
Catalytic oxidation of NO by Au2− dimers: a DFT study
Abstract
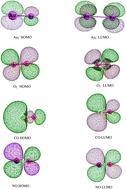
Density functional theory (DFT) calculations are performed to study the mechanistic details of NO oxidation promoted by gold dimer anions. Furthermore, we studied a full catalytic cycle producing two NO2 molecules. The reaction is explored along three possible pathways. Our theoretical results show that anionic gold dimers present catalytic activity towards NO oxidation, as indicated by the calculated low energy barriers and high exothermicities. The present results enrich our understanding of the catalytic oxidation of NO by Au-cluster based catalysts. For the first time, we have presented a systematic study on the structure and energetics of various reaction intermediates involved in NO oxidation by Au2− clusters using density functional theory (DFT).
 Please wait while we load your content...
Please wait while we load your content...

