
Identifying a non-chiral ligand for the efficient chirality transfer in carbonylation of propargylic mesylates†
Yuli
Wang
a,
Wanli
Zhang
a and
Shengming
Ma
*ab
aShanghai Key Laboratory of Green Chemistry and Chemical Process, Department of Chemistry, East China Normal University, 3663 North Zhongshan Road, Shanghai 200062, China. E-mail: masm@sioc.ac.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China
First published on 4th July 2014
Abstract
Enantioselective synthesis of optically active 2,4-disubstituted 2,3-allenoates has been realized with a very high chirality transfer efficiency using the inexpensive commercially available non-chiral DPEphos in the presence of 100–200 psi of CO at room temperature. The key for the high efficiency of this chirality transfer process has been certified as the matched coordination of Pd with DPEphos forming a relatively stable optically active square planar allenylic Pd(II) complex, which undergoes a facile carbonylation in the presence of 10 equiv. of BnOH and 100–200 psi of CO.
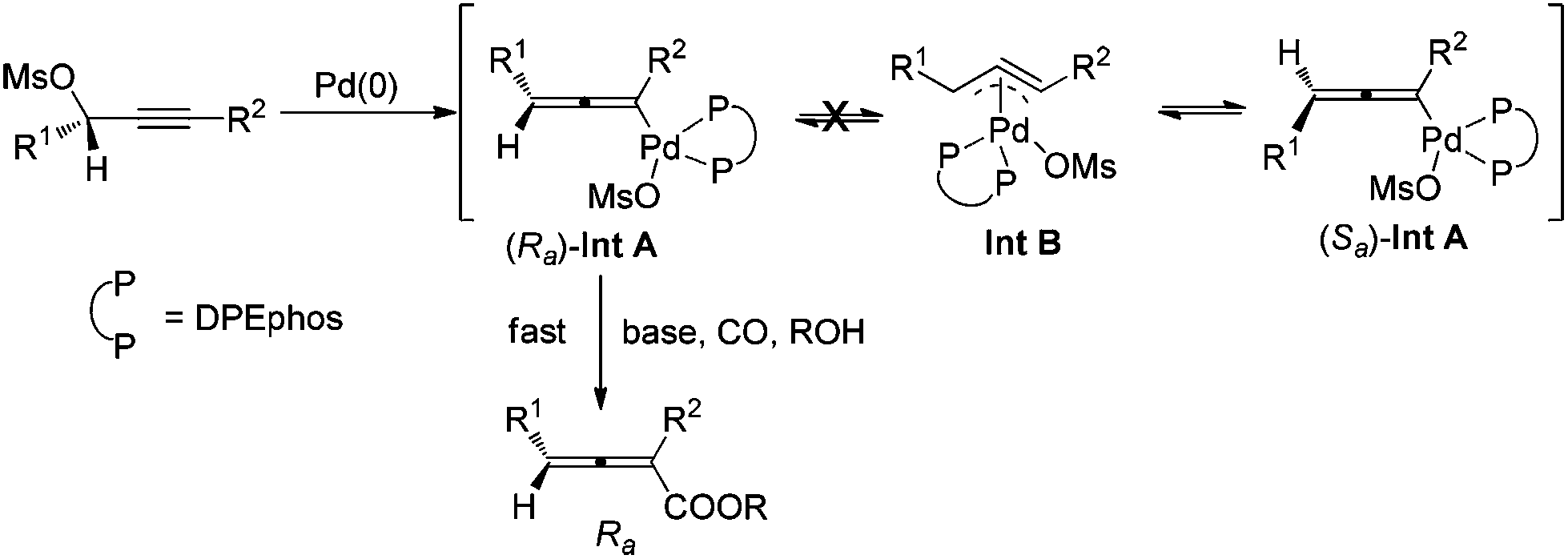
Allenes have been recognized as an important type of starting materials for organic synthesis;1 thus, asymmetric synthesis of allenes, especially 2,3-allenoates, is of high current interest in organic chemistry.2–8 Recently, it was reported that optically active 2,3-allenoates have been prepared via Pd-catalyzed carbonylation via catalytic chiral induction from the corresponding racemic starting materials, in which the racemization between (Sa)- and (Ra)-allenylic palladium complexes Int A and Int B is found to be crucial for the catalytic chiral induction strategy (Scheme 1).8 On the other hand, we have been pursuing a non-chiral ligand for a high efficiency of chirality transfer, which may importantly supplement the above mentioned reaction for R1 being an alkyl group beyond methyl (eqn (1))8 based on the following concept: a ligand for matched coordination with Pd to prevent interconversion between (Ra)-Int C and (Sa)-Int C (Scheme 1) followed by a very facile carbonylation.9
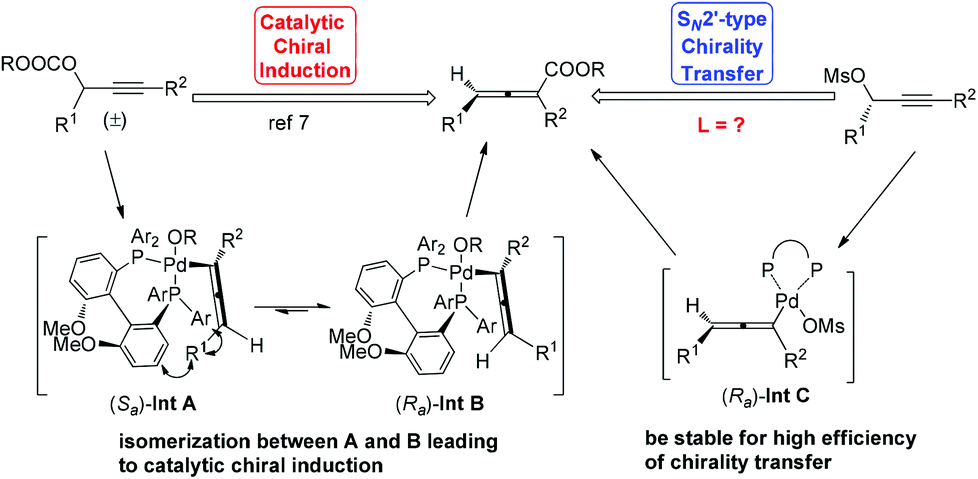 | ||
| Scheme 1 Concepts for the enantioselective synthesis of 2,3-allenoates via Pd-catalyzed carbonylation. | ||
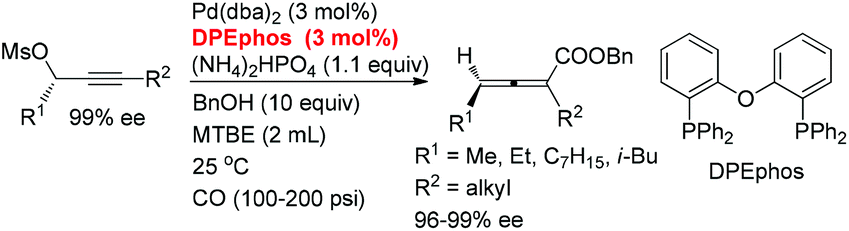
Here, we wish to report the successful identification of such a non-chiral cheap and readily available bidentate DPEphos as the ligand for the Pd-catalyzed efficient chirality transfer of enantio-enriched non-terminal propargylic mesylates forming optically active 2,3-allenoates (eqn (2)).7
 | (1) |
 | (2) |
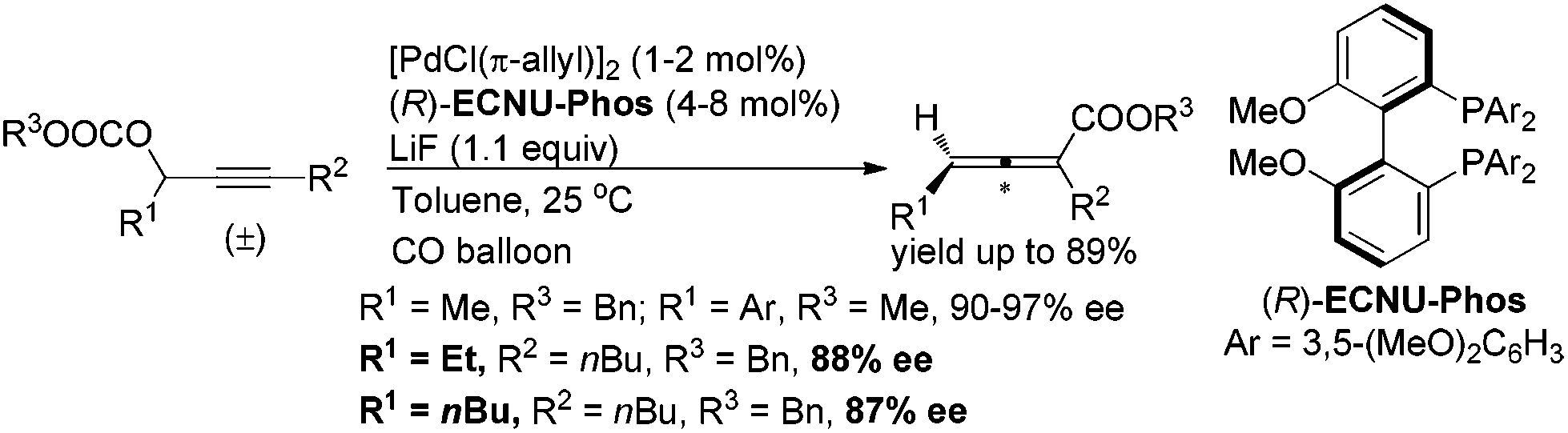
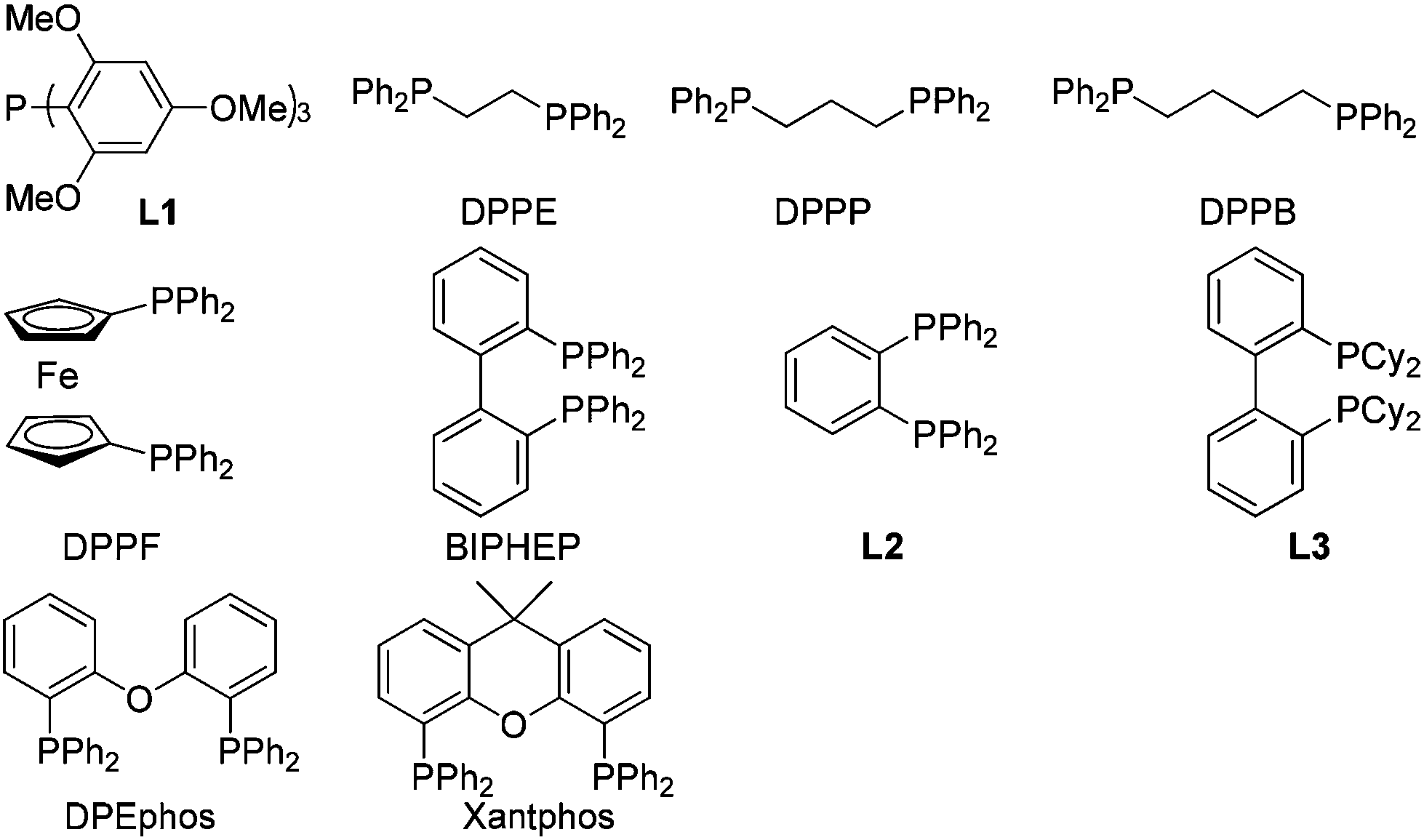
A screening of the non-chiral ligand has been carried out for the high efficiency of chirality transfer of (S)-1a:7e electron-rich tri(2,4,6-trimethoxyphenyl)phosphine (L1) was used for the reason of a better coordination ability with Pd to prevent racemization of (Ra)-Int C (Scheme 1); however, (Ra)-2a was obtained in only 26% yield and 36% ee (Table 1, entry 1). Then we started to work with bidentate ligands for the same purpose: the chirality transfer efficiency was rather low with DPPE, DPPP or DPPB as the ligand (Table 1, entries 2–4). Surprisingly, DPPF performed much better and the enantiopurity of the product reached 89% (Table 1, entry 5)! The ee value of the product further reached 93% with BIPHEP (Table 1, entry 6). Thus, we started to correlate the bite angles of some bidentate ligands with the ee of the products.10 1,2-Bis(diphenylphosphino)benzene (L2) led to a poor chirality transfer efficiency (Table 1, entry 7), and it was consistent with the result of using DPPE (Table 2, comparing entry 2 with entry 7), as the bite angles of L2 and DPPE are almost the same. Interestingly, replacing the phenyl group in BIPHEP with a more electron-donating cyclohexyl group (L3) led to the best result: the chirality transfer efficiency reached almost 100%, which must have been caused by the better electron-donating ability of this PCy2 group (Table 1, entry 8). With an increase of the bite angle, the chirality transfer efficiency was higher: when DPEphos with a bite angle of 104° was used, the optically active 2,3-allenoate (Ra)-2a was also afforded with 97% ee (Table 1, entry 9).11 However, when the bite angle became even bigger, the enantioselectivity dropped sharply: (Ra)-2a was only obtained in 70% yield with 76% ee using Xantphos with a bite angle of 108°, only 4° greater than DPEphos as the ligand (Table 1, entry 10). As the price of DPEphos is much lower than L3, it has been finally considered as the ligand of choice for further study.12
|
|
|||
|---|---|---|---|
| Entry | Ligand | Bite anglea | Yield%b of (Ra)-2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (ee%) c (ee%) |
 a Bite angles were obtained from crystal structures of bis(phosphine)PdCl2 complexes.
b Isolated yield.
c ee value of the product was determined by HPLC.
d The bite angle was obtained from a crystal structure of the bis(phosphine)Pd(1,1-dimethylallyl)tetrafluoroborate complex.
e The bite angle was obtained from a crystal structure of the bis(phosphine)PdBr2 complex.
f The bite angle was estimated based on analogy to BIPHEP.
a Bite angles were obtained from crystal structures of bis(phosphine)PdCl2 complexes.
b Isolated yield.
c ee value of the product was determined by HPLC.
d The bite angle was obtained from a crystal structure of the bis(phosphine)Pd(1,1-dimethylallyl)tetrafluoroborate complex.
e The bite angle was obtained from a crystal structure of the bis(phosphine)PdBr2 complex.
f The bite angle was estimated based on analogy to BIPHEP.
|
|||
| 1 | L1 | — | 26 (36) |
| 2 | DPPE | 8611a |
66 (24) |
| 3 | DPPP | 9111a |
73 (40) |
| 4 | DPPBd | 9411a |
75 (8) |
| 5 | DPPF | 9911a |
66 (89) |
| 6 | BIPHEP | 9211b |
77 (93) |
| 7 | L2 | 8711a |
85 (24) |
| 8 | L3 | 92 | 79 (98) |
| 9 | DPEphosd | 10411a |
82 (97) |
| 10 | Xantphosd | 10811a |
70 (76) |
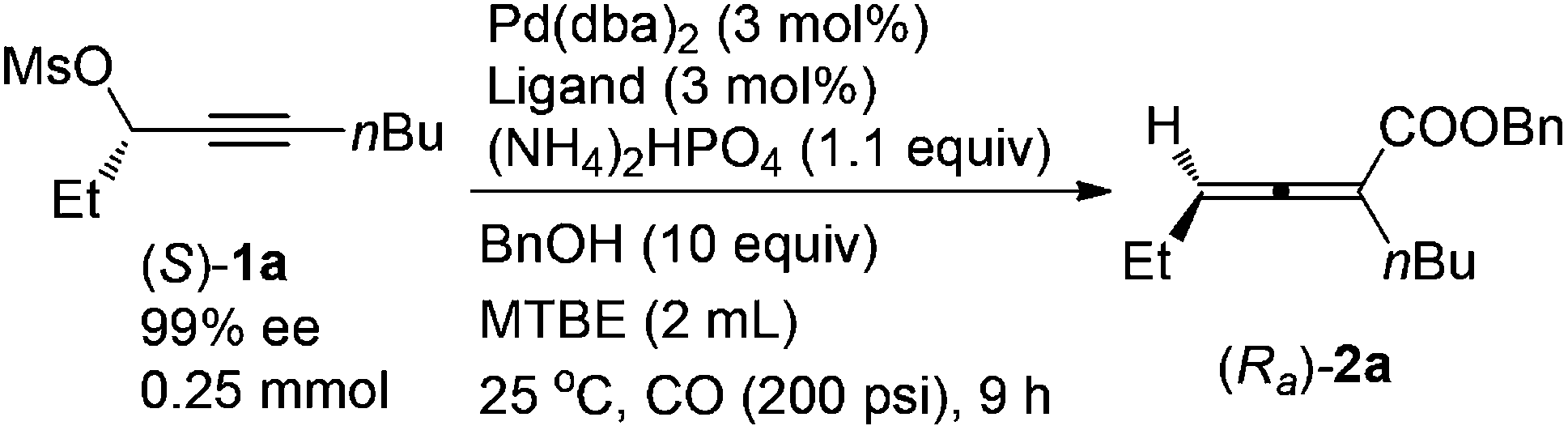
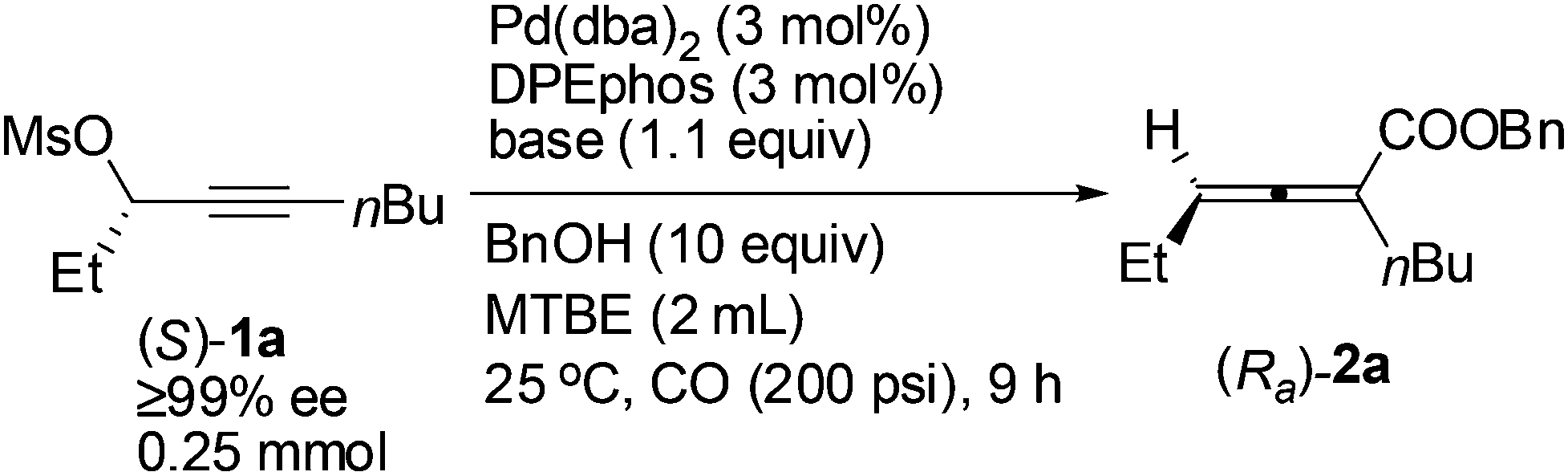
With DPEphos as the ligand, a series of bases were then examined (Table 2): using either (NH4)2HPO4 or (NH4)2CO3 led to the same chirality transfer efficiency with a slight difference of the isolated yield (Table 2, entries 1 and 2); however, the enantiopurity of (Ra)-2a dropped to 92% when K+ took the place of NH4+ (Table 2, comparing entry 2 with entry 3); (Ra)-2a could be obtained with 96% or 95% ee when NaHCO3 or KH2PO4 was used (Table 2, entries 4 and 5); interestingly, LiF, which performed best in the ECNU-Phos-induced reaction,7 led to a much lower yield (64%) albeit in 97% ee (Table 2, entry 6); the performance of organic bases was poor as compared to inorganic ones: (Ra)-2a was obtained in 75% yield with approximately 90% ee when pyridine or TEA was used (Table 2, entries 7 and 8).
A screening of the solvent was subsequently conducted. When ether, such as MTBE, 1,4-dioxane and THF, or toluene was used, the enantiopurity of (Ra)-2a was almost the same (96–97%), and the reaction in THF led to only 69% yield of (Ra)-2a (Table 3, entries 1–4). The enantiopurity of (Ra)-2a dropped to 87% with DMF as the solvent (Table 3, entry 5); the reaction proceeded in CH3CN with a very poor chirality transfer efficiency (Table 3, entry 6).
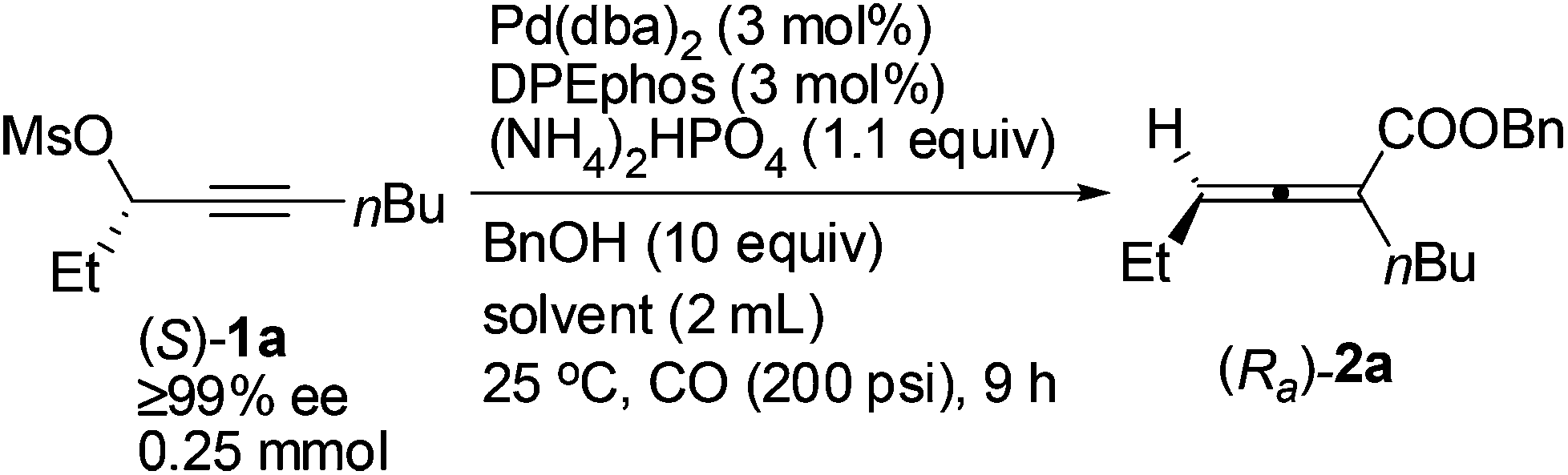
The scope of this reaction was then examined with 1 mmol of enantio-enriched non-terminal propargylic mesylates at room temperature and 200 psi of CO with 3 mol% each of Pd(dba)2 and DPEphos as the catalyst (Table 4). When the R1 group is ethyl, the enantioselectivities of the substrates with different alkyl groups were all greatly improved as compared with our previous report with (S)-Segphos as the ligand (Table 4, entries 1 and 3–7).7f And the chlorine substituted substrate (S)-1g led to the corresponding product with 97% ee (Table 4, entry 8). Furthermore, when the unsaturated groups, such as phenyl or vinyl, were introduced into R1, the corresponding products were also obtained with 98% ee, an obvious improvement again as compared to the (S)-Segphos-assisted protocol7f (Table 4, entries 9 and 10). Changing the chain length of R1 did not influence the outcome: the methyl-substituted 2,3-allenoate (Ra)-2j was obtained in 80% yield with 98% ee (Table 4, entry 11); and 2,3-allenoate (Ra)-2k with a longer n-heptyl group was also afforded in 84% yield with 96% ee (Table 4, entry 13); even the bulky groups performed very well in this process: with R1 being i-butyl, the corresponding (Ra)-2l was prepared in 85% yield with 97% ee (Table 4, entry 15). Furthermore, 4-alkyl substituted 2,3-allenoates, which were obtained with less than 90% ee unless R1 is methyl in (R)-ECNU-Phos-assisted protocol,8 were successfully produced with a very high enantioselectivity. In addition, the reaction was accelerated in the case of using (S)-1c-(S)-1g, (S)-1i and (S)-1l as the substrate, and the corresponding 2,3-allenoates were afforded with a higher enantiopurity in a much shorter time (Table 4, entries 4–8, 10 and 15, as compared with the (S)-Segphos-assisted protocol).
|
|
|||||
|---|---|---|---|---|---|
| Entry | (S)-1 | T (h) | (Ra)-2 | ||
| R1 | R2 | Yield%a | ee%b | ||
| a Isolated yield. b ee value of the product was determined by HPLC. c The reaction was carried out in the presence of 100 psi CO. d 97% purity. e 92% purity. f 98% purity. g 95% purity. | |||||
| 1 | Et | n-Bu (1a) | 9 | 86 (2a) | 98 |
| 2c | Et | n-Bu (1a) | 9 | 77d (2a) | 97 |
| 3 | Et | Me (1b) | 9 | 69e (2b) | 97 |
| 4 | Et | n-Pr (1c) | 3 | 85f (2c) | 97 |
| 5 | Et | n-C6H13 (1d) | 3 | 84 (2d) | 98 |
| 6 | Et | t-Bu (1e) | 6 | 84 (2e) | 99 |
| 7 | Et | (CH2)2CH(CH3)2 (1f) | 9 | 81 (2f) | 96 |
| 8 | Et | (CH2)4Cl (1g) | 6 | 82 (2g) | 97 |
| 9 | Et | (CH2)2Ph (1h) | 9 | 76 (2h) | 98 |
| 10 | Et | Allyl (1i) | 6 | 82 (2i) | 98 |
| 11 | Me | n-C7H15 (1j) | 9 | 80 (2j) | 98 |
| 12c | Me | n-C7H15 (1j) | 9 | 82 (2j) | 97 |
| 13 | n-C7H15 | n-Bu (1k) | 9 | 84 (2k) | 96 |
| 14c | n-C7H15 | n-Bu (1k) | 9 | 83 (2k) | 96 |
| 15 | i-Bu | n-Bu (1l) | 6 | 81g (2l) | 97 |
| 16c | i-Bu | n-Bu (1l) | 6 | 84 (2l) | 98 |
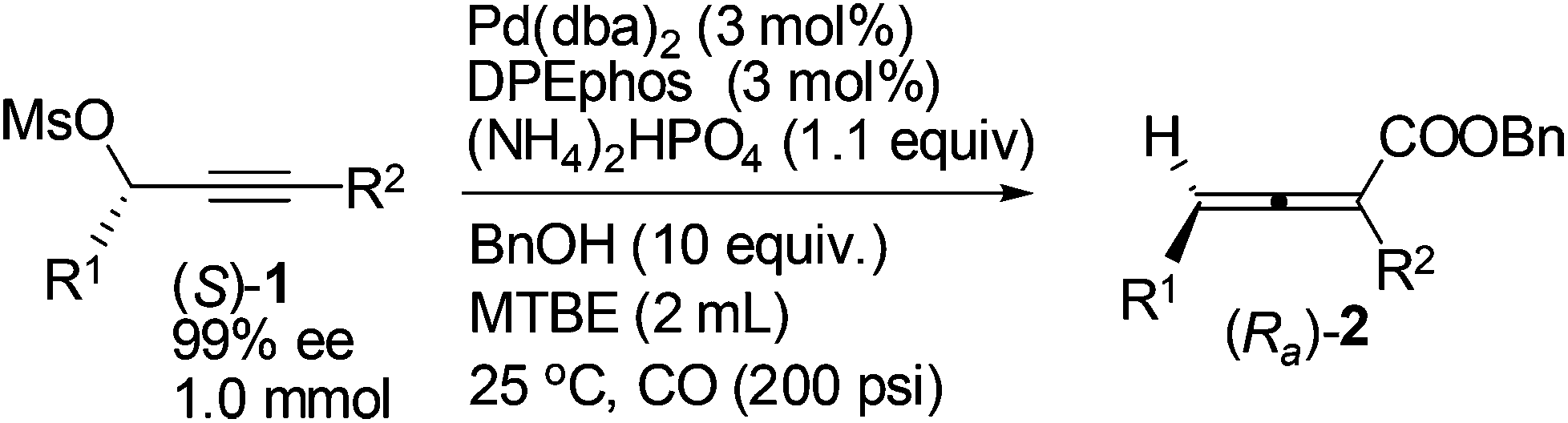
Gram scale synthesis of optically active 2,3-allenoate (Ra)-2a was also carried out: after 6 h, (Ra)-2a was obtained with 90% yield and 96% ee using just 1 mol% Pd(dba)2 and DPEphos (eqn (3)).
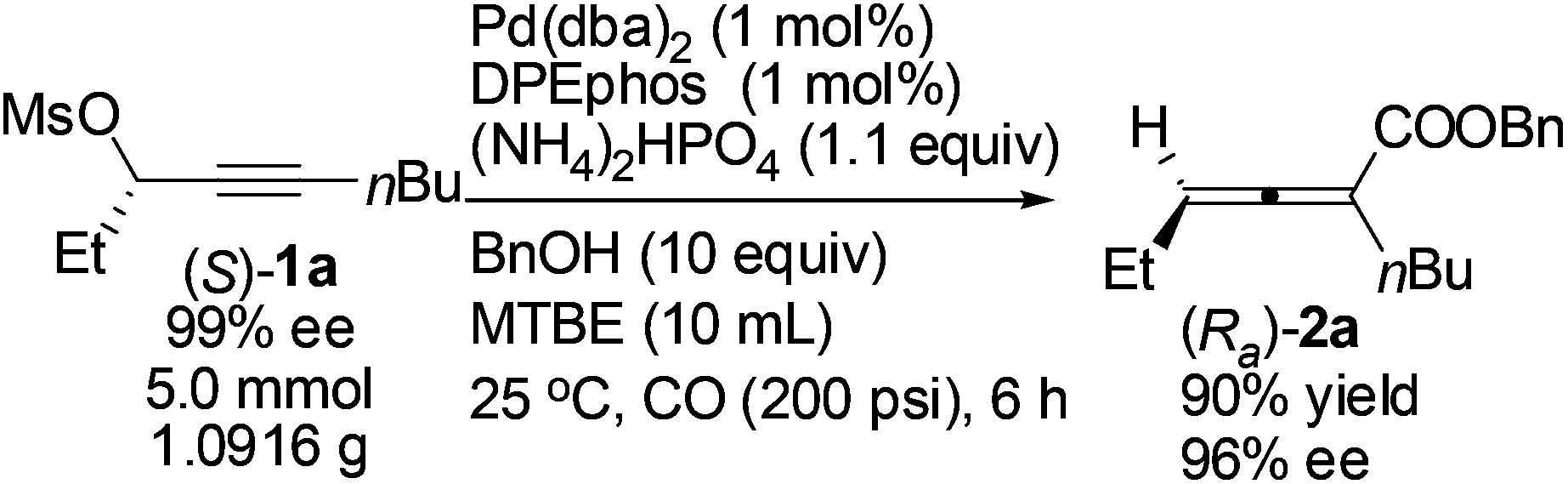 | (3) |
Optically active 2,3-allenoate (Sa)-2m was also prepared from the enantiomer (R)-1m13 in 75% yield with 99% ee, further affirming that it is a chirality transfer process with the absolute configuration of the 2,3-allenoate being determined by that of the starting propargylic mesylate and the racemization was mostly inhibited in the presence of DPEphos (eqn (4)).
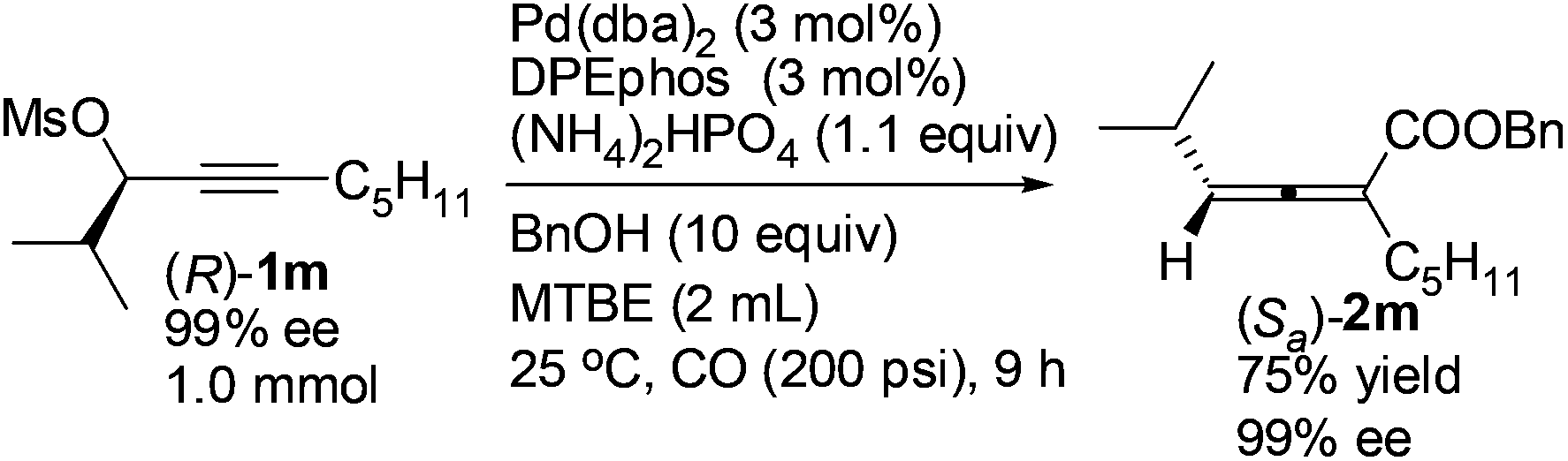 | (4) |
Reducing the loading of BnOH to 5 equiv. led to a poor result: (Ra)-2a was only afforded in 70% yield with a much lower ee (92%) (compare eqn (5) with entry 1 of Table 4).
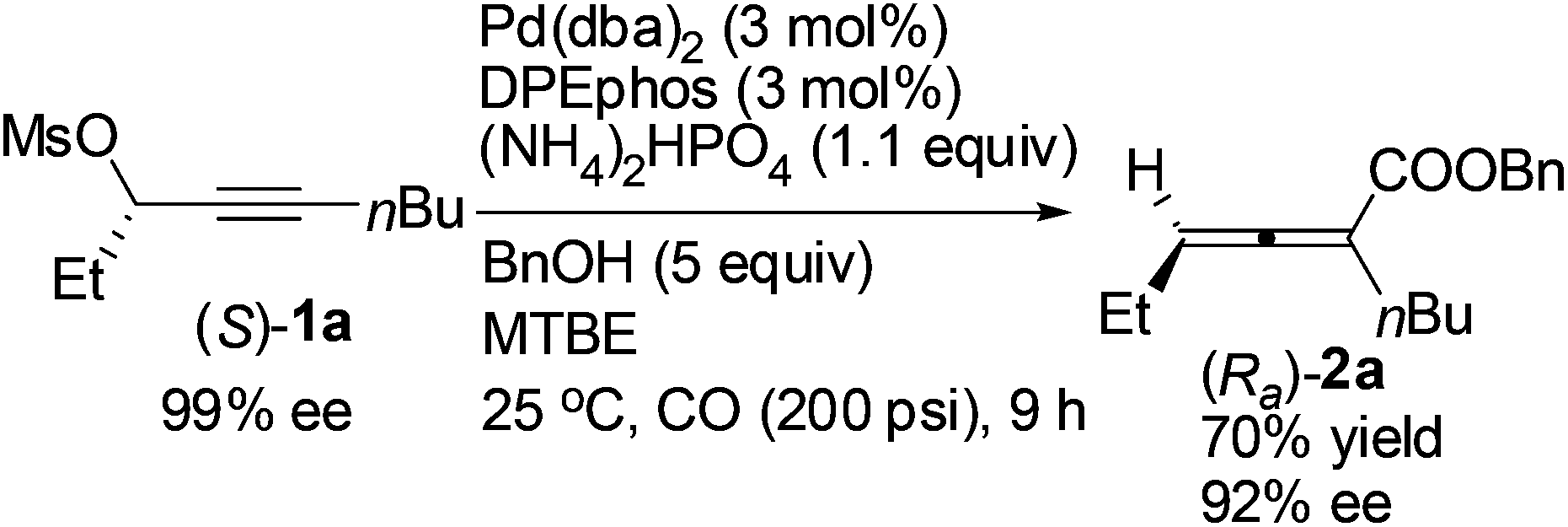 | (5) |
The effect of CO pressure has also been explored.14 As shown in Fig. 1, the ee value of (Ra)-2a increased with the CO pressure in the range of 15–100 psi. And we finally found that the reaction proceeded with the same chirality transfer efficiency even in the presence of 100 psi of CO (97% ee). Some optically active 2,3-allenoates were afforded under these conditions with almost the same results as in the presence of 200 psi of CO (Table 4, entries 2, 12, 14 and 16, compared with Table 4, entries 1, 11, 13 and 15).
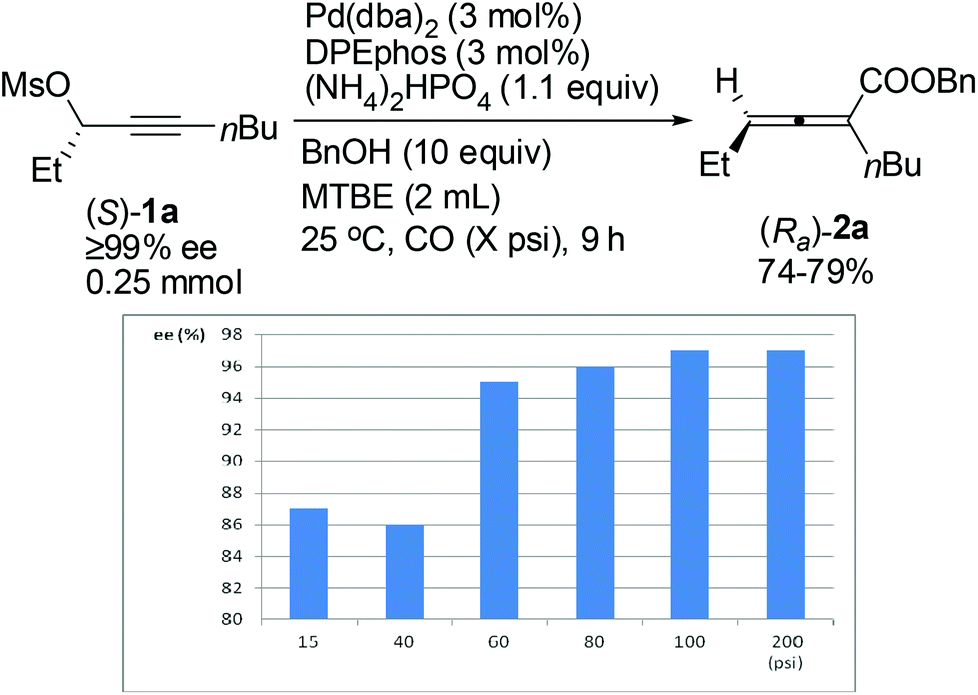 | ||
| Fig. 1 Effect of CO pressure of DPEphos-assisted carbonylation of enantioenriched (S)-1a. | ||
Based on these studies, a rationale for the high chirality transfer efficiency in this reaction has been proposed: (1) DPEphos makes the square planar complex (Ra)-Int A formed via the highly stereoselective SN2′-type oxidative addition of Pd with the optically active substrates stable enough, avoiding the racemization via η3-type Int B, which accounts for the low efficiency chirality transfer efficiency. (2) 10 equiv. of BnOH and a certain level of CO pressure are responsible for the subsequent smooth carboxylation as soon as (Ra)-Int A is generated (Scheme 2).14
 | ||
| Scheme 2 Rationale for the high chirality transfer efficiency with DPEphos. | ||
In summary, a highly effective protocol for the enantioselective synthesis of optically active 2,4-disubstituted 2,3-allenoates has been established using the inexpensive commercially available DPEphos in the presence of 100–200 psi of CO by starting from the readily available optically active propargylic alcohols. The reaction shows a very high chirality transfer efficiency, providing a very important supplement for the carbonylation of racemic propargylic carbonates using the Pd catalyst and (R)-ECNU-Phos for 4-non-methyl-alkyl-substituted 2,3-allenoates with ≥96% ee. Through control experiments, it has been unveiled that the matched coordination of Pd with DPEphos and a very fast carboxylation with BnOH and CO are the key for this highly efficient chirality transfer process by preventing racemization. Further studies in this area are being actively conducted in this laboratory.
Experimental
To a flame-dried Schlenk tube were added Pd(dba)2 (28.8 mg, 0.05 mmol) and DPEphos (27.9 mg, 0.05 mmol). Then the tube was degassed and refilled with Ar three times to ensure the complete exclusion of air. Freshly distilled MTBE (2 mL) was subsequently added under argon. The resulting Pd(0) mixture was stirred for 1 hour at room temperature.To a flame-dried tube filled with Ar were added (NH4)2HPO4 (0.7260 g, 5.5 mmol), (S)-1a (1.1021 g, 5 mmol)/MTBE (4 mL), and BnOH (5.4088 g, 50 mmol)/MTBE (4 mL). After the Pd(0) mixture prepared above was transferred by a syringe to the tube, it was placed in the Parr reactor. The air in the reactor was replaced by CO gas three times sequentially, and the Parr reactor was charged to 200 psi with CO gas. The mixture was stirred at room temperature till the reaction ended, and then the gas was vented in the hood safely. The resulting mixture was diluted with Et2O (20 mL), washed with brine (20 mL), and dried over Na2SO4. After the filtration and concentration under reduced pressure, the crude product was purified by flash chromatography on silica gel to afford (Ra)-2a (1.1750 g, 90%) as an oil [eluent: petroleum ether (b.p. 60–90 °C)–ethyl ether = 250/1]: 96% ee (HPLC conditions: Regis (S,S) Whelk-O column, hexane–i-PrOH = 200/1, 1.0 mL min−1, λ = 214 nm, tR (minor) = 11.3 min, tR (major) = 13.0 min); [α]21D = −37.8 (c = 1.21, CHCl3) [96% ee, [α]24D = −38.0 (c = 1.17, CHCl3)]; 1H NMR (300 MHz, CDCl3): δ = 7.39–7.23 (m, 5 H, Ar–H), 5.63–5.54 (m, 1 H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 5.22 (d, J = 12.6 Hz, 1 H, one proton from Bn), 5.14 (d, J = 12.6 Hz, 1 H, one proton from Bn), 2.32–2.18 (m, 2 H, CH2), 2.18–2.06 (m, 2 H, CH2), 1.49–1.23 (m, 4 H, 2 × CH2), 1.04 (t, J = 7.4 Hz, 3 H, CH3), 0.90 (t, J = 6.9 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3): δ = 209.8, 167.6, 136.5, 128.4, 127.8, 127.5, 101.1, 96.7, 66.1, 30.3, 28.1, 22.2, 21.4, 13.9, 13.2.
CH), 5.22 (d, J = 12.6 Hz, 1 H, one proton from Bn), 5.14 (d, J = 12.6 Hz, 1 H, one proton from Bn), 2.32–2.18 (m, 2 H, CH2), 2.18–2.06 (m, 2 H, CH2), 1.49–1.23 (m, 4 H, 2 × CH2), 1.04 (t, J = 7.4 Hz, 3 H, CH3), 0.90 (t, J = 6.9 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3): δ = 209.8, 167.6, 136.5, 128.4, 127.8, 127.5, 101.1, 96.7, 66.1, 30.3, 28.1, 22.2, 21.4, 13.9, 13.2.
Notes and references
- For recent reviews, see: (a) B. Alcaide, P. Almendros and T. Martínez del Campo, Chem. – Eur. J., 2010, 16, 5836 CrossRef CAS PubMed; (b) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS PubMed; (c) F. Inagaki, S. Kitagaki and C. Mukai, Synlett, 2011, 594 CAS; (d) F. López and J. L. Mascareñas, Chem. – Eur. J., 2011, 17, 418 CrossRef PubMed; (e) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed.
- For recent reviews, see: (a) M. Ogasawara, Tetrahedron: Asymmetry, 2009, 20, 259 CrossRef CAS; (b) S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384 RSC.
- For the synthesis of racemic 2,3-allenoates in a similar manner, see: (a) J. Tsuji, T. Sugiura and I. Minami, Tetrahedron Lett., 1986, 27, 731 CrossRef CAS; (b) G. E. Akpinar, M. Kus, M. Ucuncu, E. Karakus and L. Artok, Org. Lett., 2011, 13, 748 CrossRef CAS PubMed.
- For dynamic resolution of racemic 2,3-allenoates, see: (a) S. Ramaswamy, R. Hui and J. B. Jones, J. Chem. Soc., Chem. Commun., 1986, 1545 RSC; (b) J. Yu, W.-J. Chen and L.-Z. Gong, Org. Lett., 2010, 12, 4050 CrossRef CAS PubMed.
- For the enantioselective synthesis of 2,3-allenoates with stoichiometric chiral reagents, see: (a) K. Mikami and A. Yoshida, Angew. Chem., Int. Ed. Engl., 1997, 36, 858 CrossRef CAS; (b) K. Mikami and A. Yoshida, Tetrahedron, 2001, 57, 889 CrossRef CAS; (c) K. Tanaka, K. Otsubo and K. Fuji, Tetrahedron Lett., 1996, 37, 3735 CrossRef CAS; (d) J. Yamazaki, T. Watanabe and K. Tanaka, Tetrahedron: Asymmetry, 2001, 12, 669 CrossRef CAS; (e) C.-Y. Li, X.-B. Wang, X.-L. Sun, Y. Tang, J.-C. Zheng, Z.-H. Xu, Y.-G. Zhou and L.-X. Dai, J. Am. Chem. Soc., 2007, 129, 1494 CrossRef CAS PubMed; (f) C.-Y. Li, X.-L. Sun, Q. Jing and Y. Tang, Chem. Commun., 2006, 2980 RSC; (g) C.-Y. Li, B.-H. Zhu, L.-W. Ye, Q. Jing, X.-L. Sun, Y. Tang and Q. Shen, Tetrahedron, 2007, 63, 8046 CrossRef CAS.
- For the enantioselective synthesis of 2,3-allenoates from racemic starting materials via catalytic chiral induction, see: (a) H. Liu, D. Leow, K.-W. Huang and C.-H. Tan, J. Am. Chem. Soc., 2009, 131, 7212 CrossRef CAS PubMed; (b) T. Inokuma, M. Furukawa, T. Uno, Y. Suzuki, K. Yoshida, Y. Yano, K. Matsuzaki and Y. Takemoto, Chem. – Eur. J., 2011, 17, 10470 CrossRef CAS PubMed; (c) T. Inokuma, M. Furukawa, Y. Suzuki, T. Kimachi, Y. Kobayashi and Y. Takemoto, ChemCatChem, 2012, 4, 983 CrossRef CAS; (d) T. Hashimoto, K. Sakata, F. Tamakuni, M. J. Dutton and K. Maruoka, Nat. Chem., 2013, 5, 240 CrossRef CAS PubMed; (e) I. T. Crouch, R. K. Neff and D. E. Frantz, J. Am. Chem. Soc., 2013, 135, 4970 CrossRef CAS PubMed; (f) H. Qian, X. Yu, J. Zhang and J. Sun, J. Am. Chem. Soc., 2013, 135, 18020 CrossRef CAS PubMed.
- For the enantioselective synthesis of 2,3-allenoates from propargylic derivatives via chirality transfer, see: (a) G. A. Molander, E. M. Sommers and S. R. Baker, J. Org. Chem., 2006, 71, 1563 CrossRef CAS PubMed; (b) K. Kobayashi, H. Naka, A. E. H. Wheatley and Y. Kondo, Org. Lett., 2008, 10, 3375 CrossRef CAS PubMed; (c) J. A. Marshall and M. A. Wolf, J. Org. Chem., 1996, 61, 3238 CrossRef CAS; (d) J. A. Marshall, G. S. Bartley and E. M. Wallace, J. Org. Chem., 1996, 61, 5729 CrossRef CAS; (e) J. A. Marshall, M. A. Wolf and E. M. Wallace, J. Org. Chem., 1997, 62, 367 CrossRef CAS PubMed; (f) Y. Wang and S. Ma, Adv. Synth. Catal., 2013, 355, 741 CrossRef CAS.
- Y. Wang, W. Zhang and S. Ma, J. Am. Chem. Soc., 2013, 135, 11517 CrossRef CAS PubMed.
- For the mechanism of racemization of allenyl palladium complex, see: S. Ogoshi, T. Nishida, T. Shinagawa and H. Kurosawa, J. Am. Chem. Soc., 2001, 123, 7164 CrossRef CAS PubMed.
- For the bite angles obtained from crystal structures of palladium complexes, see: (a) J. E. Ney and J. P. Wolfe, J. Am. Chem. Soc., 2005, 127, 8644 CrossRef CAS PubMed and related references therein; (b) M. Ogasawara, K. Yoshida and T. Hayashi, Organometallics, 2000, 19, 1567 CrossRef CAS.
- When 1.5 mol% of [PdCl(π-allyl)]2 was used instead of Pd(dba)2 as the catalyst, (Ra)-2a was afforded in 78% yield with 94% ee.
- Recent price per gram according to J&K Chemicals: DPEphos vs. 2,2′-bis(dicyclohexyl-phosphino)-1,1′-biphenyl (L3) = 80:1890 (in CNY).
- (R)-1m was synthesized from the corresponding optically active alcohol, which was obtained via kinetic resolution from the racemic one: (a) D. Xu, Z. Li and S. Ma, Chem. – Eur. J., 2002, 8, 5012 CrossRef CAS PubMed; (b) D. Xu, Z. Li and S. Ma, Tetrahedron Lett., 2003, 44, 6343 CrossRef CAS. The absolute configuration was affirmed according to the literature: C. R. Reddy, E. Jithender and K. R. Prasad, J. Org. Chem., 2013, 78, 4251 CrossRef CAS PubMed.
- For details, see Table S1 in ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00151f |
| This journal is © the Partner Organisations 2014 |