
Ni-substituted LaMnO3 perovskites for ethanol oxidation†
Abstract
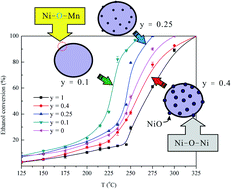
The B-site substitution of LaMnO3 perovskites by Ni was investigated under the oxidation of ethanol. Proper characterization techniques, including BET surface area measurement, XRD, FTIR, XPS, TPR, O2-TPD, and FE-SEM experiments were performed to survey the physicochemical properties of perovskites. The results reveal that up to 25% of Mn can be replaced by Ni; beyond this limit, segregated NiOx can be synthesized. Inserting Ni into the solid solution of perovskite yields unique bridging lattice oxygen sites (Ni–O–Mn) in Ni-doped LaMnO3. Based on catalytic performance in ethanol oxidation, the Ni–O–Mn sites are likely to promote ethanol conversion and the oxidation of acetaldehyde to CO2 at low reaction temperatures. The abatement of intermediates over Ni–O–Mn sites is hypothesized and a plausible reaction pathway is proposed. Moreover, the time on-stream testing revealed that the interaction between Ni and Mn is likely to enhance perovskite's thermal stability in ethanol oxidation.
 Please wait while we load your content...
Please wait while we load your content...

