One-pot synthesis of poly(2,6-dimethyl-1,4-phenylene oxide)/polystyrene alloy with CuCl2/4-dimethylaminopyridine as a versatile catalyst in water
Huan
Wang
,
Baoqing
Shentu
* and
Zhixue
Weng
State Key Lab of Chemical Engineering, Department of Chemical and biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: shentu@zju.edu.cn; Fax: +86 571 87951612; Tel: +86 571 87951612
First published on 5th November 2013
Abstract
A one-pot synthetic approach for the preparation of poly(2,6-dimethyl-1,4-phenylene oxide)/polystyrene (PPO/PS) alloy in aqueous medium was developed. The method was based on the combination of the oxidative coupling polymerization of 2,6-dimethylphenol (DMP) to form PPO in the presence of a reactive swelling agent, styrene (St), and in situ reverse atom transfer radical polymerization (RATRP) of St initiated by 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (AIBI) in alkaline aqueous solution. The complex of CuCl2 and 4-dimethylaminopyridine (DMAP) was utilized as the catalyst to catalyze the two types of polymerization mentioned above. Oxygen was employed as the sole oxidant to synthesize PPO. The introduction of St during the oxidative polymerization of DMP could increase the molecular weight of PPO. Finally thermodynamically compatible PPO/PS alloy was successfully prepared.
1. Introduction
Poly(2,6-dimethyl-1,4-phenylene oxide) (PPO) is a widely accepted engineering plastic used in a variety of fields because of its good mechanical, dimensional stabilities, low water uptake, and low dielectric characteristics.1 However, the glass transition temperature (Tg) of PPO is about 210 °C, furthermore, due to its high melt viscosity and the low oxidative stability, PPO has a problem regarding molding and it could not be used as a thermoplastic only by itself. Therefore, blending with another plastic such as polystyrene (PS) is required in order to decrease the glass transition temperature for injection processing.PPO has developed into important engineering thermoplastics in the short period of time since the discovery of the oxidative polymerization of phenols.2 The largest commercial usage of PPO is Noryl (Saudi Basic Industry Corp.) engineering resins, which are alloys of PPO and PS. Since PPO and PS are thermodynamically compatible over the complete composition range, a Noryl resin has came into the market for these days.
Synthesizing PPO alloy in water would be significance from the viewpoint of green chemistry, furthermore, PPO alloy can be separated from water easily due to its insolubility in water.3,4 While there have been only a few researches about the synthesis of PPO alloy in water. In our previous study, a one-pot synthetic method for preparing PPO/PS alloy in reactor containing aqueous medium was proposed.5 During the oxidative polymerization of DMP with potassium ferricyanide as the oxidant, the reactive styrene was used as swelling agent to increase the molecular weight of PPO. After the oxidative polymerization of DMP, styrene was in situ polymerized under the initiation of peroxide. However, during the process mentioned above, both the use of potassium ferricyanide as the oxidant and the large amount of sodium hydroxide engaged in the reaction were not adequate from the point of green process.
On the other hand, DMP could be oxidatively polymerized to give PPO in the presence of copper complex catalyst and oxygen in water medium, which would meet the demand of green chemistry.3,4 Moreover, in our previous studies of employing copper complex as catalyst in the reaction, only a little amount of NaOH was needed to synthesize PPO in water.6,7 However, bivalent copper salts are inhibitors of conventional radical polymerization, thus, a reverse atom transfer radical polymerization (RATRP) of St could be considered in this system. RATRP is based on transition metal-catalyzed atom transfer radical addition including the reversible formation of carbon–halogen bonds by transition metals in a high oxidation state, such as copper(II).8–14 The RATRP method was successfully applied to the living radical polymerization of St, MMA etc. And among all of the transition metals, copper appears to be the most robust because of its versatility. In this way, a copper complex could be selected to be the catalyst for the polymerizations of both DMP and St.
This paper reports a one-pot synthetic method of preparing PPO/PS alloy in water medium based on green chemistry. The reactive swelling agent, styrene (St) was added into the mixture to swell the polymer particles during the oxidative polymerization of DMP and increase the mobility of the oligomer chains and facilitate the oxidative polymerization at the later period, which would result in the increase of the molecular weight of PPO. And then in situ RATRP of St was initiated by 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (AIBI). The polymerization of DMP and St were both catalyzed by the complex of CuCl2 and 4-dimethylaminopyridine (DMAP). Finally thermodynamically compatible PPO/PS alloy was successfully prepared.
2. Experimental
2.1. Materials
DMP was purchased from Aldrich Co. Styrene (St) obtained from Sinopharm Chemical Reagent Co., Ltd. was distilled under reduced pressure and kept refrigerated until use. Analytically pure sodium hydroxide (NaOH), sodium chloride (NaCl), and copper(II) chloride dehydrate (CuCl2·2H2O) were supplied by Sinopharm Chemical Reagent Co., Ltd. Chemical pure sodium dodecyl sulfate (SDS) was also supplied by Sinopharm Chemical Reagent Co., Ltd. Analytically pure 4-dimethylaminopyridine (DMAP) and 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (AIBI or VA-044) were purchased from Aladdin Chemistry Co. and J&K Scientific Ltd, respectively. 2-Butanone was obtained from Hangzhou Shuanglin Chemical Reagent Factory.2.2. Preparation of PPO/PS alloy
A typical procedure for the preparation of PPO/PS alloy is as follows: DMP (3.05 g, 0.025 mol) was dissolved in water (95 mL) containing NaOH (1 g, 0.025 mol) and SDS (0.8 g, 2.5 mmol). CuCl2·2H2O (0.0171 g, 0.1 mmol) and DMAP (0.024 g, 0.2 mmol) in 5 mL water was added to the solution, and the mixture was vigorously stirred under oxygen at 50 °C for 3 h. Then St (2.6 g, 0.025 mol) was added into the reaction mixture and the oxidative polymerization continued for 21 h. After the oxidative polymerization of DMP, AIBI (0.404 g, 0.125 mmol) was added into the mixture and nitrogen was purged into the reactor to exhaust the residual oxygen. The radical polymerization of St was conducted at 85 °C for 20 h. The product was separated as powder from the reaction mixture by simple filtration after salted out.2.3. Characterizations
Fourier transform infrared (FTIR) spectroscopic analysis was carried out on a NICOLET 5700 spectrometer (Thermo Electron Corp., USA). A sample of finely crushed KBr was used as the background. The spectra were collected in the frequency range of 400–4000 cm−1 with a resolution of 4 cm−1 by averaging 16 scans.13C NMR spectra were recorded on a 400 MHz Bruker AVANCE II nuclear magnetic resonance spectrometer (Bruker BioSpin Corporation, Switzerland) using CDCl3 as the solvent.
To analyze the composition of PPO/PS blend, the product was extracted firstly by water to eliminate the water-soluble impurities, and then acetonitrile to remove the byproduct DPQ, at last 2-butanone to separate PS, then the proportion of PS and PPO could be determined respectively by measuring the weights before and after each extraction. In addition, the yield of PPO and conversion of St could be calculated according to the weights of these two polymers and the initial amounts of two monomers.
To confirm that the polymerization of St followed the rules of RATRP, a chain extension reaction was conducted. The solution containing PS after extraction was distilled under reduced pressure, and the polymer was used as macroinitiator to initiate the conventional ATRP of St in the presence of CuCl and DMAP.
The molecular weight and polydispersity of PPO and PS were determined by gel permeation chromatography (GPC, Waters 1525/2414, Waters Instrument, Milford, Massachusetts) equipped with Waters Styragel HT4/HT3/HR1 columns and a refractive index detector at 30 °C. The mobile phase was toluene and maintained at a flow rate of 1.0 mL min−1. The molecular weight was calibrated with polystyrene standards.
The glass transition temperature (Tg) of blends were determined by differential scanning calorimetry (DSC) measurements performed on a PE DSC-7 (Perkin-Elmer). The sample was heated from room temperature to 250 °C at a rate of 10 °C min−1 under N2 atmosphere.
3. Results and discussion
3.1. Synthesis and characterization of PPO/PS alloy
The synthetic procedure for the preparation of PPO/PS alloy in water medium involved adding St during the process of oxidative polymerization of DMP and the in situ polymerization of St with the initiation of AIBI after the polymerization of DMP. CuCl2/DMAP was employed as the catalyst for both the oxidative polymerization of DMP and in situ polymerization of St. Particularly, since DMAP can dissolve in water and St, CuCl2/DMAP could effectively catalyze the oxidative polymerization of oligomers and a little DMP dissolved in water as well as the ones swelled in St. Moreover, after the polymerization of DMP, reverse atom transfer radical polymerization (RATRP) of St could be in situ initiated by AIBI, and the molecular weight of PS could easily be controlled by either varying the reaction time or changing the amount of AIBI. With the same amount of initiator, molecular weights of 5100 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 were obtained when the reaction times were 7.5 h and 20 h, respectively. In addition, the yields of PPO and PS could maintain about 90% and 73%, respectively, and the amounts of impurities such as byproduct DPQ, residual catalyst and surfactant were all below 0.5% (Tables 1 and 2).
600 were obtained when the reaction times were 7.5 h and 20 h, respectively. In addition, the yields of PPO and PS could maintain about 90% and 73%, respectively, and the amounts of impurities such as byproduct DPQ, residual catalyst and surfactant were all below 0.5% (Tables 1 and 2).
| St content (wt%) | M n | M w | M w/Mn | PPO yield (%) | PS yield (%) | Impuritiesb (%) |
|---|---|---|---|---|---|---|
| a All the polymerizations of DMP were carried out in water under oxygen at 50 °C for 24 hours ([Cu(II)] = 1 mmol L−1, [DMP] = 0.25 mol L−1, [SDS] = 0.025 mol L−1, [NaOH] = 0.25 mol L−1, feeding time of St = 3 h). b Here impurities are consisted of the water-soluble impurities and the byproduct DPQ. | ||||||
| 0 | 4000 | 8800 | 2.2 | 88.63 | 72.55 | 0.34 |
| 10 | 4000 | 8900 | 2.2 | 88.96 | 73.46 | 0.33 |
| 20 | 4500 | 10200 |
2.3 | 89.45 | 71.28 | 0.37 |
| 25 | 5800 | 13900 |
2.4 | 89.99 | 73.65 | 0.40 |
| 30 | 8400 | 20300 |
2.4 | 90.34 | 72.88 | 0.38 |
| 35 | 9300 | 26000 |
2.8 | 91.20 | 71.97 | 0.30 |
| 40 | 9800 | 30100 |
3.1 | 91.76 | 72.90 | 0.32 |
| 50 | 9800 | 30100 |
3.1 | 92.00 | 74.10 | 0.29 |
| Feeding time (h) | M n | M w | M w/Mn | PPO yield (%) | PS yield (%) | Impuritiesb (%) |
|---|---|---|---|---|---|---|
| a All the polymerizations of DMP were carried out in water under oxygen at 50 °C for 24 hours with St wt% = 50% ([Cu(II)] = 1 mmol L−1, [DMP] = 0.25 mol L−1, [SDS] = 0.025 mol L−1, [NaOH] = 0.25 mol L−1). b Here impurities are consisted of the water-soluble impurities and the byproduct DPQ. | ||||||
| 1 | 7200 | 20300 |
2.8 | 91.47 | 74.36 | 0.32 |
| 1.5 | 7500 | 21200 |
2.8 | 92.08 | 71.23 | 0.31 |
| 2 | 8500 | 26000 |
3.1 | 92.10 | 70.99 | 0.36 |
| 3 | 9800 | 30100 |
3.1 | 93.28 | 72.54 | 0.35 |
| 4 | 9200 | 25600 |
2.8 | 92.90 | 71.85 | 0.44 |
| 5 | 8800 | 22300 |
2.6 | 92.78 | 73.56 | 0.33 |
| 6 | 8400 | 20200 |
2.4 | 91.84 | 73.66 | 0.29 |
| 8 | 7300 | 17200 |
2.4 | 90.77 | 72.90 | 0.43 |
| 10 | 6600 | 15100 |
2.3 | 90.14 | 73.80 | 0.33 |
| 12 | 5900 | 13700 |
2.3 | 89.25 | 74.12 | 0.40 |
| 16 | 5100 | 11500 |
2.3 | 89.00 | 73.09 | 0.34 |
| 18 | 4700 | 10000 |
2.1 | 88.60 | 74.89 | 0.39 |
| 20 | 4900 | 10000 |
2.0 | 88.78 | 72.11 | 0.38 |
The composition and molecular characteristics of the product were confirmed by FT-IR and NMR spectroscopies. Fig. 1 shows the FT-IR spectrum of PPO/PS alloy. The peaks at 3060 cm−1 and 3026 cm−1 indicate the stretching vibration of C–H bond in benzene ring, PPO and PS both possess the characteristic absorption peaks at 2924 cm−1 and 2850 cm−1 ascribed to the stretching vibration of aliphatic C–H bond and the peaks at 1601 cm−1, 1493 cm−1 and 1453 cm−1 due to the stretching vibration of benzene skeleton, the peak at 1376 cm−1 is the absorption caused by the deformation vibration of –CH3 in PPO and –CH–CH2– in PS, the peak at 1187 cm−1 is the stretching vibration of Ar–O bond, the peaks at 855 cm−1, 766 cm−1 and 697 cm−1 are assigned to the deformation vibration of C–H bond in benzene ring.15–17 The solid product contains the features of both PPO and PS, suggesting that PPO/PS alloy was obtained.
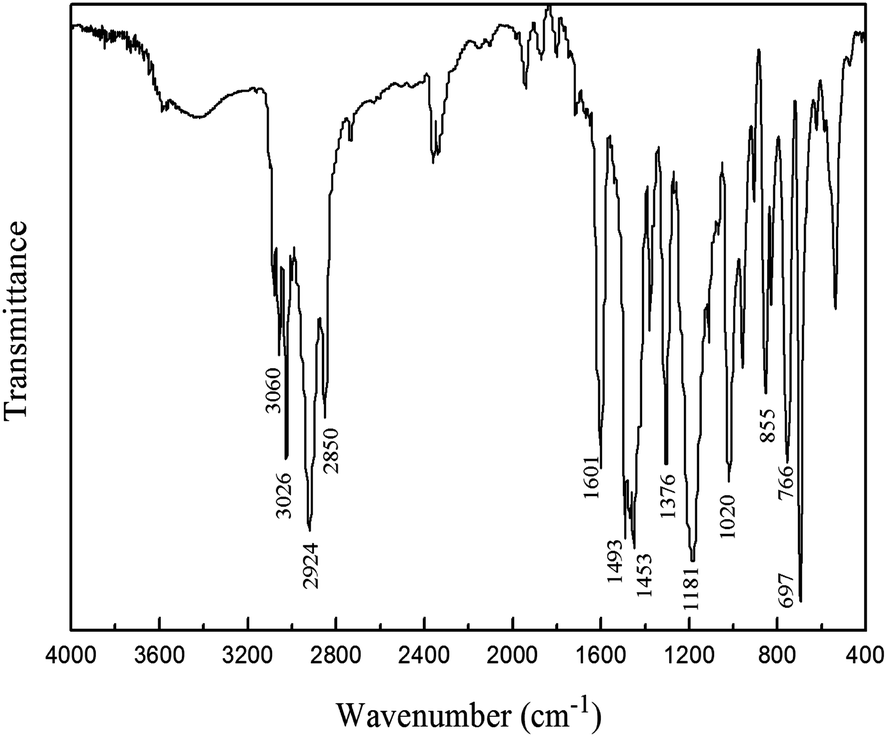 | ||
| Fig. 1 FT-IR spectrum of PPO/PS alloy. | ||
13C NMR spectrum of the polymer alloy in CDCl3 is shown in Fig. 2. The chemical shifts at 145.5, 128.2, 127.9, and 125.9 ppm are attributed to the carbons in benzene ring of PS, the signals at 154.8, 145.5, 132.3 and 114.6 ppm are assigned to the carbons in benzene ring of PPO, the peak around 46.5–41.0 ppm is ascribed to carbons in –CH2–, the peak at 40.2 ppm is attributed to the carbons in –CH–, and the chemical shift at 16.8 ppm is assigned to the carbons in –CH3 which are the substituents of benzene ring in PPO.18 All above indicate that PPO/PS alloy has been successfully synthesized.
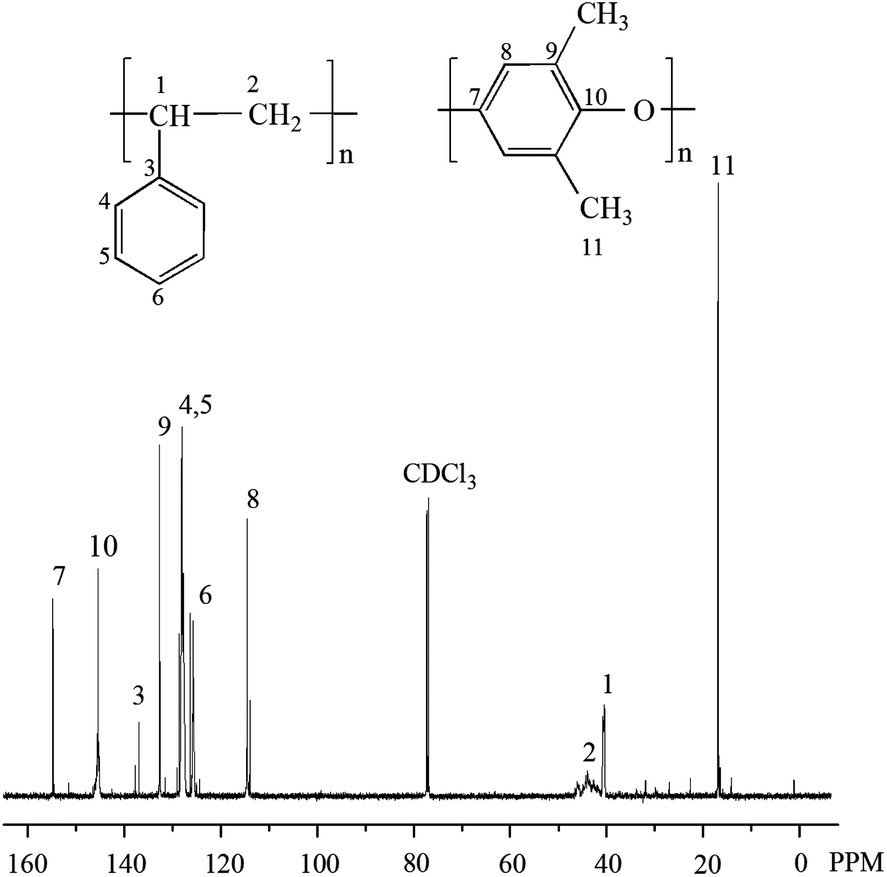 | ||
| Fig. 2 13C NMR spectrum of PPO/PS alloy in CDCl3. | ||
After extracting the product with 2-butanone for 48 h, the polymer alloy was separated into two parts. The soluble part is supposed to be PS since PS dissolves well in 2-butanone and the insoluble part is ascribed to be PPO and grafted polymers (PPO–g–PS) that may be formed during the polymerization.1913C NMR spectrum in CDCl3 of the insoluble part is shown in Fig. 3. It is found that there are only characteristic signals of PPO but no signals of PS, meaning that no PPO–g–PS was generated during the preparation of PPO/PS alloy.
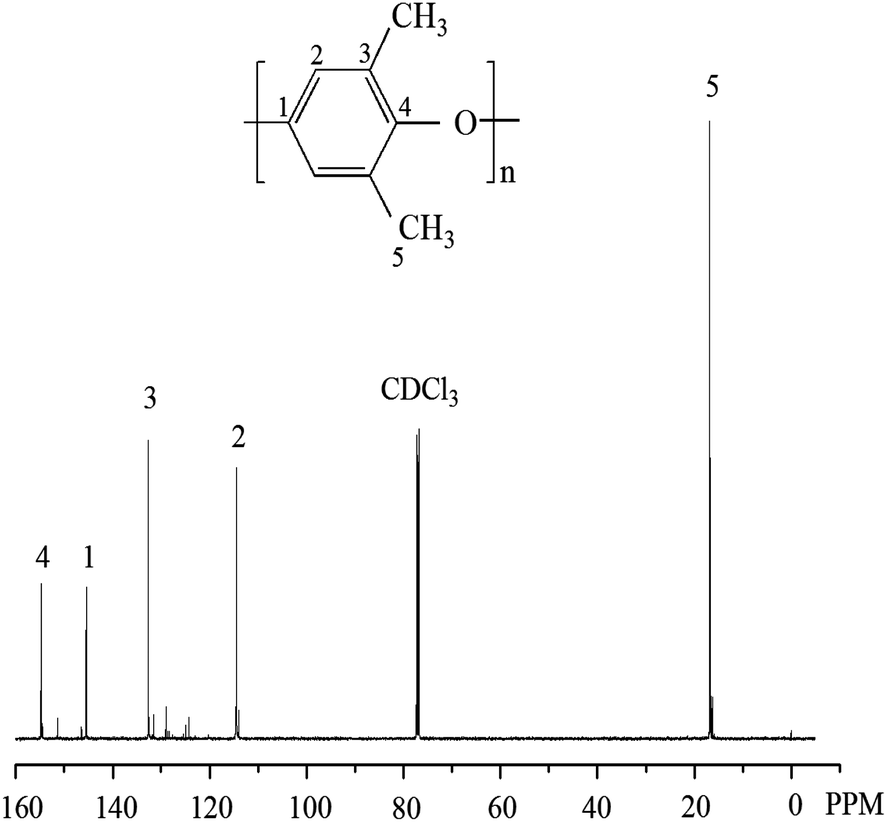 | ||
| Fig. 3 13C NMR spectrum in CDCl3 of the insoluble part in 2-butanone. | ||
Since St could undergo polymerization slowly without catalyst, how to demonstrate that CuCl2/DMAP was a versatile catalyst in this system, i.e. CuCl2/DMAP did catalyze the homopolymerization of St? The resulting PS should be able to be used as a macroinitiator for chain extension using a conventional ATRP process if the polymerization of St followed the rule of RATRP during the preparation of PPO/PS alloy.10,14 The chain extension reaction of St was carried out in bulk at 100 °C in the presence of a conventional ATRP catalyst, i.e., CuCl/DMAP, and PS (Mn = 5100, Mw/Mn = 1.10) which extracted with 2-butanone and distilled under reduced pressure was added. The Mn of the resulting PS increased up to 19300 (Mw/Mn = 1.13), which can be essentially demonstrated by the GPC curves shown in Fig. 4. No additional peak appears in Fig. 4, confirming that higher-molecular-weight PS has been formed. These results indicate that the AIBI/CuCl2/DMAP initiation system did induce living polymerization via a RATRP process during the preparation of PPO/PS alloy and CuCl2/DMAP did act as a versatile catalyst for the homopolymerizations of both monomers.
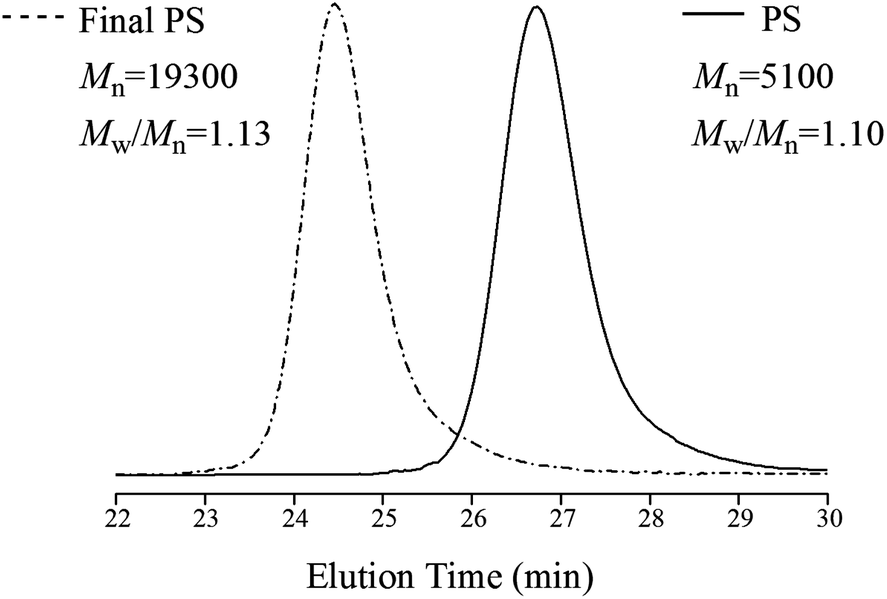 | ||
| Fig. 4 GPC curves of PS before and after chain extension reaction (the mobile phase was toluene and flow rate = 1.0 mL min−1 at 30 °C). | ||
3.2. Effect of St on Tg of PPO/PS alloy
T g value of the synthesized PPO/PS alloy was measured by DSC and the result is displayed in Fig. 5. It is found that there is only one glass transition temperature for PPO/PS alloy and its Tg is 147 °C when St content is 50 wt%, which further proves that thermodynamically compatible PPO/PS alloy has been prepared.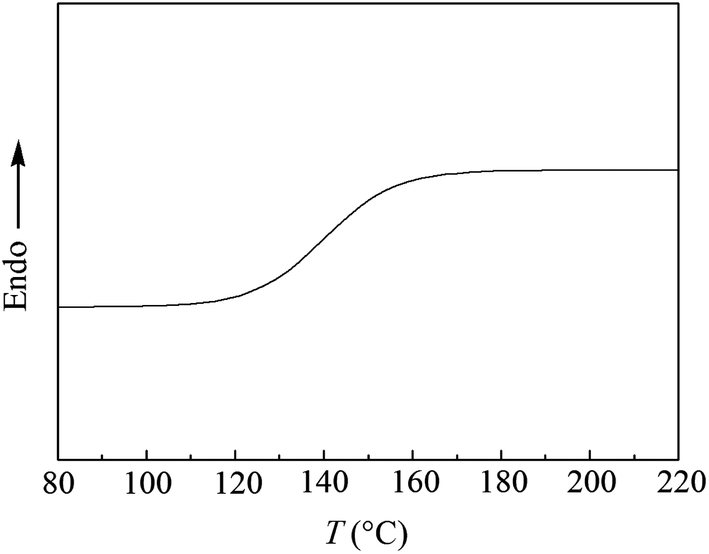 | ||
| Fig. 5 DSC curve of PPO/PS alloy (51 wt% for PPO with Mw = 30100 and 49 wt% for PS with Mw = 11800). | ||
According to Fox equation (eqn (1)) the weight percentage of PPO and PS in the PPO/PS alloy were calculated to be 51 wt% and 49 wt% based on Tg,PPO = 210 °C and Tg,PS = 100 °C,2,20 respectively, which is in good agreement with the weight measurement (51 wt% for PPO, and 49 wt% for PS).
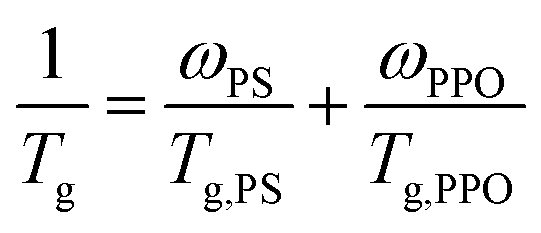 | (1) |
As reported before,5 the introduction of St could lower Tg of the PPO polymer particles, increase the mobility of the oligomer chains and facilitate the oxidative polymerization at the later period.
Two preformed polymers were blended with the same composition in comparison and similar characterization results (for FTIR, NMR, DSC) were obtained, indicating that the product synthesized using this method was exact the desired one.
3.3. Effect of St content on the molecular weight of PPO
The change of the molecular weight of PPO with St content was determined by GPC and the results are shown in Fig. 6. It is found that the molecular weight of PPO increases with the increase of styrene content. The weight–average molecular weight of PPO reaches 3.1 × 104 when styrene content is 40 wt%. However, a further increase of styrene content causes little increase of molecular weight of PPO. As stated before,5 the addition of St as the swelling agent could make the Tg of the polymer particle lower than the polymerization temperature, and PPO oligomers in the polymer particle were in the rubbery state when the St content was more than 20 wt%. Therefore, the mobility of polymer chains facilitated the oxidative polymerization at the later period, resulting in the increase of the molecular weight of PPO. The molecular weight of PPO increased with the increase of styrene content due to the further decrease of Tg of the polymer particle. Moreover, according to Fox equation, when St content is above 40 wt%, it would have little influence on the Tg of polymer particle. Thus, the molecular weight of PPO did not increase significantly when the St content was over 40 wt%.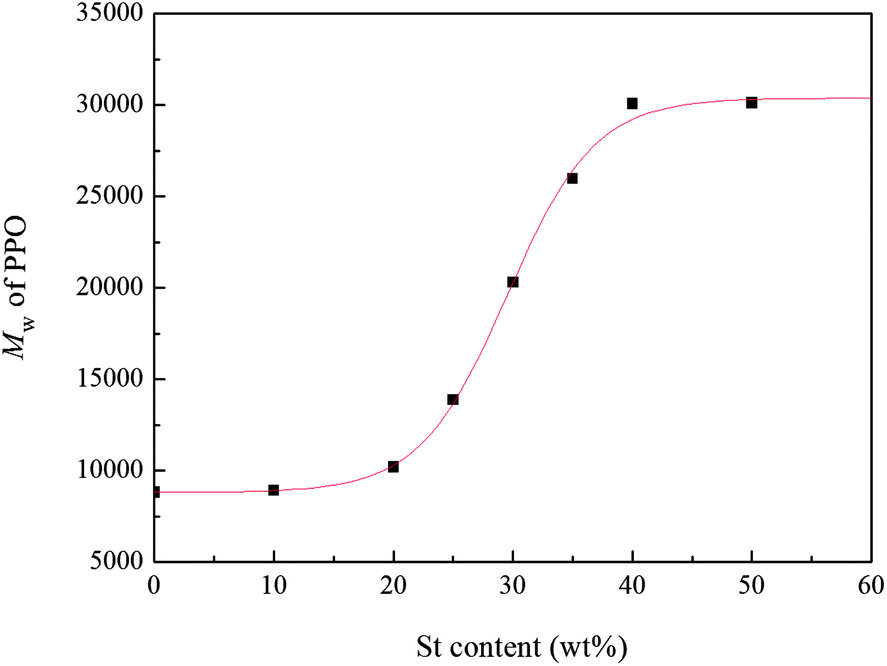 | ||
| Fig. 6 The influence of St content on the molecular weight of PPO (feeding time of St = 3 h). | ||
The feeding time of St could also affect the molecular weight of PPO, as shown in Fig. 7, when adding St at 3 h after the polymerization of DMP beginning, the molecular weight of PPO reached the highest one. This phenomenon may be explained as follows. The oxidative polymerization of DMP is influenced by two factors, one is how difficulty the oligomers and monomers could be oxidized, and the other is the mobility of oligomer chain. Since the oxidative polymerization of DMP is quite similar to condensation polymerization for the monomers quickly formed oligomers during the early period (0.5 h) of the reaction, the amount of monomers is so little that it can be negligible. A significant decrease in the oxidation potential of oligomers could be achieved with the addition of base, which make oxidation of oligomers proceed easily, and during the early stage of polymerization, the reaction rate is mainly affected by the oxidation of oligomers. When adding St during the earlier part of the polymerization, the oligomers end with –OH swelled in St and the rest part end with –ONa remained in water. The oligomers swelled in St possess higher oxidation potential than the ones end with –ONa, which makes the oxidation of them more difficult. Thus, adding St too early during the polymerization of DMP would depress the reaction rate and could not obtain the high molecular weight in turn. On the other hand, the precipitation of PPO in water plays the dominant role in inhibiting the increase of molecular weight during the later period of polymerization of DMP. Therefore, since the addition of St could lower the Tg and improve the mobility of oligomers, the later the St is added, the shorter time for it to act as the swelling agent, and the molecular weight of PPO would decrease.
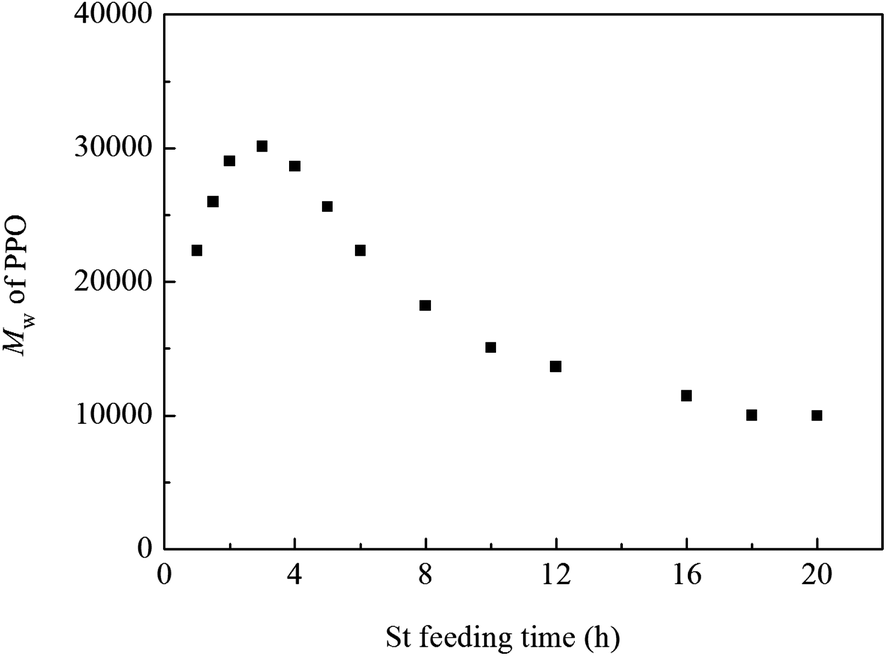 | ||
| Fig. 7 The influence of St feeding time on the molecular weight of PPO (St 50 wt%). | ||
3.4. Homopolymerizations of DMP and St in comparison
Homopolymerizations of DMP and St were conducted in the same reaction condition in comparison. For the oxidative polymerization of DMP in water, the molecular weight of the product could only reach Mn = 4000 after 24 h, and a product of Mn = 4100 was obtained after 48 h. On the other hand, the homopolymerization of St in water proceeded successfully for 20 h and the Mn of obtained PS was 10700. The results above indicate that the oxidative polymerization of DMP in aqueous solution could only give small-molecular-weight PPO without St as a swelling agent and the polymerization of St could proceed just like its homopolymerization after the synthesis of PPO during the preparation of PPO/PS alloy.
4. Conclusions
In conclusion, a one-pot synthetic method was developed to prepare PPO/PS alloy in water. The synthetic method included adding a reactive swelling agent—St during the oxidative polymerization of DMP with oxygen as the sole oxidant in alkaline aqueous solution and then in situ RATRP of St initiated by AIBI after the oxidative polymerization. Different molecular weights of PPO could be obtained by varying the amount and the feeding time of swelling agent. CuCl2/DMAP was used as a versatile catalyst for the polymerizations of both DMP and St. This method eliminated the use of a chemical oxidant, potassium ferricyanide, and the usage of sodium hydroxide was significantly reduced. Moreover, a chain extension reaction was conducted to verify that the polymerization obeyed the rules of RATRP, which indicated that CuCl2/DMAP did catalyze the polymerization of St and act as a versatile catalyst. Thus, thermodynamically compatible PPO/PS alloy has been successfully prepared in a one-pot way.Acknowledgements
We thank the Natural Science Foundation of China (20674075) and Natural Science Foundation of Zhejiang Province (Y404299) for their financial support.References
- A. S. Hay, H. S. Blanchard, G. F. Endres and J. W. Eustance, J. Am. Chem. Soc., 1959, 81, 6335–6336 CrossRef CAS.
- A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 505 CrossRef CAS.
- K. Saito, T. Tago, T. Masuyama and H. Nishide, Angew. Chem., Int. Ed., 2004, 43, 730–733 CrossRef CAS PubMed.
- K. Saito, N. Kuwashiro and H. Nishide, Polymer, 2006, 47, 6581–6584 CrossRef CAS PubMed.
- Q. Liu, B. Q. Shentu, J. H. Zhu and Z. X. Weng, Eur. Polym. J., 2007, 43, 890–894 CrossRef CAS PubMed.
- C. Gu, K. Xiong, B. Q. Shentu, W. L. Zhang and Z. X. Weng, Macromolecules, 2010, 43, 1695–1698 CrossRef CAS.
- W. L. Zhang, H. Wang, B. Q. Shentu and Z. X. Weng, J. Appl. Polym. Sci., 2011, 120, 109–115 CrossRef CAS.
- S. G. Gaynor, J. Qiu and K. Matyjaszewski, Macromolecules, 1998, 31, 5951–5954 CrossRef CAS.
- B. Liu and C. P. Hu, Eur. Polym. J., 2001, 37, 2025–2030 CrossRef CAS.
- D. Q. Qin, S. H. Qin and K. Y. Qiu, J. Appl. Polym. Sci., 2001, 81, 2237–2245 CrossRef CAS.
- A. Limer and D. M. Haddleton, Eur. Polym. J., 2006, 42, 61–68 CrossRef CAS PubMed.
- H. K. Feng and Y. Dan, J. Appl. Polym. Sci., 2006, 99, 1093–1099 CrossRef CAS.
- L. Sartore, M. Penco, S. D. Sciucca, R. Mendichi, L. D. Landro and S. D. Antone, J. Appl. Polym. Sci., 2006, 100, 4654–4660 CrossRef CAS.
- Y. Q. Xu, Q. F. Xu, J. M. Lu, X. W. Xia and L. H. Wang, Polym. Bull., 2007, 58, 809–817 CrossRef CAS.
- C. Y. Liang and S. Krimm, J. Polym. Sci., 1958, 27, 241–254 CrossRef CAS.
- C. G. Zimba and J. F. Rabolt, Macromolecules, 1989, 22, 2867 CrossRef.
- P. C. Dautenhahn and P. K. Lim, Ind. Eng. Chem. Res., 1992, 31, 463 CrossRef CAS.
- R. Wilhelm and H. Walter, Macromol. Chem., 1985, 186, 1835 CrossRef.
- F. J. Viersen, J. Renkesdma, G. Challa and J. Reedijk, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 901 CrossRef CAS.
- T. G. Fox and P. J. Flory, J. Polym. Sci., 1954, 14, 315 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |