Electron detachment dynamics of O2−(H2O): direct ab initio molecular dynamics (AIMD) approach†
Hiroto
Tachikawa
*
Division of Materials Chemistry, Graduate School of Engineering, Hokkaido University, Sapporo 060-8628, Japan
First published on 6th November 2013
Abstract
Electron detachment dynamics of the hydrated superoxide anion O2−(H2O) have been investigated by means of direct ab initio molecular dynamics (AIMD) methods. Two potential energy surfaces were examined as electronic states of oxygen molecule after the electron detachment of the hydrated superoxide anion: O2(3Σ)(H2O) and O2(1Δ)(H2O). In both electronic states, the dissociation products, O2 + H2O, were obtained. However, the internal states of O2 were essentially different from each other. On the triplet state surface, the O–O stretching mode of O2(3Σ) was excited, whereas that of O2(1Δ) was silent. The reaction mechanism was discussed on the basis of theoretical results.
1. Introduction
The superoxide anion (O2−) is one of the reactive oxygen species and plays an important role in biochemistry,1–4 in pharmacy,5–7 and in catalysis science.8–10 The O2− anion acts as a trigger in cancer generation because the triplet and singlet state oxygen molecules (expressed by O2(1Σ) and O2(1Δ)) are generated by the electron detachment. The energy difference between singlet and triplet states is 22.5 kcal mol−1. Also, the superoxide anion is known as “molecular loose cannon” that oxidizes several molecules in the cell and causes mutation in DNA and damage in protein.The O2− anion in vivo is usually solvated by a few water molecules and exists as a hydrated superoxide cluster, O2−(H2O)n (n > 0). Therefore, the reaction of the superoxide anion takes place as a hydrated cluster O2−(H2O)n. The most important reaction of O2−(H2O)n is an electron transfer reaction to biomolecule (M): O2−(H2O)n + M → O2(H2O)n + M−. In such case, two types of oxygen molecules are generated: triplet and singlet oxygen molecules.
The hydrated superoxide anion in gas phase, O2−(H2O)n (n = 1–3), have been extensively studied from both experimental and theoretical points of view. Luong et al. measured photodetachment spectra of O2−(H2O)n (n = 1–6) using photoelectron-multiple-photofragment coincidence spectroscopy.11 They found two broad peaks at electron kinetic energy (eKE) = 1.1 and 0.4 eV in case of O2−(H2O). These peaks are assigned to be the ground and first excited states of O2:
| O2−(H2O) + hν → O2(3Σ) + H2O + e−. |
| O2−(H2O) + hν → O2(1Δ) + H2O + e−. |
Namely, photo-electron detachment of O2−(H2O) gives two kinds of oxygen molecules (singlet and triplet states).
Arshadi and Kebarle determined ΔH, ΔG, and ΔS values for the formation of O2−(H2O)n (n = 1–3) by thermodynamics measurement.12 The ΔH of the formation for O2−(H2O) was measured to be −18.4 kcal mol−1. Their ab initio calculation at the MP2/6-31+G(d,p) level gave −15.5 kcal mol−1, which supports reasonably the experiment.
Ab initio calculations showed that the solvation structure of O2−(H2O) has a hydrogen bond between one-side oxygen atom of O2− and one-hydrogen.13 Also, two-hydrogen bond structure with the C2v symmetry was proposed as a stable form.14 In both forms, negative charge is fully localized on the O2− anion, suggesting that H2O molecule solvates O2− with hydrogen bond.15,16 Bork et al. studies O2(H2O)n and O3(H2O)n anionic molecular clusters (n < 12) using ab initio calculations.15 They found that anionic clusters O2−(H2O)n and O3−(H2O)n are thermally stabilized at typical atmospheric conditions for at least n = 5. The first 4 water molecules are strongly bound to the anion due to delocalization of the excess charge while stabilization of more than 4H2O is due to normal hydrogen bonding.
For a large system, O2−(H2O)n (n = 6),16–18 Antonchenko and Kryachko investigated theoretically the interaction of O2− with water hexamer using B3LYP/6-311++G(d,p) method.16,17 The solvation structure of O2− was obtained theoretically.
The solvation structures of O2−(H2O) have been thus well understood, but the mechanism of electron detachment dynamics of hydrated superoxide anion O2−(H2O) is little known, because there is no theoretical work on the reaction dynamics of the O2−(H2O) system. In particular, there is no information on the internal states of products after the photo-electron detachment of O2−(H2O).
In the present study, the electron detachment dynamics of O2−(H2O) are investigated by means of direct ab initio molecular dynamic (AIMD) method. The ground state of neutral O2 is a triplet state O2(3Σ), and the singlet state of O2(1Δ) exists as the first excited state. These states are close to each other. We focus our attention on difference of internal states of product O2 and H2O on the two potential energy surfaces (PESs).
2. Method of calculation
The direct AIMD calculation19–23 was carried out at the MP2/6-311++G(d,p) level of theory. First, the structures of the hydrated superoxide anion, O2−(H2O) were determined by static ab initio calculation. Next, the initial geometries were randomly sampled around the equilibrium point of O2−(H2O) on Franck–Condon (FC) region, and then the trajectories for neutral system O2(H2O) were run from these generated points on the assumption of vertical electron detachment. The geometries of O2−(H2O) were selected as the energy difference less than 1.0 kcal mol−1 from the equilibrium point. The electronic states of the system were monitored during the simulation. We confirmed carefully that the electronic state is kept during the reaction. A total of 30 geometrical configurations were generated around the equilibrium point of O2−(H2O). The trajectory of neutral system was propagated from the vertical electron detachment point of the neutral hydrated oxygen molecule, [O2(H2O)]ver.The velocities of atoms at the starting point were set to zero (i.e. momentum vector of each atom is zero). The equations of motion were numerically solved by the velocity Verlet algorithm method. No symmetry restrictions were applied to the calculation of the energy gradients. The time step size was chosen to be 0.10 fs, and a total of 5000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps were calculated for each dynamics calculation. The drift of the total energy was confirmed to be less than 1 × 10−3 kcal mol−1 in all trajectories (total energy condition).
000 steps were calculated for each dynamics calculation. The drift of the total energy was confirmed to be less than 1 × 10−3 kcal mol−1 in all trajectories (total energy condition).
Ab initio calculations were performed with the GAMESS,24 Gaussian 03 and 09 program packages25,26 at the CCSD and MP2/6-311++G(d,p) levels in order to obtain sufficiently accurate energetics. The derived structures and geometrical parameters of the stationary points are given in the ESI.†
3. Results
3.1. Solvation structures of superoxide anion O2−(H2O)
The optimized structures of O2−(H2O) obtained at the MP2 and CCSD/6-311++G(d,p) levels are illustrated in Fig. 1. Both levels gave the similar structures for O2−(H2O) and O2(H2O) systems. Two conformers were obtained as stable forms of O2−(H2O). One is a C2v structure where two hydrogen atoms of H2O interact equivalently with the oxygen atoms of O2−. The distance of hydrogen bond is r(H–O) = 2.032 Å at the CCSD/6-311++G(d,p) level. Another conformer is a Cs structure in which one of the hydrogen atoms of H2O interacts strongly with O2−. The distance of hydrogen bond is r(H′–O) = 1.674 Å. The CCSD calculation showed that the energy difference between C2v and Cs structures was calculated to be only 0.7 kcal mol−1: the Cs form is 0.7 kcal mol−1 more stable in energy than that of C2v form, although the energy difference is negligibly small. Hence, the electron detachment of O2−(H2O) takes place from both forms. The MP2 calculations gave the similar results for O2−(H2O): the Cs form is 0.2 kcal mol−1 more stable in energy than the C2v form after the zero-point energy correction. These energetics are in good agreement with previous calculations.27–30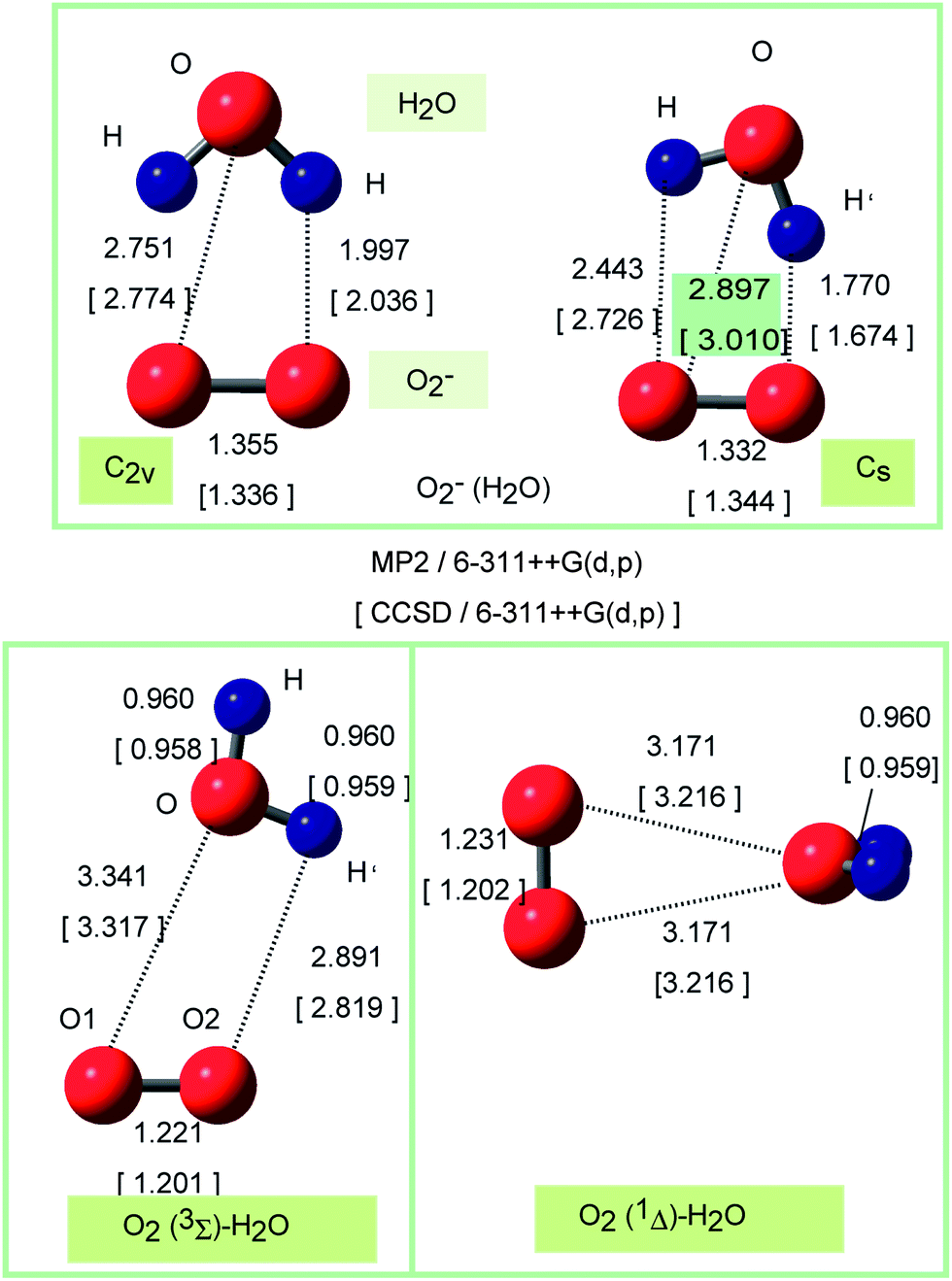 | ||
| Fig. 1 Optimized structures of hydrated superoxide anions, O2−(H2O) (upper), triplet (left) and singlet states (right) of neutral complex, O2(H2O), calculated at the CCSD and MP2/6-311++G(d,p) levels. The bond distances obtained at the CCSD/6-311++G(d,p) level are given in parenthesis. Bond lengths are in Å. | ||
Next, triplet and singlet states of O2(H2O) were fully optimized at the CCSD method. The structures of O2(1Δ)H2O were given in Fig. 1 (lower). At the singlet state (lower right panel in Fig. 1), the oxygen atom of H2O interacts directly with the oxygen atoms of O2. The oxygen atom of H2O directs to the center of mass of O2(1Δ). The interatomic distance was 3.216 Å, which is significantly longer than those of O2−(H2O) (2.774 Å for C2v and 3.010 Å for Cs). The binding structure of triplet state is different from that of singlet state (lower left panel in Fig. 1): the O–H bond interacts with two oxygen atoms of O2(3Σ). The intermolecular distance was r(H′–O2) = 2.819 Å, which is slightly shorter than those of O2(1Δ)H2O. Table 2 shows the Mulliken atomic charges of O2(1Δ)H2O calculated by MP2 and CCSD methods. The MP2 calculations represented reasonably the structure and electronic states of O2(1Δ)H2O.
| Molecule | Atom | CCSD | MP2 |
|---|---|---|---|
| O2 | O | 0.01 | 0.01 |
| O | 0.01 | 0.01 | |
| H2O | O | −0.52 | −0.54 |
| H | 0.25 | 0.26 | |
| H | 0.25 | 0.26 |
3.2. Electron detachment dynamics of O2−(H2O) to triplet potential energy surface
Snapshots of O2(H2O) on the triplet potential energy surface (PES) are illustrated in Fig. 2. At time zero, the structure of [O2(3Σ)(H2O)]ver corresponds to that of the equilibrium structure of O2−(H2O). After the electron detachment, the water molecule rotates itself and leaves rapidly from O2. This is due to the fact that the repulsive interaction between proton of H2O (H′) and oxygen atom of O2 is generated suddenly generated after the detachment. The oxygen–oxygen bond of O2 vibrated strongly after the electron detachment.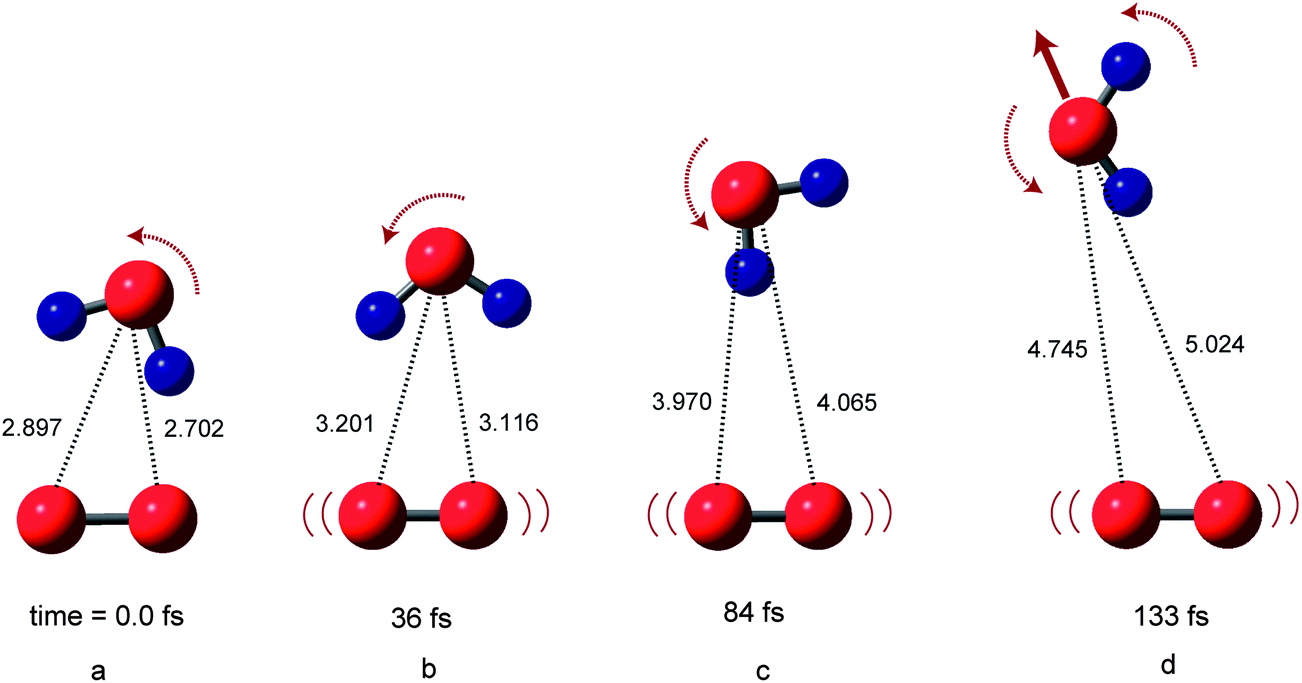 | ||
| Fig. 2 Snapshots of sample trajectory of O2(H2O) on the triplet state potential energy surface (PES) following electron detachment of O2−(H2O) (Cs structure). Bond lengths are in Å. Dynamics calculation were carried out at the MP2/6-311++G(d,p) level. Trajectory was started from the optimized geometry of O2−(H2O). | ||
At 36 fs (point b), the water molecules are located at R1 = 3.116 Å and R2 = 3.201 Å. At 84 fs (point c), R1 and R2 were 4.065 and 3.970 Å, respectively. The snapshots indicate that the water molecule leaves monotonically from the O2 molecule after the electron detachment without the complex formation. The relative translational energy between O2 and H2O was calculated to be 5.0 kcal mol−1.
3.3. Electron detachment dynamics of O2−(H2O) to singlet potential energy surface
Snapshots of O2(H2O) on the singlet PES are illustrated in Fig. 3. At time zero, the structure of [O2(3Σ)(H2O)]ver corresponds to that of the equilibrium structure of O2−(H2O). After the electron detachment, the trajectory of H2O is very similar to that of the triplet state: namely, the water molecule rotates and leaves rapidly from O2. At 57 fs (point b), the water molecules are located at R1 = 3.470 Å and R2 = 3.454 Å. At 107 fs (point c), R1 and R2 were 4.403 and 4.156 Å, respectively. The snapshots indicate that the water molecule leaves monotonically from the O2 molecule after the electron detachment without the complex formation. Thus, the translational energies of H2O⋯O2 on both singlet and triplet PESs are similar to each other. However, the oxygen–oxygen bond of O2 was silent on the singlet PES after the electron detachment. The relative translational energy between O2 and H2O was calculated to be 4.4 kcal mol−1, which is close to that of triplet state (5.0 kcal mol−1). | ||
| Fig. 3 Snapshots of sample trajectory of O2(H2O) on the singlet state potential energy surface (PES) following electron detachment of O2−(H2O) (Cs structure). Bond lengths are in Å. Dynamics calculation were carried out at the MP2/6-311++G(d,p) level. Trajectory was started from the optimized geometry of O2−(H2O). | ||
3.4. Potential energies of the systems
Time evolution of potential energy of triplet state O2(H2O) after the vertical electron detachment of O2−(H2O) is plotted in Fig. 4 (dashed line). The zero level of the energy corresponds to the total energy of [O2(3Σ)(H2O)]ver at the vertical electron detachment point on the triplet potential energy surface. After the electron detachment, the potential energy of O2(H2O) decreases suddenly to −33.6 kcal mol−1 at 4.1 fs. The next energy minima were appeared at 11.8, 20.1, and 28.3 fs. The intervals are Δt = 7.7, 8.3, and 8.2 fs. Average time period is 〈τ〉 = 16.2 fs. This vibration is originated mainly from the O–O vibrational stretching mode of O2(3Σ): namely, the mode is enhanced due to the difference of equilibrium distances between O2− and O2(3Σ) after the detachment.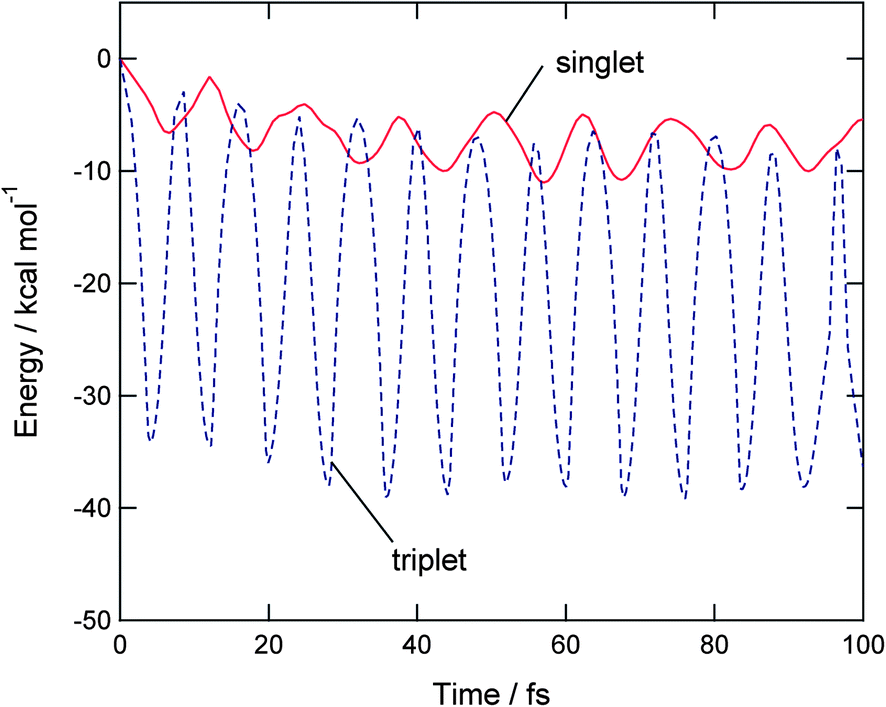 | ||
| Fig. 4 Time evolutions of potential energies of O2(3Σ)H2O and O2(1Δ)H2O following electron detachment of O2−(H2O) (Cs structure). | ||
Time evolution of potential energy of singlet state O2(H2O) after the vertical electron detachment of O2−(H2O) is given in Fig. 4 (real line). First, the energy decreased to −6.8 kcal mol−1 at 6.3 fs. The next energy minimum appeared at 18.0 fs with the energy of −8.5 kcal mol−1. At longer time propagation, the energy minima are located around −10 kcal mol−1, which is significantly shallower than the lowest energy in the triplet state (−38 kcal mol−1 as shown in dotted line). The vibration of potential energy is mainly originated from a O–O stretching mode of O2(1Δ) after the electron detachment.
Thus, the shapes of time evolutions of potential energies were essentially different from each other, although the products are the same on both PESs: namely, the oxygen and water molecules: O2 + H2O. The potential energy of triplet state vibrates largely as a function of time. The energy is minimized up to −38 kcal mol−1. On the other hand, the singlet state has a small amplitude of the potential energy, and the energy is minimized to only −10 kcal mol−1. The large amplitude on the triplet state surface is originated mainly from the O–O stretching vibration of O2(3Σ) after the electron detachment. On the singlet PES, time evolution of potential energy is originated from vibration of O2(1Δ), as will be discussed in later section.
Fig. 5 shows time evolutions of the relative distances between H2O and O2, and the internal modes of O2 and H2O. The intermolecular distances increase linearly as a function of time (Fig. 5A), indicating that the water molecule leaves gradually from O2 after the electron detachment. The translational energies for singlet and triplet states were estimated to be 4.4 and 5.0 kcal mol−1, respectively.
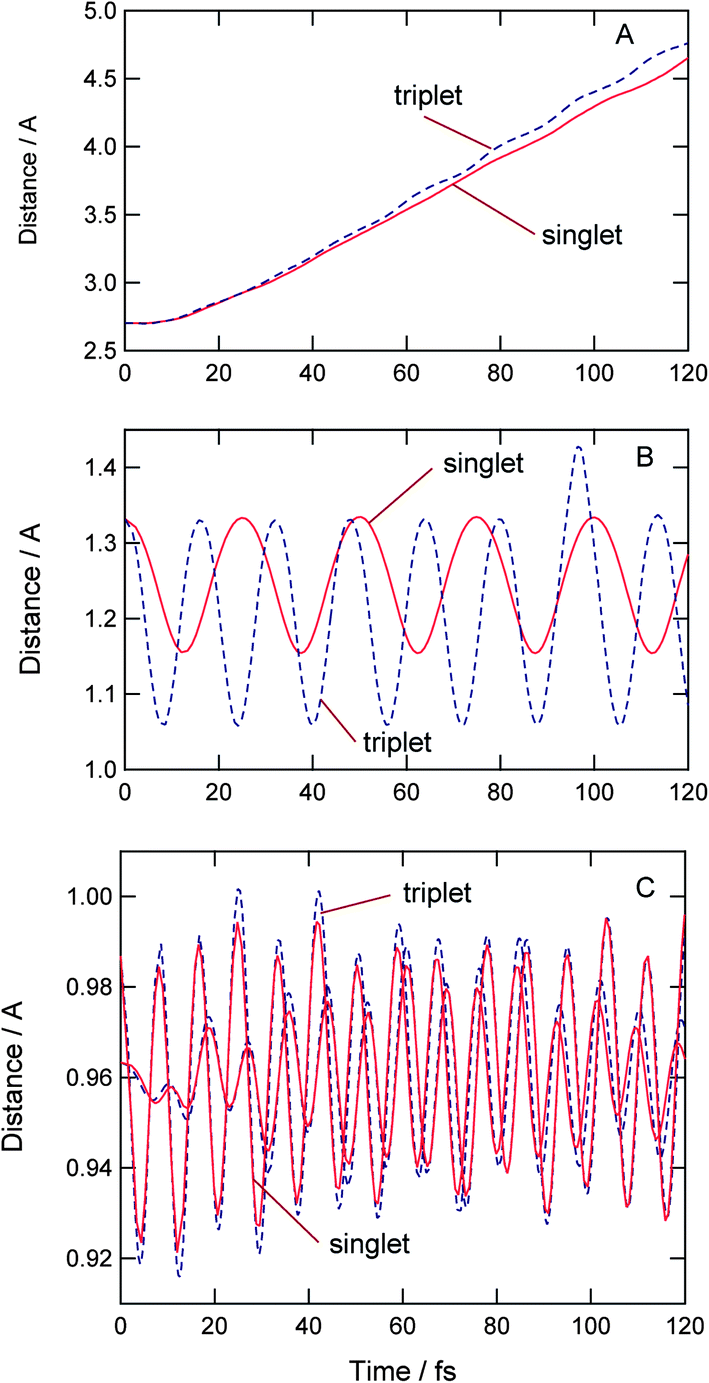 | ||
| Fig. 5 Time evolutions of distance between H2O and O2 (A), O–O stretching modes of O2 (B) and O–H stretching modes of H2O (C). | ||
Thus, the translational energies for both states were similar to each other. On the other hand, the internal states of O2 are different: the O–O stretching mode of O(3Σ) is excited.
The intramolecular distances of O–O bond of O2 are plotted in Fig. 5B. After the electron detachment to the singlet state, the O–O bond vibrated periodically with a time period of 25 fs. This is assigned to the O–O vibration of O2(1Δ). On the triplet PES, the vibrational period of O2 is estimated to be 15.7 fs, which is the vibration of O2(3Δ).
Fig. 5C shows time evolutions of O–H stretching mode of the product H2O. The amplitude of H2O on the triplet PES is larger than that on the singlet PES. This is due to the fact that the repulsive impact on triplet PES is larger than that on the singlet PES.
3.5. Energetics
Energy diagram for O2(H2O) after the electron detachment of O2−(H2O) is given in Fig. 6. The energy level in the left side denoted by O2− + H2O is a summation of total energies of O2− plus water molecule. Superoxide anion is solvated by the water molecules as O2− + H2O → O2−(H2O) + ΔEsolv(−). The first hydration energy of O2− was ΔEsolv(−) = 0.92 eV. This energy of O2− was experimentally measured to be 0.85 ± 0.05 eV (ref. 15, 31 and 32) which is in good agreement with the present value (0.92 eV). | ||
| Fig. 6 Energy diagram of O2−(H2O)/O2−(H2O) system calculated at the MP2/6-311++G(d,p) level. Each value are given in Table 1. | ||
Electron detachment of free O2− gives two low-lying electronic states of O2: triplet and singlet states of O2 molecule. The present calculation shows the vertical electron detachment energies of free O2− to O2(3Σ) and O2(1Δ) are 0.34 and 1.47 eV, respectively.
In case of O2−(H2O), the vertical electron detachment energies of O2− are largely blue-shifted to 1.53 eV (triplet) and 2.54 eV (singlet). After the electron detachment of O2−(H2O)n, the solvation structure of [O2−(H2O)]ver will be relaxed and the hydration molecules are re-oriented around the O2 molecule. The relaxation energies on the triplet and singlet state PESs are ΔEstable(triplet) = 0.69 eV and ΔEstable(singlet) = 0.44 eV. The relaxation energy of triplet state is larger than that of singlet state. The solvation energies of O2(3Σ) and O2(1Δ) by a water molecule are only ΔEsolv = 0.04 and 0.11 eV, respectively, indicating that the interaction energy of neutral O2 molecules with water molecules is negligibly small. The small solvation energy causes the dissociation of water molecules from O2(H2O) complexes. These results indicate that PES is open for the dissociation channel because the O2(H2O) complex is weakly bound on the PES.
3.6. Electron detachment dynamics of O2−(H2O) with C2v structure
Direct AIMD calculations were carried out for the C2v structure of O2−(H2O) for comparison. The results were given in ESI (Fig. S1 and S2†). The similar results were obtained from the C2v structure of O2−(H2O). The dissociation product was the main channel. Relative translational energies for singlet and triplet states were 2.0 and 4.2 kcal mol−1, respectively. Therefore, it can be concluded that photo-electron detachment of superoxide anion interacting with water molecule gives a dissociation product.3.7. Effects of initial configurations on the reaction dynamics
In the present calculations, a total of 30 trajectories were run from the selected geometrical configurations generated around the equilibrium points. To check the effects of initial configurations on the product and reaction dynamics, all trajectories were checked carefully. A part of the results were given in ESI (Fig. S3†). All trajectories gave the similar results and the same products (O2 + H2O).4. Discussion
4.1. Summary
In the present study, electron detachment dynamics of hydrated superoxide anion, O2−(H2O), have been investigated by direct AIMD method at the MP2/6-311++G(d,p) level. Low-lying triplet and singlet states of O2(H2O) were examined as potential energy surfaces after the electron detachment. The results can be summarized as follows. On both PESs, the product is O2 + H2O, meaning that dissociation is main channel after the electron detachment of O2−(H2O). This is due to the fact that O2(3Σ) and O2(1Δ) are shallow trapped by H2O and both PESs are open for the dissociation channels.However, the internal states of product are different from each other. On the triplet PES, the O–O stretching mode of O2(3Σ) is excited in vibrational energy after the electron detachment. Also, the O–H stretching mode of H2O is slightly excited due to the repulsive interaction between oxygen atom of O2 and proton of H2O at time zero. On the other hand, the O–O stretching mode of O2(1Δ) is silent. This difference is an important information on the experimental detection of reaction channels.
4.2. Comparison with the experiments
Luong et al. investigated photoelectron detachment dynamics of O2−(H2O)n (n = 1–4) using photoelectron-multiple-photofragment coincidence spectroscopy.11 They found dissociation products after the photo-irradiation to O2−(H2O): O2−(H2O) + hν → O2 + H2O + e−. However, the mechanism of this process was not clearly understood. The present calculations suggest that dissociation occurs easily without complex formation after the electron detachment of O2−(H2O). The specific dynamics is originated from the weakly bound of O2 with the water molecule (0.04 eV for triplet and 0.11 eV for singlet). Also, FC region of O2−(H2O) does not overlap with the neutral complex region. Hence, the trajectory shows that the H2O molecule is dissociated directly from O2.Luong et al. measured the photoelectron spectra of O2−(H2O). They found that two broad peaks are appeared at eKE = 1.1 and 0.4 eV. The present calculation assigns theoretically that these peaks originated from
| O2−(H2O) + hν → O2(3Σ) + H2O eKE = 1.1 eV | (1) |
| O2−(H2O) + hν → O2(1Δ) + H2O eKE = 0.4 eV. | (2) |
In the present study, O2–H2O 1:1 complex was only examined as a hydrated superoxide anion. The calculations showed that dissociation channel, O2 + H2O, is main product following the electron detachment of the complex anion. In case of O2− in liquid water, the O2− ion is solvated by four water molecules as the first solvation shell, O2−(H2O)4. If the electron detachment of O2− takes place in liquid water, the solvent re-orientation will be observed and the solvent dissociation will be prohibited due to the bulk solvent. Finally, a hydrated O2−H2O complex is formed as a product.
4.3. Reaction model
Schematic illustration of the electron detachment process of O2−(H2O) is given in Fig. 7. The lowest PES means anion solvation of O2−(H2O) at time zero. The arrows indicate the electron detachment process to the vertical points of neutral complex. Two electronic states are concerned as the neutral state dynamics: lower triplet state and higher singlet state PESs. The lower and upper potential energy surfaces correspond to the triplet and singlet electronic states, respectively, along the dissociation coordinate. On both PESs, the trajectories pass rapidly on the intermediate complex regions and the dissociation product was obtained. | ||
| Fig. 7 Schematic illustration of model of electron detachment of O2−(H2O). | ||
Acknowledgements
The author acknowledges partial support from a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (JSPS) (Grant number 2455000102). This work is partially supported by a Grant-in-Aid for Scientific Research on Innovation Areas “Evolution of Molecules in Space” (Grant number 2510800413).References
- L. Saleh and C. Plieth, Nat. Protoc., 2010, 5, 1635–1641 CrossRef CAS PubMed.
- A. Kanamori, M. M. Catrinescu, A. Mahammed, Z. Gross and L. A. Levin, J. Neurochem., 2010, 114, 488–498 CrossRef CAS PubMed.
- C. E. M. Berger, H. K. Dattaa and B. R. Horrocks, Phys. Chem. Chem. Phys., 2011, 13, 5288–5297 RSC.
- A. Cossarizza, R. Ferraresi and L. Troiano, Nat. Protoc., 2009, 4, 1790–1797 CrossRef CAS PubMed.
- V. V. Barbakadze, K. G. Mulkidzhanyan, M. I. Merlani, L. M. Gogilashvili, L. S. Amiranashvili and E. K. Shaburishvili, Pharm. Chem. J., 2011, 44, 604–607 CrossRef CAS.
- F. Jiang, Y. Zhang and G. J. Dusting, Pharmacol. Rev., 2011, 63, 218–242 CrossRef CAS PubMed.
- G. Hasko, J. Linden and B. Cronstein, Nat. Rev. Drug Discovery, 2008, 7, 759–770 CrossRef CAS PubMed.
- K. Zhang, X. N. Hu, J. B. Liu, J. J. Yin, S. A. Hou, T. Wen, W. W. He, Y. L. Ji, Q. Wang and X. C. Wu, Langmuir, 2011, 27, 2796–2803 CrossRef CAS PubMed.
- G. Granados-Oliveros, E. A. Paez-Mozo, F. M. Ortega, M. Piccinato, F. N. Silva, C. L. B. Guedes, E. Di Mauro, M. F. da Costa and A. T. Ota, J. Mol. Catal. A: Chem., 2011, 339, 79–85 CrossRef CAS PubMed.
- G. Krylova, N. M. Dimitrijevic, D. V. Talapin, J. R. Guest, H. Borchert, T. Rajh and E. V. Shevchenko, J. Am. Chem. Soc., 2010, 132, 9102–9110 CrossRef CAS PubMed.
- A. K. Luong, T. G. Clements, M. Sowa Resat and R. E. Continetti, J. Chem. Phys., 2001, 114, 3449–3452 CrossRef CAS.
- M. Arshadi and P. Kebarle, J. Phys. Chem., 1970, 74, 1483–1488 CrossRef CAS.
- Q. L. Wang, D. Z. Chen, X. W. Liu and L. F. Zhang, Comput. Theor. Chem., 2011, 966, 357–363 CrossRef CAS PubMed.
- J. Li, H. Hou and B. Wang, J. Phys. Chem. A, 2009, 113, 800–804 CrossRef CAS.
- N. Bork, T. Kurten, M. B. Enghoff, J. O. P. Pedersen, K. V. Mikkelsen and H. Svensmark, Atmos. Chem. Phys., 2011, 11, 7133–7142 CrossRef CAS.
- V. Y. Antonchenko and E. S. Kryachko, Chem. Phys., 2006, 327, 485–493 CrossRef CAS PubMed.
- V. Y. Antonchenko and E. S. Kryachko, J. Phys. Chem. A, 2005, 109, 3052–3059 CrossRef CAS PubMed.
- J. C. Li, H. Hou and B. S. Wang, J. Phys. Chem., 2009, 113, 800–804 CrossRef CAS.
- H. Tachikawa, J. Phys. Chem. A, 2011, 115, 9091–9096 CrossRef CAS PubMed.
- H. Tachikawa, Phys. Chem. Chem. Phys., 2011, 13, 11206–11212 RSC.
- H. Tachikawa, Chem. Phys. Lett., 2011, 513, 94–98 CrossRef CAS PubMed.
- H. Tachikawa and T. Fukuzumi, Phys. Chem. Chem. Phys., 2011, 13, 5881–5887 RSC.
- H. Tachikawa and A. J. Orr-Ewing, J. Phys. Chem. A, 2008, 112(391), 11575–11580 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision B.04, Gaussian, Inc., Wallingford, CT, 2004, p. 374 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009, p. 389 Search PubMed.
- J. M. Weber, J. A. Kelley, S. B. Nielsen, P. Ayotte and M. A. Johnson, Science, 2000, 287, 2461 CrossRef CAS.
- J. M. Weber, J. A. Kelley, W. H. Robertson and M. A. Johnson, J. Chem. Phys., 2001, 114, 2698 CrossRef CAS.
- A. J. Bell and T. G. Wright, Phys. Chem. Chem. Phys., 2004, 6, 4385 RSC.
- W. S. Sheu and M. F. Chiou, J. Phys. Chem. A, 2011, 115, 99–104 CrossRef CAS PubMed.
- M. A. Buntine, D. J. Lavrich, C. E. Dessent, M. G. Scarton and M. A. Johnson, Chem. Phys. Lett., 1993, 216, 471 CrossRef CAS.
- H. D. Babcock and L. Herzberg, Astrophys. J., 1948, 108, 167 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electron detachment dynamics of O2−(H2O) (C2v structure) and optimized structures. See DOI: 10.1039/c3ra45753b |
| This journal is © The Royal Society of Chemistry 2014 |