The exploration of Kemp's triacid (KTA) as the core for the synthesis of 3-fold symmetric 23-cyclophane, 22-cyclophane and novel linker directed designs†
Santhosh Reddy Nainia,
Subramania Ranganathan*a,
Jhillu Singh Yadav*a,
K. V. S. Ramakrishna*b,
G. Gayatric,
G. Narahari Sastry*c,
K. Basu Royd and
N. Shamala*d
aDiscovery Laboratory, Indian Institute of Chemical Technology, Hyderabad, 500 007, India. E-mail: srgiict@gmail.com; Fax: +91 2719 0757; Tel: +91 90000 90847
bCenter for NMR, Indian Institute of Chemical Technology, Hyderabad, 500 007, India
cMolecular Modeling Group, Organic Chemical Sciences, Indian Institute of Chemical Technology, Hyderabad, 500 007, India
dDepartment of Physics, Indian Institute of Science, Bangalore, 560 012, India
First published on 6th November 2013
Abstract
For the first time, two units of KTA have been linked to three units of cyst-di-OMe. The reaction is noteworthy since it involves the formation of six amide bonds leading to a three-fold symmetric 23-cyclophane (3) harboring a cluster of three S–S bridges. The major product is a di-imide (4), arising from the interaction of a cystine NH with a neighbouring activated ester. A third reaction of tethering KTA with a single cyst-di-OMe unit afforded the flexible compound 6 and, with benzidine, the novel linker directed 7 with orthogonally disposed anchor modules.
Introduction
Sulphur clusters arising from both cystine1 and cysteine display a wide range of functions. They are ubiquitous and amongst those important systems are iron–sulphur clusters,2a–c transferrins,2d metallothionein,2b neurotoxins,2e heat stable enterotoxins2f and channel blockers.3 Recently they have attracted much attention under the name “inhibitory cystine knot (ICK)” which are found in plants, anti microbial peptides and venoms.4 Besides, their notable affinity for complexation with silver5 has found application in radio immunotherapy6 and those complexes also possess strong antibacterial activity.7Generally, sulphur clusters are modular in nature and either form part of a metal lattice or are embedded in a protein scaffold and play a critical role in the cell cycle.
To the best of our knowledge, three fold symmetric closely spaced sulphur clusters8 are not known although we consider that such systems would be an addition to the widening interest in this domain. The synthetic goal was to attach 3 cystine chains to two units of cis,cis-1,3,5-trimethyl cyclohexane 1,3,5-tricarboxylic acid, Kemp's acid, harboring 3 axially oriented non-racemizable carboxyl groups built on a cyclohexane framework. The present work reports, inter alia, the synthesis of 23-cyclophane 3 and 22-cyclophane 4.
The synthesis of 3 and 4 became feasible because of the closely spaced COOH groups in KTA. The role of KTA in the design and synthesis of unusual structures and properties continue to elicit wide interest as highlighted by the following brief account.
cis,cis-1,3,5-Trimethylcyclohexane–1,3,5-tricarboxylic acid (Kemp's triacid, KTA) (1) occupies a unique position in organic chemistry.9 The potential of 1 has not been fully explored, possibly because of its high cost and the difficulties associated with its preparation. The original procedure to 1 from 3,5,7-trimethyl adamantanol10 is not viable because of the unavailability of the precursor. An alternative route from mesitoic acid, though tedious, makes possible the preparation of KTA in gram quantities.11,12 1H NMR studies show that the carboxylic acids are axially oriented in 1 however, the orientation of the trianion is completely equatorial.11
The proximity of the COOH units in 1 enable the ready formation of closely related constructs that are amenable for further elaboration and play a pivotal role in the chemistry of 1. The key reaction of III is the one step transformation to the imide (Scheme 1).9,13
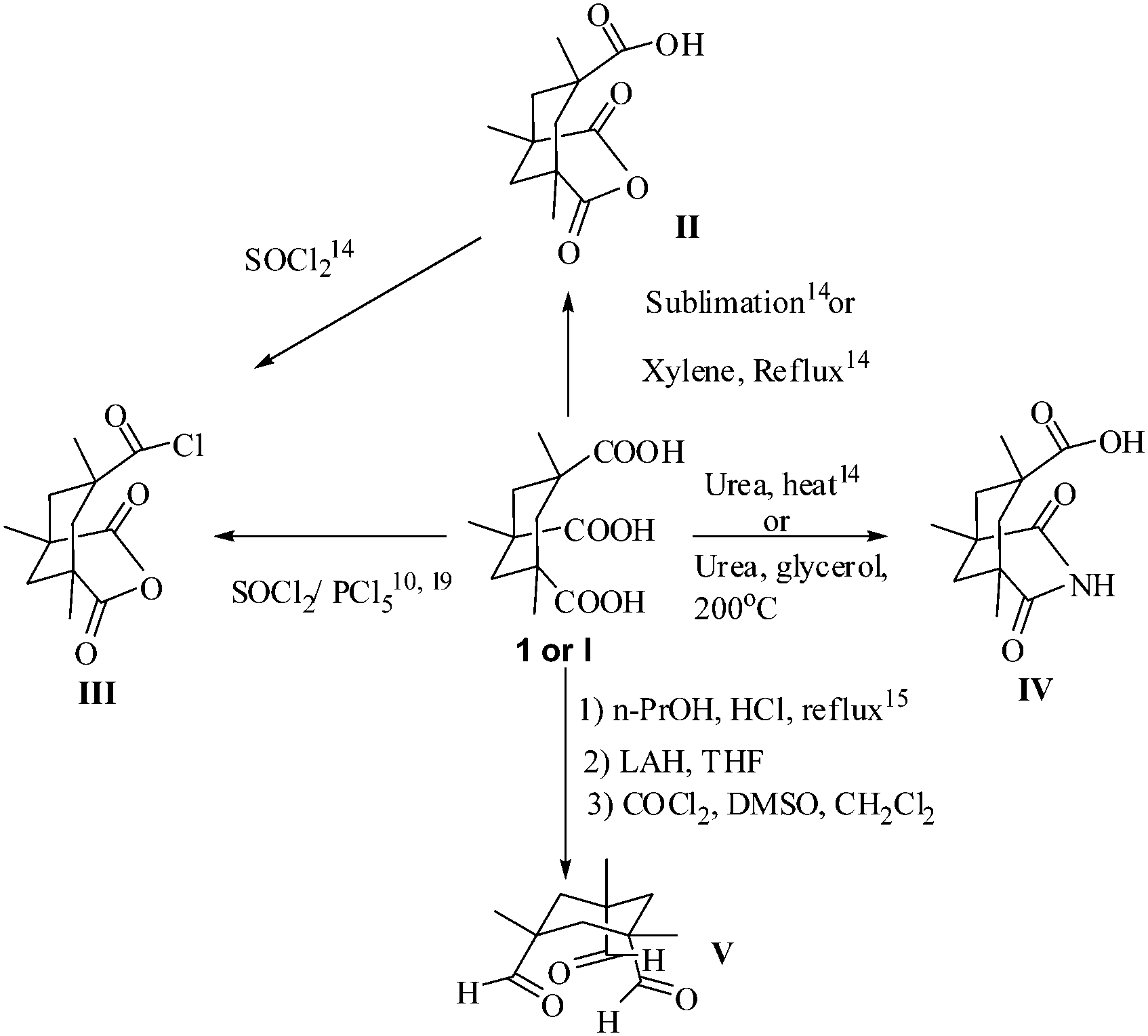 | ||
| Scheme 1 Key transformations of Kemp's triacid. | ||
Much of the notable reactions of 1 are based on a variety of aromatic 1,ω-diamines that readily link two units of III or 1 to form a unique cleft harboring a crown of two carboxylic groups that face each other,13 which are excellent hosts for code bases, distally placed nitrogen lone pairs as in pyrazine and DABCO and a variety of others.11
The creative use of Kemp's triacid (1 or I), implies the generation of a self reproducing system from a template crafted from IV by amidation of a linker harboring an adenosine unit.14 The consequence of the close placement of the CO groups is reflected in a novel π2s + π2s + π2s addition of V in CDCl3 to wurtzilane.15
1 is an excellent anchor for the crafting of clusters in proteins, sugars, polymers, supramolecules and in the design of novel materials.16,17 Although much remains to be done, the principle has already been illustrated. Simple capping of 1 can lead to cage structures as illustrated with alanine ester anchored on a benzene scaffold at the 1, 3 and 5 position carbons.18 A remarkable property of KTA (1) is that it can withstand a very wide range of transformations which makes it a very versatile synthon.19
Results and discussion
The challenge in tethering two units of KTA with 3 units of cyst-di-OMe (2) by normal peptidation with the formation of 6 amide bonds and three closely spaced S–S bridges arises in the isolation of the major product 4 (55%), formed by the interaction of one of the NH groups of the linker with a neighbouring activated ester to form a two fold symmetric cystine linked di-imide. The desired 23-cyclophane 3, having a three-fold axial symmetry and a two-fold equatorial symmetry, was prepared in a 9% yield (Scheme 2). | ||
| Scheme 2 The synthesis of triple and double linked modules from cyst-di-OMe (2) and KTA (1). | ||
The successful preparation of closely spaced, three fold axially symmetric, sulphur cluster 3 is likely to generate interest in several areas20 ranging from metal complexation to drug candidates due to the integration of the chemistry of cystine clusters and KTA.10
The complexity of the formation of 3 is noteworthy since it involves the generation of six covalent bonds from six activated esters. The formation of 4 is rationalised in Scheme 3.
 | ||
| Scheme 3 Rationalisation of the formation of 4. | ||
A practical observation of potential significance is the comparison of the three fold symmetric systems 3 and 8, previously reported by us,21 by linking three cyst-di-OMe units with two units of mesitoyl chloride. We found that the S–S cluster 8 is practically insoluble in all solvents except in DMSO, whereas the cluster 3 from KTA and analogues are soluble in most organic solvents, thus making further elaboration possible (Fig. 1).
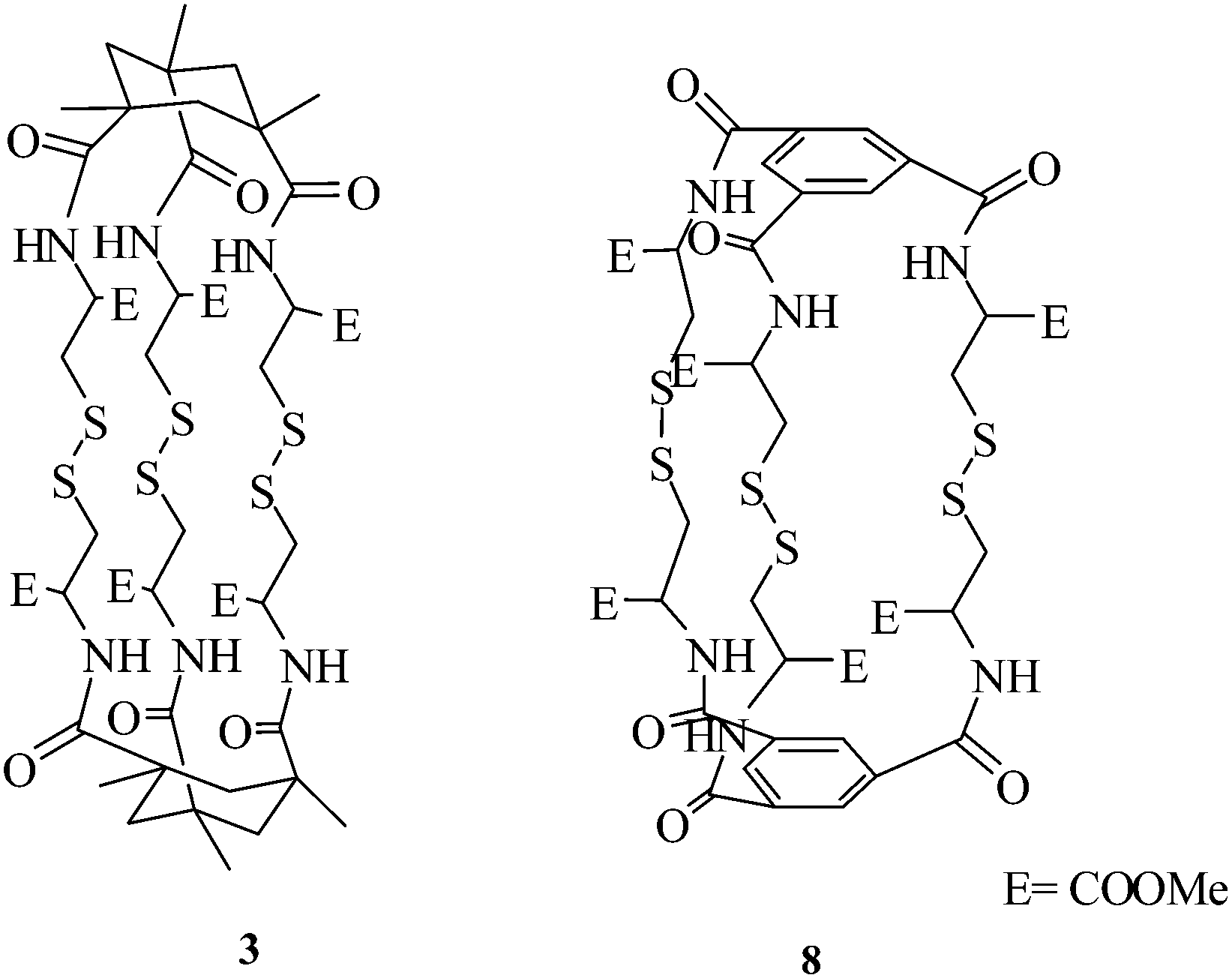 | ||
| Fig. 1 Comparison of compounds 3 and 8. | ||
Tethering of 1 with a single unit of cyst-di-OMe was achieved with 5 (readily prepared from 1 and SOCl2). Thus, the reaction of cyst-di-OMe with equivalent amounts of 5 afforded 6 in 63% yield (Scheme 4).
The structural assignment of 6 is supported by MS and 1H NMR analyses. The rigid linker benzidine, with equivalent amounts of 5, gave 85.5% of 7 (Scheme 5).
The structural assignment of 7 is supported by MS, 1H NMR spectroscopy and X-ray crystallography. The 1H NMR spectrum seems to indicate a twist in the aromatic rings. They are displayed as a clean doublet for 4 protons at 8.76 ppm and a marginally broad peak for the other 4 aromatic protons at 7.25 ppm. The presence of 7 in the preparation of KTA embedded linear supramolecular structures is proved by the extended MS spectrum of 7, which shows peaks at 629 (M + H+) (90%), 646 (M + H3O+) (100%), 1275 (2M + H3O+) (100%), 1903 (3M + H3O+) (30%).
X-ray crystal structure of 7
A solution of 7 in a minimum amount of DMSO was kept aside for one week to give suitable light yellow crystals. The ORTEP diagram of 7, presented in Fig. 2, shows the novel orthogonally disposed cyclohexyl units, clearly directed by the benzidine linker. | ||
| Fig. 2 ORTEP diagram of the Kemp-benzidine (7) molecule with 50% ellipsoid probability. | ||
Kemp-benzidine 7 crystallizes in the monoclinic space group P21/c, with half of the molecule and a co-crystallized solvent molecule dimethyl sulfoxide (DMSO) in the asymmetric unit (Fig. 3).
 | ||
| Fig. 3 The packing of molecules in the Kemp-benzidine (7) crystal. | ||
The solvent molecule (DMSO) in the crystal was located from a difference Fourier map. The co-crystallized solvent molecule, DMSO, has a positional disorder at the atomic site of the sulphur atom, which was thus treated with partial occupancy. All the hydrogen atoms (H2, H3, H4, H5, H6, H101, H102, H131, H132, H151, H152), except those of the CH3-groups at C12, C14 and C17, were located from difference Fourier maps. The hydrogen atoms of the CH3-groups at C12, C14 and C17, were fixed geometrically in an idealized position and allowed to ride with the respective carbon atoms, to which each was bonded, in the final cycles of refinement. The hydrogen atoms of the disordered solvent molecule (DMSO) were geometrically fixed in an idealized position. The final R value was R1 = 0.0534 (wR2 = 0.1643) for 4682 observed reflections with |Fo| ≥ 4σFo and for 301 parameters. The details of the crystal data and structure refinement for Kemp-benzidine are given in the ESI Table.† The packing profile of 7 is given above.
The ORTEP profile of 7 presented in Fig. 2 is noteworthy, since it shows, to the best of our knowledge, the first example of a linker directed orthogonal placement of the COOH groups in the cyclohexane units. In the numerous examples cited (vide supra), the linking of KTA to distally placed lone pairs results in a cavity where the cyclohexyl COOH groups face each other forming a canopy. Chloroform solutions of 3 and 4 (1 eq.), when mixed with methanolic AgClO4 (2 eq.), gave dark brown silver salts as seen in the MS spectra by the clean appearance of characteristic doublet peaks at, respectively, 1319, 1321 and 1051,1053 (M + Ag+). Extended scans did not show further Ag+ complexation.
NMR studies on 3 and 4
NMR spectra (1D and 2D experiments) of compounds 23-cyclophane (3) and 22-cyclophane (4) were obtained at 500 and 600 MHz (1H), as well as at 125 and 150 MHz (13C). The structure and stereochemistry of the compounds were characterized by double quantum filtered spectroscopy (DQFCOSY). The nuclear overhauser spectroscopy (NOESY) spectra presented in Fig. 4 and 5 show the spatial connectivity that exists between several protons (mixing time 500 milli seconds). Total correlation spectroscopy (TOCSY) spectra (see ESI†) which enabled the assignment of the peaks (Table 1). | ||
| Fig. 4 The NOESY spectrum of 23-cyclophane (3). | ||
 | ||
| Fig. 5 The NOESY spectrum of 22-cyclophane (4). (1) NH/CβH (2) NH/H-3(eq) (3) NH/C2–CH3 (4) CαH/Cβ2H. (5) H-1(eq)/C2–CH3 (6) H-1(eq)/C6–CH3 (7) H-5(axi)/C6–CH3 (8) H-1(ax)/H-3(ax) (9) H-3(eq)/C4–CH3. (10) H-3(ax)/H-5(ax). | ||
| Atom pairs | Distances (Å) | Atom pairs | Distances (Å) |
|---|---|---|---|
| Cβ1H2 to Cβ11H2 | 1.8 | H3{eq}toC2CH3 | 2.30 |
| NH to Cα1H | 3.03 | H3{eq}to H3{axi} | 1.82 |
| NH to Cβ11H2 | 2.63 | H1{eq}toH1{axi} | 1.83 |
| NH to H1{eq} | 2.07 | H1{eq} to C6CH3 | 2.56 |
| Cα2H to Cβ2H2 | 3.08 | H1{eq} to C2CH3 | 2.82 |
| Cα2H to Cβ21H2 | 2.93 | H5{eq} to H5{axi} | 1.79 |
| Cα1H to Cβ1H2 | 2.48 | H5{eq} to C4CH3 | 2.50 |
| Cα1H to Cβ21H2 | 2.23 | H1{axi} to C6CH3 | 2.83 |
| Cβ2H2 to Cβ21H2 | 1.78 | H1{axi} to C2CH3 | 2.48 |
23-Cyclophane
The 600 MHz 1H NMR spectrum of compound 23-cyclophane (3) taken in CDCl3 shows very clearly a three-fold axial symmetry, as well as a two-fold equatorial symmetry. Thus, all the three linkers are spatially equivalent from the point of view of NMR. The 6 α-protons and 6 NH protons are observed and the signals of the α-proton are a doublet of doublets of doublets as a result of their coupling with the amide and the β-protons. The methyl protons attached to the cyclohexane ring appear as a singlet. Due to spectral overlaps, it was not possible to get individual chemical shifts for several of the cyclohexyl protons. NOE correlations between the amide and the cyclohexyl protons, and the amide and the methyl protons were noticed. All the spectral data are in agreement with 23-cyclophane.22-Cyclophane
The NH protons appear as a doublet at 5.86 ppm, the Cα1H protons as a clean doublet of doublets of doublets (ddd) at 5.94 ppm, the Cα2H protons as a doublet of doublets at 5.74 ppm, the Cβ1H2, Cβ11H2, Cβ2H2, Cβ21H2 protons as two sets of doublet of doublets (dd), the ester peaks at 3.7 and 3.69, the cyclohexane ring protons as a triplet of doublets (td, δ 3.02, 3.34, 2.04), and the methyl protons attached to the cyclohexane ring as singlets (Fig. 5). The characteristic NOEs between the axial protons H-1/H-3, H-3/H-5 and H-1/H-5 and the equatorial protons H-1/C-6 Me, H-3/C-6 Me and H-5/C-6 Me suggest that protons H-1, H-3 and H-5 are in the same plane. This was further supported by the NOE correlation between H-3(eq)/C-2 Me and H-3(eq)/C-4 Me, NH/H-3(eq), NH/C-2 Me, NH/Cβ1H and Cα1H/Cβ2H (Fig. 6). | ||
| Fig. 6 Representive characteristic NOE correlations in 4. | ||
The energy minimised structures of 3 and 4 (Fig. 7) are in excellent agreement with the NMR observations.
 | ||
| Fig. 7 Energy minimized structures of 23-cyclophane (3) and 22-cyclophane (4) at the B3LYP/6-31G level of theory using the G09 programme.22 | ||
The nature of the bonding in these closely spaced S–S bridges and the potential of 3, 4 and 7 to widen the domain of sulphur clusters and the synthesis of KTA embedded supramolecular structures are currently being pursued.
Conclusion
The preparation of novel closely placed “sulphur clusters” 3 and 4 form a notable addition to the vibrant domain of such modules. The formation of mono silver complexes from 3 and 4 is of interest in radiology. Compound 7 is the first example of a linker directed orthogonal placement of the cyclohexyl units of KTA, which has potential for the formation of dendrimeric and supramolecular structures.Experimental section
General
Melting points were recorded on a Fisher-John's apparatus and are uncorrected. Infrared spectra were recorded on a Thermo Nicolet Nexus 670 spectrometer as KBr pellets and the prominent bands are expressed in cm−1. 1H and 13C NMR spectra were recorded on Varian Gemini 200, Bruker Avance 300, Varian Inova 400, Inova 500, and Bruker-Avance 600 NMR spectrometers. Chemical shifts are expressed in δ (parts per million) with TMS at 0.0000 as the internal reference. The ESI-MS spectra were obtained on a micromass QUATTRO-LC instrument, and the HRMS spectra on a QSTAR XL instrument. Reactions were monitored, when possible, by TLC. Silica gel G (Merck) was used for the TLC, and silica gel (100–200 mesh) for the chromatographic columns. Columns were generally made from a slurry in chloroform and products were eluted with a mixture of chloroform–methanol.I: Cystine-di-OMe dihydrochloride (2): To a suspension of L-cystine (6 g, 25 mmol) in dry methanol (200 ml), a steady stream of dry HCl was passed for 5 h, concentrated to 30 ml, refrigerated, filtered and crystallized from methanol–ether to afford 6.43 g (75%) of (2) m.p 165–175 °C.
IIa: Reaction of Kemp's triacid (1) with cystine-di-OMe: To an ice cooled and stirred solution of Kemp's triacid (0.258 g, 1 mmol) in CH2Cl2 (10 ml), drops of DMF (1 ml) were added followed by EDCI (0.862 g, 4.5 mmol), HOBt (0.609 g, 4.5 mmol) and DIPEA (0.623 ml, 3.6 mmol, d = 0.742). After 10 minutes, L-cystine-di-OMe free base [freshly prepared from L-cystine-di-OMe·2HCl (2) by using sat. Na2CO3 extraction with CH2Cl2 (3 × 25 ml), treatment with Na2SO4 and evaporation, 74% yield] (0.402 g, 1.5 mmol) in CH2Cl2 (5 ml) was added. The reaction mixture was left stirring for 2 days, quenched with sat. NH4Cl, diluted with CHCl3 (10 ml), washed with 1 N HCl (5 ml), sat. NaHCO3 (5 ml) and brine (5 ml), dried with Na2SO4 and evaporated to give 0.320 g of crude product which was chromatographed on silica gel. Elution with CHCl3–MeOH (97.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5) afforded 0.260 g (55%) of 4, mp: 110–117 °C and 0.055 g (9%) of 3 mp: 251–259 °C (dec).
:1.5) afforded 0.260 g (55%) of 4, mp: 110–117 °C and 0.055 g (9%) of 3 mp: 251–259 °C (dec).
23-Cyclophane (3)
1H NMR (500 MHz, CDCl3): δ = 7.62 (d, J = 7.9 Hz, 6H, amideNH), 4.94 (m, 6H; CαH), 3.76 (s, 18H, COOMe), 3.07 (dd, J = 3.8, 14.6 Hz, 6H, CβH1), 2.93 (dd, J = 8.8, 14.6 Hz, 6H, CβH2), 2.75 (d, J = 15.7 Hz, 6H, cyclohexyl CH2), 1.36 (s, 18H, CH3), 1.28–1.25 (m, 6H, cyclohexyl CH2). 13C NMR (CDCl3, 125 MHz): δ = 178.1, 170.9, 52.6, 42.7, 40.9, 37.4, 31.6, 29.7 ppm. IR (cm−1): 3749, 1745, 1644, 1549, 1532, 1466, 1213, 748, 667. ESI-MS (m/z) (%): 1235 (M + Na+) (100%). HRMS (ESI): m/z calcd for C48H72N6O18 S6Na: 1235.3134; found: 1235.3119.22-Cyclophane (4)
1H NMR (600 MHz, CDCl3): δ = 5.86 (d, J = 9.5 Hz, 2H, amideNH), 5.74 (dd, J = 5.3, 1.0 Hz, 2H, Cα2H), 4.94 (ddd, J = 3.6, 9.6, 13.0 Hz, 2H, Cα1H), 3.70, 3.69 (s, s, 12H, COOMe), 3.47 (dd, J = 15.3, 5.3 Hz, 2H, Cβ2H2), 3.40 (dd, J = 14.3, 3.7 Hz, 2H, Cβ1H2), 3.02 (dt, J = 13.8, 2.1 Hz, 2H, H3{eq}), 2.88 (dd, J = 15.3, 5.3 Hz, 2H, Cβ21H2), 2.56 (dd, J = 14.3, 12.4 Hz, 2H, Cβ11H2), 2.34 (dt, J = 15.2, 2.1 Hz, 2H, H1{eq}), 2.04 (dt, J = 13.2, 2.1 Hz, 2H, H5{eq}), 1.57 (m, 2H, H1{axi}) 1.41 (d, J = 13.2 Hz, 2H, H5{axi}), 1.36 (s, 6H, C6CH3), 1.26 (s, 6H, C2CH3),1.24 (s, 6H, C4CH3), 1.06 (d, J = 13.8 Hz, 2H, H3{axi}). 13C NMR (CDCl3, 125 MHz): δ = 176.2, 175.5, 174.6, 171.6, 169.8, 54.3,52.8, 52.7, 50.9, 44.3, 43.7, 43.6,42.4, 40.6, 40.1, 37.5, 37.4, 31.9, 25.9, 25.2 ppm. IR (cm−1): 3360, 1745, 1683, 1519, 1461, 1213, 751, 667. ESI-MS (m/z) (%): 967 (M + Na+) (100%). HRMS (ESI): m/z calcd for C40H56N4O14 S4Na: 967.2571; found: 967.2573.IIb: Reaction of 3 with AgClO4, preparation of mono silver complex (3·Ag): in complete darkness, a solution of 3 (0.005 g, 0.00413 mmol, 1 eq.) in chloroform (1 ml) was admixed with AgClO4 (0.002 g, 0.00826 mmol, 2 eq.) in MeOH (1 ml), the mixture was stirred for 2 h and the clear solution was evaporated to afford the complex as a dark brown compound 3.Ag.
Silver complex: ESI-MS (m/z) (%): 1319, 1321 (M + Ag+) (20%). HRMS (ESI): m/z calcd for C48H72N6O18AgS6: 1319. 2273; found: 1319. 2251.
IIc: Reaction of 4 with AgClO4, preparation of mono silver complex (4·Ag): in complete darkness, a solution of 4 (0.005 g, 0.0053 mmol, 1 eq.) in chloroform (1 ml) was mixed with AgClO4 (0.0022 g, 0.0106 mmol, 2 eq.) in MeOH (1 ml), the mixture was stirred for 2 h and the clear solution was evaporated to afford the complex as the dark brown compound 4·Ag.
Silver complex: ESI-MS (m/z) (%): 1051, 1053 (M + Ag+) (70%) HRMS (ESI): m/z calcd for C40H56N4O14AgS4: 1051.1721; found: 1051.1727.
IIIa: Reaction of 1 with thionylchloride, isolation of anhydride acid chloride 5: a solution of 1 (0.129 g, 0.5 mmol) in SOCl2 (3 ml) was refluxed for 4 h, the residue was evaporated and crystallized from toluene to afford 0.130 g (100% yield) of 5.
IIIb: Reaction of 5 with cystine-di-OMe·2HCl, formation of 6: under nitrogen, to a stirred solution of 5 (0.129 g, 0.5 mmol) in pyridine (5 ml), cystine-di-OMe·2HCl (0.085 g, 0.25 mmol) was added and the reaction mixture was kept at 90 °C overnight. The reaction mixture was evaporated, the residue dissolved in sat. NaHCO3, acidified with 2 N HCl and extracted with CHCl3 to give 0.113 g of 6 (63%). 1H NMR (500 MHz, CDCl3) δ = 5.65 (m, 2H, CαH), 3.78, (s, 6H, COOMe), 2.63 (m, 4H, CβH), 2.5, 2.3, 2.05 (brs, brs, brs, 6H), 1.65 (brs, 2H), (cyclohexyl CH2), 1.4–0.95 (m, 18H and 4H, CH3 and cyclohexyl CH2). IR (cm−1): 1728, 1685, 1461, 1214, 1090, 751, 667. ESI-MS (m/z) (%): 735 (M + Na+) (100%). HRMS (ESI): m/z calcd for C32H44 N2O12NaS2: 735.2276; found: 735.2228 (Scheme 4).
 | ||
| Scheme 4 Rationalisation of the formation of 6. | ||
IIIc: Reaction of 5 with benzidine, formation of 7: under nitrogen, to a stirred solution of 5 (0.1295 g, 0.5 mmol) in pyridine (5 ml), benzidine (0.092 g, 0.5 mmol) and catalytic amounts of DMAP were added and the reaction mixture was kept at 90 °C overnight. The reaction mixture was evaporated, the residue dissolved in sat. NaHCO3, acidified with 2 N HCl and extracted with EtOAc to give 0.134 g of 7 (85.5%). mp: 285–292 °C (dec). 1H NMR (500 MHz, DMSO-d6): δ = 7.65 (d, J = 11 Hz, 4H, aromatic H), 7.25 (br s, 4H, aromatic H), 2.6 (d, J = 14.6 Hz, 2H), 2.2 (d, J = 14 Hz, 2H), 1.55 (d, J = 15 Hz, 2H), 1.35 (d, J = 14 Hz, 4H), 1.25 (d, J = 15 Hz, 2H) (cyclohexyl CH2), 1.2 (s, 18H, CH3); 13C NMR (DMSO-d6, 125 MHz): δ = 177.6, 176.2, 139.2, 136.1, 79.2, 43.1, 42.1, 41.2, 40.1, 30.6, 25.5 ppm. IR (cm−1): 2923, 2853, 1710,1550, 1461, 1214, 1025, 1004, 753, 667. ESI-MS (m/z) (%): 629 (M + H+) (100%). HRMS (ESI): m/z calcd for C36H40N2O8Na: 651.2696; found: 651.2677 (Scheme 5).
 | ||
| Scheme 5 The reaction of 5 with benzidine. | ||
X-ray diffraction study of 7
The data was collected on a BRUKER AXS KAPPA APEXII CCD with MoKα (0.71073 Å) radiation at low temperature (200 K) for Kemp-benzidine 7 and in phi and omega scan type mode. The structure was solved by using direct methods in SHELXS. Initially the structure was refined against F2 isotropically, followed by full matrix anisotropic least-squares refinement using SHELXL-97.23Acknowledgements
Financial support from CSIR, New Delhi is gratefully acknowledged.Notes and references
- S. Ranganathan, K. M. Muraleedharan, M. Vairamani, A. C. Kunwar and A. Ravisankar, Chem. Commun., 2002, 314–315 RSC.
- (a) H. Beinet, R. H. Holm and E. Munck, Science, 1997, 277, 653 CrossRef; (b) W. Braun, M. Vasak, A. H. Robbins, C. D. Stout, G. Wagner, H. R. Kagi and K. Wuthrich, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 10124 CrossRef CAS PubMed; (c) Iron–sulfur proteins, ed. W. Lowenberg, Academic Press, New York, 1973, vol. I and II Search PubMed; (d) N. A. Peterson, B. F. Anderson, G. B. Jameson, J. W. Tweedie and E. N. Baker, Biochemistry, 2000, 39, 6625 CrossRef CAS PubMed; (e) B. M. Olivera, W. R. Gray, R. Zeikus, J. M. McIntosh, J. Varga, J. Rivier, V. Santos and L. J. Cruz, Science, 1985, 230, 1338 CAS; (f) L. Ozoki, T. Sato, H. Kubota, Y. Hata, Y. Katsube and Y. Shimonishi, J. Biol. Chem., 1991, 266, 5934 Search PubMed.
- M. Price-Carter, M. S. Hull and D. P. Goldenburg, Biochemistry, 1998, 37, 9851–9861 CrossRef CAS PubMed; M. Price-Cartier, G. Badaj and D. P. Goldberg, Biochemistry, 2002, 41, 3507–3519 CrossRef.
- K. H. Gowd, K. D. Blais, K. S. Elmslie, A. M. Steiner, B. M. Olivera and G. Bulaj, Peptide Science, 2012, 98, 212–223 CrossRef CAS PubMed.
- A. S. Craig, R. Kataky, D. Parker, H. Adams, N. Bailey and H. Schneider, J. Chem. Soc., Chem. Commun., 1989, 1870–1872 RSC.
- J. R. Murphy, R. Kataky, D. Parker, M. A. W. Eaton, A. T. Millican, A. Harrison and C. Walker, J. Chem. Soc., Chem. Commun., 1989, 792 RSC; J. P. L. Cox, K. J. Jankowski, D. Parker, R. Kataky, M. A. W. Eaton, N. R. A. Beelay, A. T. Millican, K. Millar, B. A. Boyce, A. Harrison and C. Walker, J. Chem. Soc., Chem. Commun., 1989, 796 Search PubMed.
- S. Y. Liau, D. C. Read, W. J. Pugh, J. R. Furr and A. D. Russell, Lett. Appl. Microbiol., 1997, 25, 279–283 CAS; G. P. Ellis, Antimicrobial Activity and action of Silver, Progress in Medicinal Chemistry, vol. 31, ch. 7 Search PubMed.
- For a comprehensive review of “Compounds containing S
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond”, G. W. Kutney and K. Turnbull, Chem. Rev., 1982, 82(4), 334–354 CrossRef.
S bond”, G. W. Kutney and K. Turnbull, Chem. Rev., 1982, 82(4), 334–354 CrossRef. - J. Rebek Jr, Science, 1987, 235, 1478–1484 CAS.
- D. S. Kemp and K. S. Petrakis, J. Org. Chem., 1981, 46, 5140–5143 CrossRef CAS.
- J. Rebek Jr, B. Askew, M. Killoran, D. Nemeth and F. T. Lin, J. Am. Chem. Soc., 1987, 109, 2426–2431 CrossRef.
- A. Steitz Jr, J. Org. Chem., 1968, 33, 2978–2979 CrossRef.
- J. Rebek Jr, L. Marshall, R. Wolak, K. Parris, M. Killoran, B. Askew, D. Nemeth and N. Islam, J. Am. Chem. Soc., 1985, 107, 7476–7481 CrossRef.
- J. Rebek Jr, Angew. Chem., Int. Ed. Engl., 1990, 29, 245–255 CrossRef.
- H. Izumi, O. Setokuchi and Y. Shimizu, J. Org. Chem., 1997, 62, 1173–1175 CrossRef CAS.
- P. Kocis, O. Issakova, N. F. Sepetov and M. Lebl, Tetrahedron Lett., 1995, 36(37), 6623–6626 CrossRef CAS.
- A. Szczepanska, J. L. Espartero, A. J. Moreno-vargas, A. T. Carmona and I. Robina, J. Org. Chem., 2007, 72, 6776–6785 CrossRef CAS PubMed.
- R. D. Ionescu and T. Frejd, Chem. Commun., 2001, 1088–1089 RSC.
- F. M. Menger, J. Bian and V. A. Azov, Angew. Chem., Int. Ed., 2002, 41(14), 2581–2584 CrossRef CAS.
- C. Boulegue, H. J. Musiol, V. Prasad and L. Moroder, Chem. Today, 2006, 24, 4 Search PubMed.
- S. Ranganathan, P. Venkateshwarlu, S. M. Babu, N. S. Reddy, S. J. Basha, A. V. S. Sarma, D. Vijay and G. N. Sastry, Tetrahedron, 2010, 66, 3923–3929 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- G. M. Sheldrick, SHELXS-97, A Program for Automatic Solution of Crystal Structures, University of Göttingen, Göttingen, 1997 CrossRef PubMed; G. M. Sheldrick, SHELXL-97, A program for crystal structure refinement, University of Göttingen, Göttingen, 1997 CrossRef PubMed; G. M. Sheldrick, Acta Cryst., 2008, A64, 112–122 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of compounds 3, 4 and 7, TOCSY spectra of compounds 3 and 4 and X-ray crystallographic data of compound 7. CCDC 927209. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra44327b |
| This journal is © The Royal Society of Chemistry 2014 |