Stereoselective synthesis of C-sulfonylated aziridines from halomethyl phenyl sulfone and N-tert-butanesulfinyl imines†
Ya Li*,
Haiji Huang,
Zhen Wang,
Fan Yang,
Desheng Li,
Bo Qin and
Xinfeng Ren*
Department of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, 333 Longteng Road, Shanghai 201620, China. E-mail: ya.li@sues.edu.cn; renxf@sues.edu.cn; Fax: +86-21-67791214; Tel: +86-21-67791220
First published on 6th November 2013
Abstract
A highly efficient and stereoselective synthesis of C-sulfonylated aziridines is developed by using a one-step aza-Darzens reaction. When sodium bis(trimethylsilyl)amide (NaHMDS) was used as the base, bromomethyl phenyl sulfone reacted with N-tert-butanesulfinyl imines to afford the 2-sulfonylated aziridine products in very good yields and with stereoselectivities up to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
As the smallest saturated nitrogen-containing heterocyclic compound, aziridine has been receiving considerable attention since its first synthesis at the end of 19th century.1 Due to the intrinsically high reactivity of these three-membered heterocycles, aziridines are being used extensively in the synthesis of a wide variety of bioactive nitrogen-containing compounds.1,2 C-heteroatom-substituted aziridines, i.e., aziridines with the heteroatom substituent (such as halo atom, sulfonyl, alkoxy, nitro, and phosphoryl) directly bonded to the carbon atom(s) of the three-membered ring, represent a valuable subclass of aziridines.3 Owing to the existence of the heteroatom substituents, this kind of aziridines possess unique chemical reactivities,3 and they have been used for the synthesis of many other aziridine derivatives and diverse types of ring-opening products.3,4
The sulfonyl group is well-known for imparting biological activity to a lot of natural and unnatural products.5 Therefore, the C-sulfonylated aziridine is of great structural and synthetic interests. Taking advantage of the basis that the aziridines can undergo regioselective ring-opening reactions3,4 and sulfones can be used for versatile synthetic transformations,6 the C-sulfonylated aziridines are valuable synthetic intermediates in the preparation of bioactive nitrogen-containing molecules.7 Therefore, the synthesis of C-sulfonylated aziridines have drawn a lot of endeavors.6,8 A method reported by Carlier and coworkers used a cascade Michael addition–intramolecular substitution reaction between 1-bromoalkene sulfones and primary amines.8a Deprotonation of N-Bus (Bus = tert-butylsulfonyl)-protected terminal aziridines to generate an aziridinyl anion followed by trapping with benzenesulfonyl fluoride could also access the target compounds.8c,8d However, both methods are only limited to specific compounds. Addition of chloromethyl phenyl sulfone to N-aryl imines for the synthesis of C-sulfonylated aziridines was also developed; however, the substrates were limited only to aryl imines, and the N-aryl protecting group in the substrate makes it difficult to further functionalize the nitrogen atom.7b
As to the asymmetric synthesis of C-sulfonylated aziridines, it has proven challenging and only a few examples have been reported.9 Recently, Bew and coworkers developed an expedient synthesis of C-sulfonylated aziridines from O-acylated (S)-N-(α-methylbenzyl)-N-(2-(phenylsulfonyl)ethyl)-hydroxylamine, based on an intramolecular ring-closure strategy.9a Good yield and moderate stereoselectivity was obtained. However, the O-acylated reactant is not easily accessible and its preparation requires a several-step synthesis starting from (S)–N-(α-methylbenzyl)hydroxylamine, which will limit its practical use. The reaction between N-hydroxy-N-phenylpivalamide and vinylsulfonylbenzene catalyzed by chiral quaternary salts also afforded the C-sulfonylated aziridine product.9b However, this reaction suffers from low yields and poor enatioselectivities. A general method for the highly stereoselective synthesis of C-sulfonylated aziridines, and, especially, the asymmetric synthesis of C-sulfonylated di- and tri-substituted aziridines, is still highly desirable. Herein we report a highly stereoselective synthesis of C-phenylsulfonylated aziridines via a one-step aza-Darzens reaction of halomethyl phenyl sulfone with Ellman's N-tert-butanesulfinyl imines.10
Firstly, chloromethyl phenyl sulfone 1 was chosen as the nucleophile.11 Using imine 2a as the model compound, the aza-Darzens reaction between compound 1 and imine 2a was examined at different reaction conditions. Into an equimolar mixture of sulfone 1 and imine 2a in THF at −70 °C was slowly added a THF solution of LiHMDS (Table 1, entry 1). The reaction mixture was kept at −70 °C for 0.5 h and then quenched with 1 N HCl. A complex mixture of the addition products between (phenylsulfonyl)bromomethyl anion and compound 2a and aziridine stereoisomers was obtained according to HPLC-MS analysis. Surprisingly, when DMF was used as the solvent, this reaction proceeded smoothly and the target product 4a‡ was obtained at 68% yield and with 7:1 stereoselectivity (entry 2). Further optimization using different base and solvent was tried and no improvement was observed (entry 3, Table 1). Given the fact that the bromide ion was a better leaving group, commercially available bromomethylsulfone 3 was used as the nucleophile. Fortunately, its reaction with imine 2a afforded the product 4a at a better yield and a higher stereoselectivity (entry 4–6). After a quick survey of the reactant ratio, the product was obtained at a yield up to 83% and with a diastereoselectivity up to 50:1 (entry 6).
|
|||
|---|---|---|---|
| Entry | Reaction conditionsb | Yield of 4aa | 4a/other diastereomers |
| a Isolated yield.b Determined by HPLC-MS analysis of the crude product.c The molar ratio 2a:3:base = 1.0:1.2:1.2.d Not determined. |
|||
| 1 | 1, LiHMDS, THF, −70 °C | Trace | Nd |
| 2 | 1, LiHMDS, DMF, −50 °C | 68% | 7:1 |
| 3 | 1, LiHMDS, toluene, −70 °C | 23% | Nd |
| 4 | 3, LiHMDS, THF, −70 °C | 78% | 50:1 |
| 5 | 3, NaHMDS, DMF, −50 °C | 45% | Nd |
| 6c | 3, NaHMDS, THF, −70 °C | 83% | 50:1 |

With the optimized reaction conditions in hand (Table 1, entry 6), the scope and limitation of this method was examined. As shown in Table 2, this reaction can be applied to aromatic and aliphatic imines, both with good stereoselectivities. Aromatic imines with electron-withdrawing substituents, such as chloro or the trifluoromethyl group (Table 2, entry 2–4), can often deliver the target products at better yields and with higher stereoselectivities, compared to those with electron-donating substituents (entry 7–9). However, the ortho-chloro substituent resulted in a comparably lower yield of 55% (entry 5), which indicates that steric hindrance may exert an effect on this reaction. Heteroaromatic imines, 2-furyl- and 2-pyridyl imines, were also tried. However, the desired products were not obtained, possibly due to the labile nature of the resultant products under specified reaction conditions. For enolizable aliphatic imines, although the reaction was carried out under strong basic conditions, this reaction proceeded smoothly giving the corresponding aziridine products in moderate yields (Table 2, 53% and 54% yield for 4l and 4m, respectively). Among all the four possible aziridine products, only two aziridine diastereomers were detected by HPLC-MS for all entries except entry 4l. Besides, all the aziridine products listed in Table 2 can be easily purified from the crude reaction mixtures by flash column chromatography.
|
|||
|---|---|---|---|
| Entry | Product | Yield of 4a | 4/other diastereomersb |
| a Isolated yield.b Determined by HPLC-MS analysis of the crude product. | |||
| 1 | 4a, R = C6H5 | 83% | 50:1 |
| 2 | 4b, R = p-Cl-C6H4 | 67% | 4.8:1 |
| 3 | 4c, R = 3,4-ClC6H3 | 75% | 43:1 |
| 4 | 4d, R = m-CF3-C6H4 | 77% | 32:1 |
| 5 | 4e, R = o-Cl-C6H4 | 55% | 5.5:1 |
| 6 | 4f, R = 4-Br-C6H4 | 76% | 6.0:1 |
| 7 | 4g, R = p-MeO-C6H4 | 61% | 3.3:1 |
| 8 | 4h, R = p-(CH3)2C-C6H4 | 63% | 7.0:1 |
| 9 | 4i, R = m-CH3-C6H4 | 62% | 8.4:1 |
| 10 | 4j, R = 2-naphthyl | 66% | 5.0:1 |
| 11 | 4k, R = styryl | 65% | 40:1 |
| 12 | 4l, R = isopropyl | 53% | 7.8:1.1:1 |
| 13 | 4m, R = isobutyl | 54% | 24:1 |
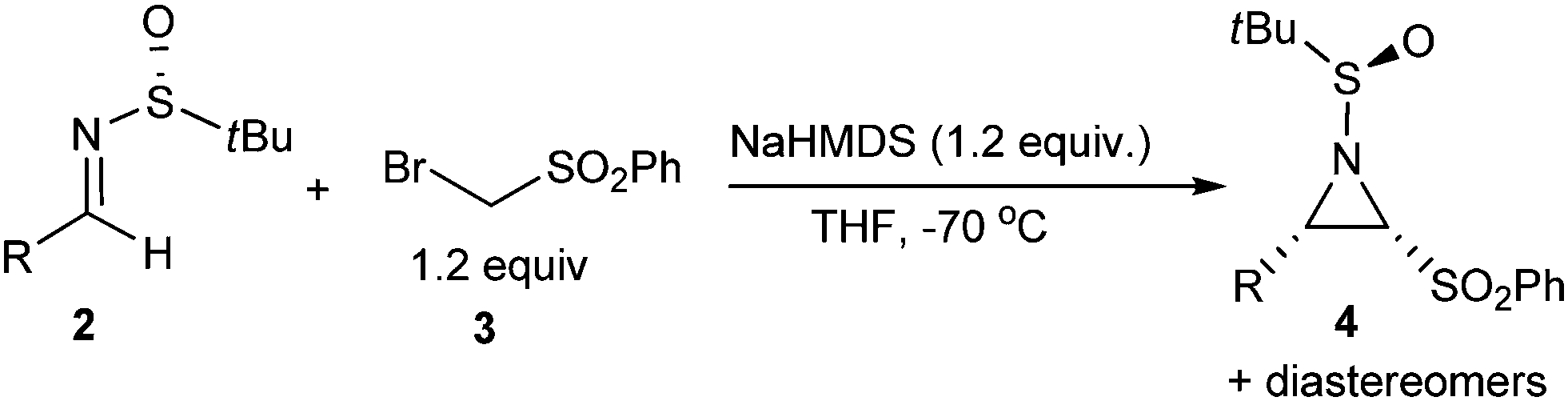
The absolute configuration of sulfinamide 4a was determined by single-crystal X-ray analysis (Fig. 1), and the configurations of others were assigned by analogy. The high cis-selectivity for compound 4 can be explained by a six-membered chairlike transition state, in which the metal coordinates with one of the oxygen of the sulfonyl group and the sulfinyl imine nitrogen atom. The sulfinyl imine is assumed to adopt the s-cis conformation in the transition state, as indicated by recent studies that the sulfinyl oxygen in a s-cis arrangement with respect to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond is the most stable conformation for the N-(phenylsulfinyl) imine.12 Thus, the approach of the nucleophile to the less hindered face of the imine would lead to the observed stereoselectivity as shown in Fig. 2. A similar transition state was also reported for the asymmetric synthesis of aziridine 2-phosphonates from diethyl iodomethylphosphonate and enantiopure sulfinimines.3c
N bond is the most stable conformation for the N-(phenylsulfinyl) imine.12 Thus, the approach of the nucleophile to the less hindered face of the imine would lead to the observed stereoselectivity as shown in Fig. 2. A similar transition state was also reported for the asymmetric synthesis of aziridine 2-phosphonates from diethyl iodomethylphosphonate and enantiopure sulfinimines.3c
 | ||
| Fig. 1 X-ray crystal structure of 4a. | ||
 | ||
| Fig. 2 Transition state for the formation of 4. | ||
Next, several sulfinylketimines were investigated, and they all reacted smoothly under the same reaction conditions. As shown in Scheme 1, the electronic withdrawing/donating nature of the substituent on the aromatic ring has little effect on this reaction, and very good yield was obtained for each case (Scheme 1). However, when ketimine 5d derived from acetophenone was subjected to the reported reaction conditions, no desired product was obtained, which may be due to its low reactivity and enolization.
 | ||
| Scheme 1 Synthesis of tri-substituted aziridines 5. | ||
Selectivie C–N bond cleavage is very useful in the preparation of valuable synthetic intermediates. When compound 4a was treated with BF3·OEt2 in CH2Cl2 solvent, the carbon-skeleton rearranged product 6 was obtained in 61% yield (Scheme 2).13 The formation of enamines via ring-opening of aziridines is important both in terms of aziridine chemistry and the applications of enamines in organic synthesis.14 The structure of compound 6 was determined by single-crystal X-ray analysis (Fig. 3).
 | ||
| Scheme 2 C–N Bond cleavage of aziridine 4a. | ||
 | ||
| Fig. 3 X-ray crystal structure of 6. | ||
N-Sulfonyl activation is often desired because aziridines with N-sulfonyl activation often afford superior reactivity and regiospecificity.3,4 Therefore, introduction this activating group by oxidation of compound 4 with MCPBA was attempted. This oxidation reaction proceeded smoothly and the corresponding oxidation products 7a§ and 7b were obtained in 87% and 85% yields, respectively (Scheme 3). Efforts were also tried to remove the tert-butyl sulfinyl group under acidic (such as HCl in methanol, or TFA in acetone)10 or strong basic (such as CH3MgBr in THF)3b conditions. However, the substrate 4a decomposed gradually under the above specified reaction conditions.
 | ||
| Scheme 3 Oxidation of compound 4a to the N-sulfonylated 7. | ||
To further explore the chemical reactivity of the C-sulfonylated aziridine motif, compound 7b was investigated under typical Suzuki cross-coupling reaction conditions (Scheme 4). This reaction proceeded smoothly, giving the corresponding coupling product 8 in good yield, which indicates the tolerance of the chemical functionalities to the reaction conditions.
 | ||
| Scheme 4 Suzuki coupling reaction of compound 7b. | ||
In conclusion, a highly stereoselective and efficient synthesis of 2-sulfonylated aziridines via a one-step aza-Darzens reaction was developed. Reaction of (R)-(N-tert-butylsulfinyl)aldimines with bromomethyl phenyl sulfone affords the corresponding aziridines products at very good yields and with high stereoselectivities (up to 50:1). This method can be applied to both aromatic and aliphatic imines. The synthetic applications of C-sulfonylated aziridines are under further investigation in our lab.
Acknowledgements
Support of our work by the National Natural Science Foundation of China (21102089), Innovation Program of Shanghai Municipal Education Commission (12YZ155), and Innovation Program of University Students in Shanghai Higher Education Institutions (cs1204001), is gratefully acknowledged.Notes and references
- (a) J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247 RSC; (b) D. Tanner, Angew. Chem., Int. Ed., 1994, 33, 599 CrossRef; (c) U. M. Lindström and P. Somfai, Synthesis, 1998, 109 CrossRef; (d) H. M. I. Osborn and J. Sweeney, Tetrahedron: Asymmetry, 1997, 8, 1693 CrossRef CAS.
- (a) B. Zwanenburg and P. ten Holte, Top. Curr. Chem., 2001, 216, 93 CrossRef CAS; (b) W. McCoull and F. A. Davis, Synthesis, 2000, 1347 CrossRef CAS.
- (a) N. De Kimpe, R. Verhe, L. De Buyck and N. Schamp, J. Org. Chem., 1981, 46, 2079 CrossRef CAS; (b) F. A. Davis, T. Ramachandar and Y. Wu, J. Org. Chem., 2003, 68, 6894 CrossRef CAS PubMed; (c) F. A. Davis, Y. Wu, H. Yan, W. McCoull and K. R. Prasad, J. Org. Chem., 2003, 68, 2410 CrossRef CAS PubMed; (d) M. Mihara, Y. Ishino, S. Minakata and M. Komatsu, J. Org. Chem., 2005, 70, 5320 CrossRef CAS PubMed; (e) T. Ooi, M. Takahashi, K. Doda and K. Maruoka, J. Am. Chem. Soc., 2002, 124, 7640 CrossRef CAS PubMed; (f) S. Fioravanti, L. Pellacani, S. Stabile, P. A. Tardella and R. Ballini, Tetrahedron Lett., 1997, 38, 3309 CrossRef CAS; (g) G. Verniest, F. Colpaert, E. Van Hende and N. De Kimpe, J. Org. Chem., 2007, 72, 8569 CrossRef CAS PubMed.
- G. S. Singh, M. D'hooghe and N. De Kimpe, Chem. Rev., 2007, 107, 2080 CrossRef CAS PubMed.
- (a) C.-B. Xue, X.-T. Chen, X. He, J. Roderick, R. L. Corbett, B. Ghavimi, R.-Q. Liu, M. B. Covington, M. Qian, M. D. Ribadeneira, K. Vaddi, J. Trzaskos, R. C. Newton, J. J.-W. Duan and C. P. Decicco, Bioorg. Med. Chem. Lett., 2004, 14, 4453 CrossRef CAS PubMed; (b) H. Tamamura, Y. Koh, S. Ueda, Y. Sasaki, T. Yamasaki, M. Aoki, K. Maeda, Y. Watai, H. Arikuni, A. Otaka, H. Mitsuya and N. Fujii, J. Med. Chem., 2003, 46, 1764 CrossRef CAS PubMed; (c) S. Nakatani, K. Hidaka, E. Ami, K. Nakahara, A. Sato, J.-T. Nguyen, Y. Hamada, Y. Hori, N. Ohnishi, A. Nagai, T. Kimura, Y. Hayashi and Y. Kiso, J. Med. Chem., 2008, 51, 2992 CrossRef CAS PubMed; (d) S. N. Raja, B. W. Surber, J. Du and J. L. Cross, J. Labelled Compd. Radiopharm., 2009, 52, 98 CrossRef CAS.
- N. S. Simpkins, Sulphones in Organic Synthesis, Pergamon Press, 1993 Search PubMed.
- (a) A. Viso, R. Fernández de la Pradilla, M. Ureña, R. H. Bates, M. A. del Águila and I. Colomer, J. Org. Chem., 2012, 77, 525 CrossRef CAS PubMed; (b) V. Reutrakul, V. Prapansiri and C. Panyachotipun, Tetrahedron Lett., 1984, 25, 1949 CrossRef CAS; (c) R. E. Dabby, J. Kenyon and R. F. Mason, J. Chem. Soc., 1952, 4881 RSC; (d) M. Julia and D. Arnould, Bull. Soc. Chim. Fr., 1973, 743 CAS; (e) J.-M. Gaillot, Y. Gelas-Mialhe and R. Vessiere, Chem. Lett., 1983, 1137 CrossRef CAS; (f) T. Satoh, M. Ozawa, K. Takano and M. Kudo, Tetrahedron Lett., 1998, 39, 2345 CrossRef CAS.
- (a) P. Carlier, Y. Gelas-Mialhe and R. Vessiere, Can. J. Chem., 1977, 55, 3190 CAS; (b) S. Calet and H. Alper, Tetrahedron Lett., 1986, 27, 2739 CrossRef CAS; (c) D. M. Hodgson, P. G. Humphreys and J. G. Ward, Org. Lett., 2005, 7, 1153 CrossRef CAS PubMed; (d) D. M. Hodgson, S. P. Hughes, A. L. Thompson and T. D. Heightman, Org. Lett., 2008, 10, 3453 CrossRef CAS PubMed; (e) M. S. Valle, A. Tarrade-Matha, P. Dauban and R. H. Dodd, Tetrahedron, 2008, 64, 419 CrossRef CAS; (f) B. Bachowska and T. Zujewska, Monatsh. Chem., 2001, 132, 849 CrossRef CAS; (g) M. Makosza, T. Glinka, S. Ostrowski and A. Rykowski, Chem. Lett., 1987, 61 CrossRef CAS; (h) S. Ostrowski and M. Makosza, Tetrahedron, 1988, 44, 1721 CrossRef CAS.
- (a) S. P. Bew, D. L. Hughes, V. Savic, K. M. Soapi and M. A. Wilsona, Chem. Commun., 2006, 3513 RSC; (b) J. Aires-de-Sousa, S. Prabhakar, A. M. Lobo, A. M. Rosa, M. J. S. Gomes, M. C. Corvo, D. J. Williams and A. J. P. White, Tetrahedron: Asymmetry, 2001, 12, 3349 CrossRef; (c) V. D. B. Bonifácio, C. González-Bello, H. S. Rzepa, S. Prabhakar and A. M. Lobo, Synlett, 2010, 145 Search PubMed.
- For the applications of butanesulfinyl imines in synthesis, see: (a) J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984 CrossRef CAS PubMed; (b) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS PubMed.
- For additions of fluoromethyl phenyl sulfone to butanesulfinyl imines, see: (a) J. Liu and J. Hu, Chem.–Eur. J., 2010, 16, 11443 CrossRef CAS PubMed; (b) Y. Li, C. Ni, J. Liu, L. Zhang, J. Zheng, L. Zhu and J. Hu, Org. Lett., 2006, 8, 1693 CrossRef CAS PubMed.
- Y. Arroyo, A. Meana, J. F. Rodríguez, M. A. Sanz-Tejedor, I. Aloson and J. L. García Ruano, J. Org. Chem., 2009, 74, 4217 CrossRef CAS PubMed.
- For ring-opening reaction of N-(aziridin-2-ylmethylene)hydrazines to functionalized enamines, see: Z. Zhang, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 5334 RSC.
- For the applications of enamines in organic synthesis, see: (a) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed; (b) R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and copies of the 1H and 13C NMR spectra of the new compounds. CCDC 938808 (for 4a), 966482 (for 6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra44174a |
| ‡ Typical experimental procedure for the synthesis of 2-sulfonylated cis-aziridine 4a: NaHMDS (1.2 equiv, 1.2 mmol, 1.0 mol L−1 in THF) was added to a mixture of the imine 2a (1.0 mmol) and bromomethyl phenyl sulfone (1.2 equiv, 1.2 mmol) in THF (3.0 mL) at −70 °C. Reaction mixture was stirred over 0.5 h. Then half saturated NH4Cl–H2O (10 mL) was added at −70 °C and the quenched reaction mixture was extracted three times with ethyl acetate (20 mL × 3). The combined organic layers were dried over anhydrous MgSO4. Evaporation of the solvent afforded the crude product, which was subject to flash chromatography to give pure aziridine 4a (302 mg, 83%). Compound 4a: white solid, mp 122.7–128.6 °C (from ethyl acetate/hexane); [α]25D − 7.0 (c 1.06 in CHCl3); IR (film, νmax/cm−1): 1448, 1333, 1157, 1081, 741, 690; 1H NMR (400 MHz, CDCl3) δ 7.64–7.56 (m, 1H), 7.54–7.49 (m, 2H), 7.46–7.38 (m, 4H), 7.37–7.30 (m, 3H), 4.00 (d, J = 6.5 Hz, 1H), 3.64 (d, J = 6.5 Hz, 1H), 1.26 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 138.4, 134.0, 129.4, 129.0, 128.9, 128.6, 128.2, 128.1, 57.8, 54.5, 38.4, 22.4; ESI (m/z) 364.1 (M+ + 1), 386.1 (M+ + 23); HRMS (ESI) calcd. For C18H21NO3S2Na (M + Na+): 386.0855, Found 386.0855. |
| § Typical experimental procedure for oxidation of compound 4a to N-sulfonylated product 7a: compound 4a (364 mg, 1 mmol) was dissolved in 5 mL dichloromethane, then 1.2 mmol MCPBA was added at 0 °C. The reaction mixture was stirred at 0 °C for 4 h and was then 2 N Na2CO3 (4 mL) was added. The reaction mixture was extracted three times with ethyl acetate (20 mL × 3). The combined organic layers were dried over anhydrous MgSO4. Evaporation of the solvent afforded the crude product, which was subject to flash chromatography to give the corresponding aziridine 7a (330 mg, 87%). Compound 7a: white solid, mp 136.8–138.1 °C (from ethyl acetate/hexane); [α]25D + 47.7 (c 0.66 in CHCl3); IR(film, νmax/cm−1): 1449, 1324, 1160, 1128, 765, 713; 1H NMR (400 MHz, CDCl3) δ 7.65–7.57 (m, 1H), 7.54 (dd, J = 8.4, 1.2 Hz, 2H), 7.42 (t, J = 7.9 Hz, 2H), 7.40–7.29 (m, 5H), 4.19 (d, J = 6.7 Hz, 1H), 4.10 (d, J = 6.7 Hz, 1H), 1.60 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 137.7, 134.2, 129.0, 128.8, 128.7, 128.5, 128.2, 128.1, 60.9, 57.8, 45.8, 24.0; ESI (m/z) 402.1 (M+ + 23); HRMS (ESI) calcd. For C18H21NO4S2Na (M + Na+): 402.0804, Found 402.0802. |
| This journal is © The Royal Society of Chemistry 2014 |