 mA g−1). | ||
Fig. 6b shows the cycling performance of Fe2O3/G(H) at a current density of 200 mA g−1. The material shows a relatively stable capacity of ∼900 mA h g−1 in the first 100 cycles. The capacity slowly decreases to 554 mA h g−1 in the second 100 cycles, and increases from 200 to 400 cycles. Similar phenomenon has also been observed in other transition metal oxide anodes and the increase of discharge capacity upon cycling can be generally attributed to the reversible formation/dissolution of a polymeric gel-like film resulting from electrolyte degradation.28–30 After 450 cycles at 200 mA g−1, a capacity of 1049 mA h g−1 can be maintained. This value is significantly higher than the theoretical specific capacity of graphite anodes (372 mA h g−1). The Coulombic efficiency stabilises at 99% from the fifth cycle onward. For comparison, the electrochemical performance of bare Fe2O3 nanocrystals is also studied. The morphology of bare Fe2O3 nanocrystals is similar to that of Fe2O3 nanoparticles in Fe2O3/G(H) in terms of size and crystallinity. However, they show quite poor cycling performances (Fig. 6c); after 50 cycles at 200 mA g−1, the discharge capacity is only 320 mA h g−1. Interestingly, although Fe2O3/G(P) and Fe2O3/G(H) show a similar Fe2O3-nanocrystal-on-graphene structure and graphene content, the cycling performance of Fe2O3/G(P) is much poorer. The capacity decreases continuously from the beginning, and a low capacity of 372 mA h g−1 is obtained after 50 cycles at 200 mA g−1. These results clearly demonstrate that both the highly crystallized Fe2O3 nanocrystals and conductive graphene sheets are indispensible for the extended cycle life of Fe2O3/G(H).
The rate performance was further investigated by cycling the Fe2O3/G(H) electrode at various current densities (200–2000 mA g−1). The material exhibits a reversible capacity of 948 mA h g−1 at 200 mA g−1. When the current density is increase to 2000 mA g−1, a high capacity of 634 mA h g−1 can still be retained (nearly 67% of the capacity at 200 mA g−1), which is still much higher than the theoretical capacity of graphite. Remarkably, when the current is reduced to 200 mA g−1 after the high rate test, a capacity of 877 mA h g−1 can be resumed, exhibiting excellent rate performance when applied as anode materials for LIB applications.
4. Conclusions
In conclusion, we have successfully developed a facile one-step hydrothermal method for the synthesis of highly crystallized Fe2O3 nanocrystals on graphene sheets using L-arginine as the hydrolysis-controlling agent. The synthesised nanocomposites exhibit significantly enhanced cycling performance than bare Fe2O3 nanocrystals and poorly crystallized Fe2O3-graphene nanocomposites. Our finding clearly demonstrates the importance of high crystallinity in achieving high cycling performance in LIB applications, which may also applicable to other metal oxide-graphene nanocomposite based anode materials.Acknowledgements
The authors acknowledge the Australian Research Council for financial support. We also acknowledge the facilities, the scientific and technical assistance from Centre for Microscopy and Microanalysis at the University of Queensland.Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- J. S. Chen, T. Zhu, X. H. Yang, H. G. Yang and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 13162–13164 CrossRef CAS PubMed.
- B. Wang, J. S. Chen, H. B. Wu, Z. Y. Wang and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 17146–17148 CrossRef CAS PubMed.
- L. Zhou, H. Y. Xu, H. W. Zhang, J. Yang, S. B. Hartono, J. Zou and C. Z. Yu, Chem. Commun., 2013, 49, 8695–8697 RSC.
- X. Xu, R. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988–4991 CrossRef CAS PubMed.
- X. J. Zhu, Y. W. Zhu, S. Murali, M. D. Stollers and R. S. Ruoff, ACS Nano, 2011, 5, 3333–3338 CrossRef CAS PubMed.
- M. Zhang, B. H. Qu, D. N. Lei, Y. J. Chen, X. Z. Yu, L. B. Chen, Q. H. Li, Y. G. Wang and T. H. Wang, J. Mater. Chem., 2012, 22, 3868–3874 RSC.
- Y. Q. Zou, J. Kan and Y. Wang, J. Phys. Chem. C, 2011, 115, 20747–20753 CAS.
- N. Kang, J. H. Park, J. Choi, J. Jin, J. Chun, I. G. Jung, J. Jeong, J. G. Park, S. M. Lee, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2012, 51, 6626–6630 CrossRef CAS PubMed.
- B. Koo, H. Xiong, M. D. Slater, V. B. Prakapenka, M. Baasubramanian, P. Podsiadlo, C. S. Johnson, T. Rajh and E. V. Shevchenko, Nano Lett., 2012, 12, 2429–2435 CrossRef CAS PubMed.
- J. Ma, X. D. Zhang, K. Z. Chen, G. C. Li and X. D. Han, J. Mater. Chem. A, 2013, 1, 5545–5553 CAS.
- Y. Nuli, R. Zeng, P. Zhang, Z. P. Guo and H. K. Liu, J. Power Sources, 2008, 184, 456–461 CrossRef CAS PubMed.
- D. Larcher, C. Masquelier, D. Bonnin, Y. Chabre, V. Masson, J. B. Leriche and J. M. Tarascon, J. Electrochem. Soc., 2003, 150, A133–A139 CrossRef CAS PubMed.
- S. Y. Liu, J. Xie, Q. Pan, C. Y. Wu, G. S. Cao, T. J. Z. And and X. B. Zhao, Int. J. Electrochem. Sci., 2012, 7, 354–362 CAS.
- D. Z. Chen, W. Wei, R. N. Wang, J. C. Zhu and L. Guo, New J. Chem., 2012, 36, 1589–1595 RSC.
- S. Bai, S. Q. Chen, X. P. Shen, G. X. Zhu and G. X. Wang, RSC Adv., 2012, 2, 10977–10984 RSC.
- S. B. Yang, Y. Sun, L. Chen, Y. Hernandez, X. L. Feng and K. Mullen, Sci. Rep., 2012, 2, 427 Search PubMed.
- G. W. Zhou, J. L. Wang, P. F. Gao, X. W. Yang, Y. S. He, X. Z. Liao, J. Yang and Z. F. Ma, Ind. Eng. Chem. Res., 2013, 52, 1197–1204 CrossRef CAS.
- X. D. Huang, K. Qian, J. Yang, J. Zhang, L. Li, C. Z. Yu and D. Y. Zhao, Adv. Mater., 2012, 24, 4419–4423 CrossRef CAS PubMed.
- H. Zhang, X. J. Lv, Y. M. Li, Y. Wang and J. H. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- J. J. Zhang, Y. F. Sun, Y. Yao, T. Huang and A. S. Yu, J. Power Sources, 2013, 222, 59–65 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- W. J. Yu, P. X. Hou, F. Li and C. Liu, J. Mater. Chem., 2012, 22, 13756–13763 RSC.
- J. J. Zhang, T. Huang, Z. L. Liu and A. S. Yu, Electrochem. Commun., 2013, 29, 17–20 CrossRef CAS PubMed.
- S.-L. Kuo, W.-R. Liu, C.-P. Kuo, N.-L. Wu and H.-C. Wu, J. Power Sources, 2013, 244, 552–556 CrossRef CAS PubMed.
- S. Grugeon, S. Laruelle, L. Dupont and J. M. Tarascon, Solid State Sci., 2003, 5, 895–904 CrossRef CAS.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dolle, L. Dupont and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A627–A634 CrossRef CAS PubMed.
- L. Zhou, H. B. Wu, T. Zhu and X. W. Lou, J. Mater. Chem., 2012, 22, 827–829 RSC.
| This journal is © The Royal Society of Chemistry 2014 |