
Catalytic stereospecific allylation of protected hydrazines with enantioenriched primary allylic amines†
Yong
Wang
,
Jing-Kun
Xu
,
Yonghong
Gu
* and
Shi-Kai
Tian
*
Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: ygu01@ustc.edu.cn; tiansk@ustc.edu.cn; Tel: (+86) 551-6360-0871
First published on 7th July 2014
Abstract
An unprecedented allylation reaction of protected hydrazines with enantioenriched allylic amines has been developed in a stereospecific manner with net stereoretention. A wide variety of protected hydrazines underwent palladium/acid-catalyzed allylation with highly enantioenriched primary allylic amines to give structurally diverse N-allylhydrazines in moderate to excellent yields with extremely high regioselectivity and complete retention of the configuration. Importantly, the reaction exhibits high atom-economy by yielding ammonia as the sole byproduct.
Introduction
The hydrazine moiety is not only present in some biologically relevant compounds such as the agents for treating HCV and HIV-1 infections,1 but also serves as a foundation for a broad range of chemical transformations such as reduction, addition, and substitution.2 Thus, much attention has been paid to the introduction of the hydrazine moiety into target compounds, for which a common strategy is the derivatization of protected hydrazines.3 In this regard, it is particularly noteworthy that a versatile allyl moiety can be directly attached to the nitrogen atoms of protected hydrazines through transition metal-catalyzed allylic substitution.4,5 Although the asymmetric allylation of protected hydrazines has been realized in the presence of chiral transition metal complexes for the synthesis of enantioenriched N-allylhydrazines, the allylic components are limited to a couple of symmetrical (α-substituent = γ-substituent) allylic esters or carbonates.6 To the best of our knowledge, there is no previous report on the asymmetric allylation of protected hydrazines with unsymmetrical (α-substituent ≠ γ-substituent) allylic electrophiles.7When compared to traditional allylic electrophiles such as allylic halides and alcohol derivatives, allylic amines have been far less explored in coupling directly with nucleophiles through allylic C–N bond cleavage despite their wide application in protective group chemistry.8 Whereas the amino group is a very poor leaving group, sporadic studies have demonstrated that transition metals can catalyze the direct substitution of allylic amines with a few types of nucleophiles.9–11 Particularly, primary allylic amines are able to serve as effective allylic components, wherein the leaving NH2 group has a mass of 16 amu.9 It should be pointed out that primary allylic amines can be prepared following synthetic routes not longer than allylic halides and alcohol derivatives, and their basicity allows purification in large quantities by simple extractive procedures instead of routine chromatography.9a,b Moreover, enantioenriched primary allylic amines are readily accessible by resolution of the racemic mixtures with tartaric acids.9a,b,12 In the course of exploring the synthetic utilities of C(sp3)–N bond cleavage9,13 as well as protected hydrazines,14 we found that palladium/acid could catalyze the stereospecific allylation of protected hydrazines with highly enantioenriched primary allylic amines. Importantly, the reaction provides facile access to new enantioenriched N-allylhydrazines with excellent ee and exhibits high atom-economy by yielding ammonia as the sole byproduct.
Results and discussion
Since the Boc group has been frequently employed to protect hydrazines, we selected an N-Boc hydrazine, carbazate 2a, as a model substrate to undergo asymmetric allylation with enantioenriched primary allylic amine 1a (94% ee). A number of common palladium sources (5 mol%) and ligands were examined in the reaction performed in dioxane at 80 °C (Table 1, entries 1–8). While most of them failed to catalyze the reaction, to our delight, the combined use of Pd(OAc)2 and racemic BINAP led to the formation of N-allylhydrazine 3a in 35% yield with 93% ee, >99% α-selectivity, and complete retention of alkene geometry (Table 1, entry 6). Assuming that a catalytic amount of an acid could activate the leaving NH2 group but meanwhile would not remove the Boc group, we added a Brønsted acid or a Lewis acid (5 mol%) to the reaction mixture and found that the use of p-toluenesulfonic acid not only improved the yield to 92% but also avoided the erosion of enantiopurity (Table 1, entry 11). Replacing dioxane with another common organic solvent proved fruitless to improve the yield further, and moreover, decreased the enantiopurity (Table 1, entries 12–14). Finally, both optically active (R)-BINAP and (S)-BINAP were examined for comparison. While the use of (R)-BINAP gave a higher yield than that of (S)-BINAP, in both cases the chirality was completely transferred with the same sense of chirality as substrate 1a (Table 1, entries 15 and 16). Clearly, the stereochemistry of the reaction was completely controlled by the substrate rather than by the reagent. Taken together, we decided to use racemic BINAP, which is less expensive than the optically active one, as the ligand in the reaction since it resulted in complete retention of the configuration as well as an excellent yield (Table 1, entry 11).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Pd] | Ligand | Acid | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: amine 1a (0.30 mmol), carbazate 2a (0.36 mmol), [Pd] (5 mol%), ligand (if any, 10 mol%), acid (if any, 5 mol%), solvent (0.30 mL), 80 °C, 2 h. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. d 20 mol% PPh3 was used. | ||||||
| 1 | Pd2(dba)3 | None | None | Dioxane | 0 | — |
| 2 | Pd(PPh3)4 | None | None | Dioxane | Trace | — |
| 3 | Pd(OAc)2 | PPh3d | None | Dioxane | Trace | — |
| 4 | Pd(OAc)2 | Xantphos | None | Dioxane | Trace | — |
| 5 | Pd(OAc)2 | dppb | None | Dioxane | Trace | — |
| 6 | Pd(OAc)2 | BINAP | None | Dioxane | 35 | 93 |
| 7 | Pd(OAc)2 | TMEDA | None | Dioxane | 0 | — |
| 8 | Pd(OAc)2 | BINOL | None | Dioxane | Trace | — |
| 9 | Pd(OAc)2 | BINAP | B(OH)3 | Dioxane | 86 | 92 |
| 10 | Pd(OAc)2 | BINAP | ZnCl2 | Dioxane | 46 | 93 |
| 11 | Pd(OAc)2 | BINAP | TsOH | Dioxane | 92 | 94 |
| 12 | Pd(OAc)2 | BINAP | TsOH | DME | 83 | 92 |
| 13 | Pd(OAc)2 | BINAP | TsOH | MeCN | 85 | 91 |
| 14 | Pd(OAc)2 | BINAP | TsOH | Toluene | Trace | — |
| 15 | Pd(OAc)2 | (R)-BINAP | TsOH | Dioxane | 90 | 94 |
| 16 | Pd(OAc)2 | (S)-BINAP | TsOH | Dioxane | 76 | 94 |
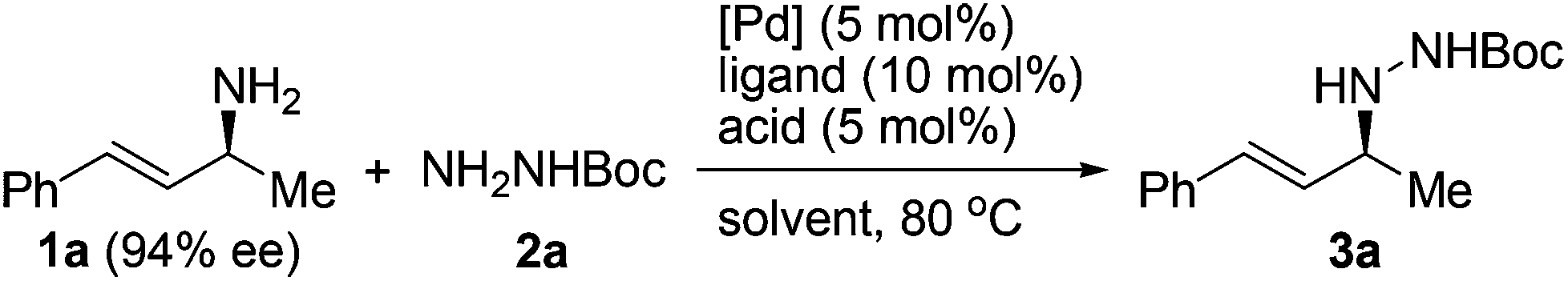
A variety of protected hydrazines were examined in the allylation reaction with highly enantioenriched unsymmetrical primary allylic amines in the presence of 5 mol% Pd(OAc)2, 10 mol% racemic BINAP, and 5 mol% p-toluenesulfonic acid (Table 2).15 To our delight, N-alkyl-N′-Boc hydrazines were allylated by enantioenriched primary allylic amine 1a in a stereospecific manner with extremely high regioselectivity, albeit the reaction gave lower yields than that with carbazate 2a (Table 2, entries 1–5). In sharp contrast, the reaction with an N-aryl-N′-Boc hydrazine, a less reactive nitrogen nucleophile, failed to take place under the standard conditions (Table 2, entry 6). A few other alkoxycarbonyl-, acyl-, and phosphoryl-protected hydrazines also served as suitable nitrogen nucleophiles, the reaction of which with allylic amine 1a gave the corresponding N-allylhydrazines in good to excellent yields with complete retention of configuration (Table 2, entries 7–11).16 Nevertheless, the desired allylation product was not obtained at all from the reaction of allylic amine 1a with phenylhydrazine (2l) in that it was oxidized quickly by air to give hydrazone 4a (Table 2, entry 12).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1, R1, R2, R3 | 2, R4, R5 | 3 | Yieldb (%) | eec (%) | |
| 1 | 3 | |||||
a Reaction conditions: amine 1 (0.30 mmol), protected hydrazine 2 (0.36 mmol), Pd(OAc)2 (5 mol%), racemic BINAP (10 mol%), TsOH (5 mol%), dioxane (0.30 mL), 80 °C, 2 h.
b Isolated yield.
c Determined by HPLC analysis on a chiral stationary phase.
d PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHC(Me)NNHPh (4a) was obtained in 86% yield. CHC(Me)NNHPh (4a) was obtained in 86% yield.
|
||||||
| 1 | 1a, Ph, H, Me | 2a, H, Boc | 3a | 92 | 94 | 94 |
| 2 | 1a, Ph, H, Me | 2b, 1-Propyl, Boc | 3b | 63 | 94 | 94 |
| 3 | 1a, Ph, H, Me | 2c, Allyl, Boc | 3c | 60 | 94 | 94 |
| 4 | 1a, Ph, H, Me | 2d, Propargyl, Boc | 3d | 40 | 94 | 94 |
| 5 | 1a, Ph, H, Me | 2e, PhCH2, Boc | 3e | 77 | 94 | 94 |
| 6 | 1a, Ph, H, Me | 2f, Ph, Boc | 3f | 0 | 94 | — |
| 7 | 1a, Ph, H, Me | 2g, H, CO2Et | 3g | 96 | 94 | 94 |
| 8 | 1a, Ph, H, Me | 2h, H, Cbz | 3h | 99 | 94 | 94 |
| 9 | 1a, Ph, H, Me | 2i, H, COPh | 3i | 97 | 94 | 94 |
| 10 | 1a, Ph, H, Me | 2j, H, COMe | 3j | 95 | 94 | 94 |
| 11 | 1a, Ph, H, Me | 2k, H, PO(OEt) 2 | 3k | 80 | 94 | 94 |
| 12 | 1a, Ph, H, Me | 2l, H, Ph | 3l | 0d | 94 | — |
| 13 | 1b, 2-ClC6H4, H, Me | 2a, H, Boc | 3m | 80 | 96 | 96 |
| 14 | 1c, 2-Naphthyl, H, Me | 2a, H, Boc | 3n | 84 | 90 | 90 |
| 15 | 1d, Cyclohexyl, H, Me | 2a, H, Boc | 3o | 82 | 97 | 97 |
| 16 | 1e, Ph, Me, Me | 2a, H, Boc | 3p | 63 | 96 | 96 |
| 17 | 1f, Ph, H, Et | 2a, H, Boc | 3q | 81 | 99 | 99 |
| 18 |
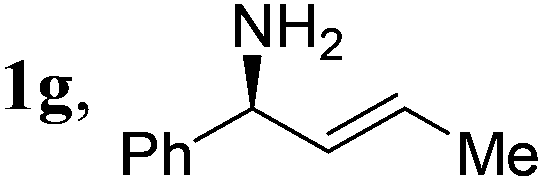
|
2a, H, Boc | 3a | 90 | 96 | 96 |
| 19 | 1h, Ph, H, Ph | 2a, H, Boc | 3r | 74 | 98 | 0 |
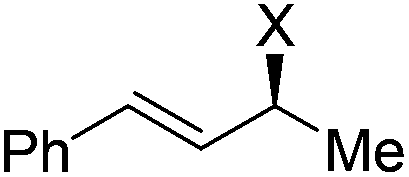
|
||||||
| 20 | 1i, X = NHCH2Ph | 2a, H, Boc | 3a | 63 | 94 | 94 |
| 21 | 1j, X = N(CH2CH2)2O | 2a, H, Boc | 3a | 74 | 94 | 94 |
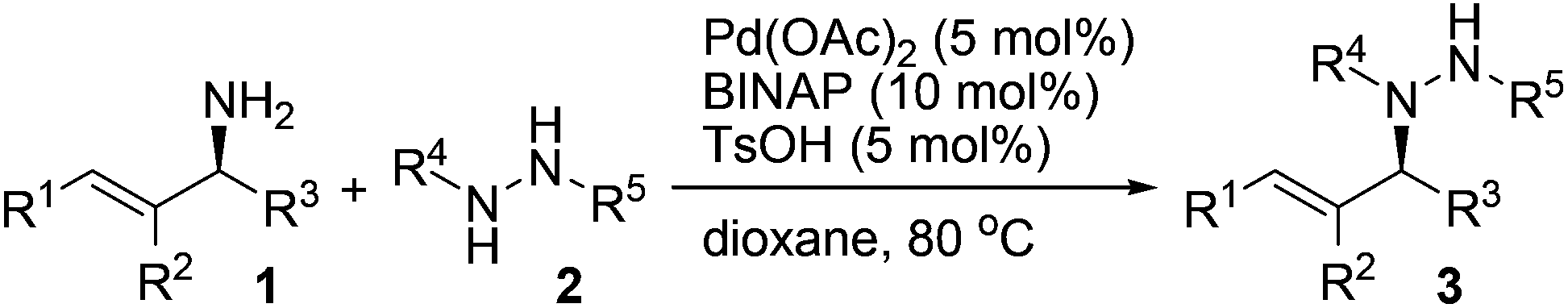
A range of enantioenriched unsymmetrical primary allylic amines smoothly underwent allylation with carbazate 2a in a stereospecific manner under the standard conditions (Table 2, entries 13–18). In line with typical allylic substitution,5 the regioselectivity was determined by the steric and electronic properties of the α- and γ-substituents in the allylic amines. When the α-substituent was an alkyl group and the γ-substituent was an aryl group or a bulkier alkyl group, the reaction proceeded in an α-selective fashion (Table 2, entries 1–17). In sharp contrast, the reaction proceeded in a γ-selective fashion when the α-substituent was an aryl group and the γ-substituent was an alkyl group (Table 2, entry 18). The γ-selectivity could arise from both maximizing conjugation and minimizing steric hindrance prior to the C–N bond formation between the π-allylpalladium intermediate and the protected hydrazine (see below). Nevertheless, effective chirality transfer was not applicable to a symmetrical allylic amine due to the symmetry of the π-allyl unit in the resulting π-allylpalladium intermediate (Table 2, entry 19). Moreover, replacing the NH2 group in the allylic amine either with a monoalkylamino group or with a dialkylamino group led to a lower yield but still with complete retention of configuration (Table 2, entries 20 and 21).
The optically active N-allylhydrazines we obtained could undergo a range of chemical transformations with complete retention of enantiopurity under appropriate conditions.17 For example, exposure of Boc-protected N-allylhydrazine 3a (94% ee) to trifluoroacetic acid at room temperature smoothly removed the Boc group to give salt 5a, which could be converted to acyl-protected N-allylhydrazine 3ivia routine acylation (Scheme 1). On the other hand, compound 3a underwent propargylation under basic conditions to give trisubstituted hydrazine 3d with extremely high regioselectivity. Moreover, treatment of N-allylhydrazine 3j (94% ee) with diphenyl diselenide, ammonium persulfate, and trifluoromethanesulfonic acid at room temperature led to the formation of functionalized tetrahydropyrazole 6a in 76% yield with >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and complete retention of enantiopurity.18
:1 dr and complete retention of enantiopurity.18
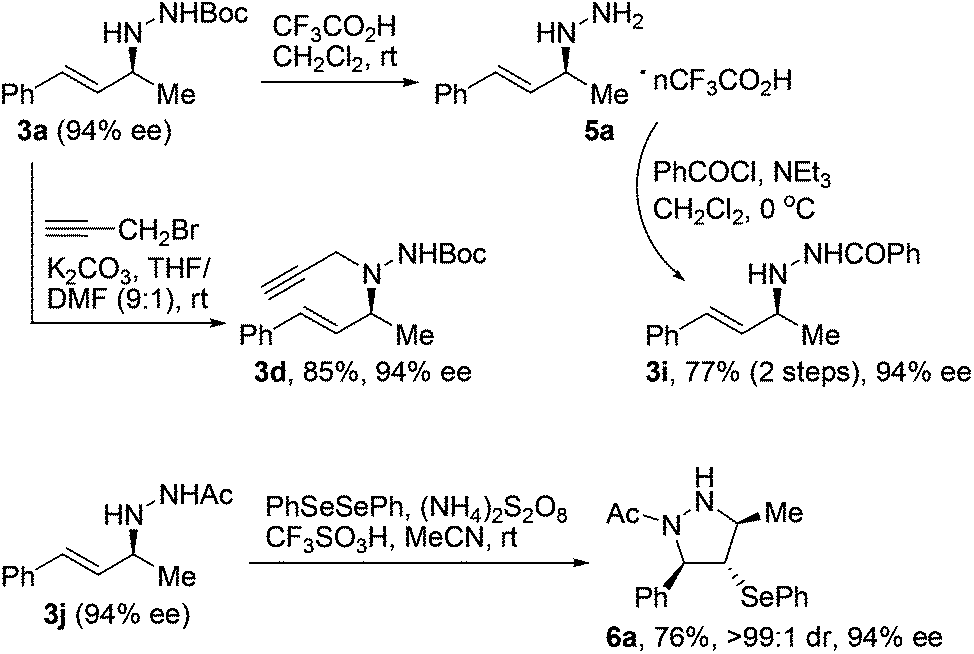 | ||
| Scheme 1 Transformations of N-allylhydrazines. | ||
Our experimental results and previous relevant mechanistic studies allow us to propose the following catalytic cycle for the stereospecific allylation of protected hydrazines with enantioenriched allylic amines (Scheme 2).5,9c,e Initially, palladium(0) (PdLn) is generated in situ from Pd(OAc)2 through reduction by the protected hydrazine and/or the phosphine ligand. Palladium(0) cleaves the allylic C–N bond in allylic amine 1, the NH2 group of which is activated by TsOH, with inversion of configuration to give π-allylpalladium 7, which releases ammonia to yield a more electrophilic species, π-allylpalladium 8.19 Nucleophilic attack of protected hydrazine 2 on the allylic carbon of π-allylpalladium 8 proceeds with inversion of configuration to give N-allylhydrazine 3 and regenerate palladium(0) and TsOH to continue the catalytic cycle. The configuration has been inverted twice in the whole process, and consequently, N-allylhydrazine 3 is produced with net stereoretention. It is noteworthy that the reaction proceeds with complete retention of configuration because no racemization occurs with π-allylpalladium 8via Pd–Pd-exchange under the standard reaction conditions.5c–e Moreover, the regioselectivity is determined by the steric and electronic properties of the R1 and R3 groups, and the attack of protected hydrazine 2 on π-allylpalladium 8 prefers to take place at the allylic position having less steric hindrance and/or leading to a higher degree of conjugation. When R1 = R3, the reaction will lose enantiopurity completely due to the symmetry of the π-allyl unit in the π-allylpalladium intermediate.
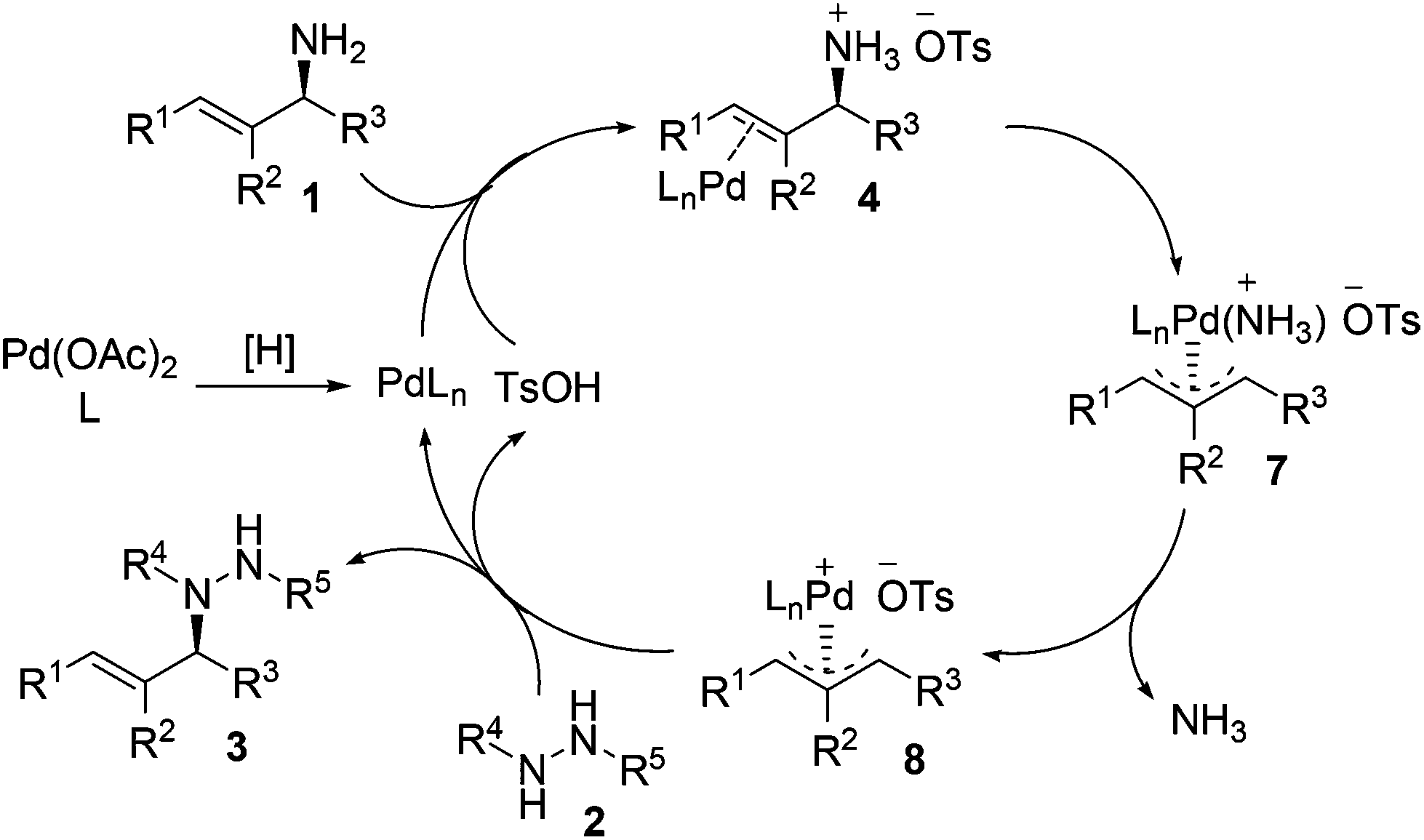 | ||
| Scheme 2 Proposed catalytic cycle. | ||
Conclusions
In summary, we have developed, for the first time, a stereospecific allylation reaction of protected hydrazines with enantioenriched allylic amines. A wide variety of protected hydrazines underwent allylation with highly enantioenriched primary allylic amines in the presence of 5 mol% Pd(OAc)2, 10 mol% racemic BINAP, and 5 mol% p-toluenesulfonic acid to give structurally diverse N-allylhydrazines in moderate to excellent yields with extremely high regioselectivity and complete retention of configuration. Importantly, the reaction exhibits high atom-economy by yielding ammonia as a sole byproduct. Moreover, this study adds a useful entry to the synthetic applications of enantioenriched primary allylic amines through C–N bond cleavage.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21232007 and 21172206) and the National Key Basic Research Program of China (2014CB931800).Notes and references
- For examples, see: (a) R. Zhang, J. P. Durkin and W. T. Windsor, Bioorg. Med. Chem. Lett., 2002, 12, 1005 CrossRef CAS PubMed; (b) A. Raja, J. Lebbos and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2003, 1, 857 CrossRef PubMed.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry Part B: Reactions and Synthesis, Springer, New York, 5th edn, 2007 Search PubMed.
- For a review, see: U. Ragnarsson, Chem. Soc. Rev., 2001, 30, 205 RSC.
- For examples on the regioselective allylation of protected hydrazines, see: (a) R. Matunas, A. J. Lai and C. Lee, Tetrahedron, 2005, 61, 6298 CrossRef CAS; (b) S. Tšupova and U. Mäeorg, Org. Lett., 2013, 15, 3381 CrossRef PubMed and references cited therein.
- For reviews on transition metal-catalyzed allylic substitution, see: (a) J. Tsuji, Acc. Chem. Res., 1969, 2, 144 CrossRef CAS; (b) B. M. Trost, Tetrahedron, 1977, 33, 2615 CrossRef CAS; (c) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed; (d) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (e) U. Kazmaier and M. Pohlman, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004, p. 531 Search PubMed.
- (a) T. Yamagishi, M. Ohnuki, T. Kiyooka, D. Masui, K. Sato and M. Yamaguchi, Tetrahedron: Asymmetry, 2003, 14, 3275 CrossRef CAS; (b) R. J. Kloetzing and P. Knochel, Tetrahedron: Asymmetry, 2006, 17, 116 CrossRef CAS; (c) D. Popa, R. Marcos, S. Sayalero, A. Vidal-Ferran and M. A. Pericàs, Adv. Synth. Catal., 2009, 351, 1539 CrossRef CAS; (d) H. Yuan, Z. Zhou, J. Xiao, L. Liang and L. Dai, Tetrahedron: Asymmetry, 2010, 21, 1874 CrossRef CAS; (e) K. E. Lutz and R. J. Thomson, Angew. Chem., Int. Ed., 2011, 50, 4437 CrossRef CAS PubMed.
- Certain enantioenriched N-allylhydrazines have been reported to be synthesized by catalytic asymmetric additions to azodicarboxylates. See: (a) S. Bertelsen, M. Marigo, S. Brandes, P. Dinér and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 12973 CrossRef CAS PubMed; (b) J. Wang, J. Chen, C. W. Kee and C.-H. Tan, Angew. Chem., Int. Ed., 2012, 51, 2382 CrossRef CAS PubMed.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., Hoboken, NJ, 4th edn, 2007 Search PubMed.
- (a) M.-B. Li, Y. Wang and S.-K. Tian, Angew. Chem., Int. Ed., 2012, 51, 2968 CrossRef CAS PubMed; (b) X.-S. Wu, Y. Chen, M.-B. Li, M.-G. Zhou and S.-K. Tian, J. Am. Chem. Soc., 2012, 134, 14694 CrossRef CAS PubMed; (c) M.-B. Li, H. Li, J. Wang, C.-R. Liu and S.-K. Tian, Chem. Commun., 2013, 49, 8190 RSC; (d) X.-T. Ma, Y. Wang, R.-H. Dai, C.-R. Liu and S.-K. Tian, J. Org. Chem., 2013, 78, 11071 CrossRef CAS PubMed; (e) T.-T. Wang, F.-X. Wang, F.-L. Yang and S.-K. Tian, Chem. Commun., 2014, 50, 3802 RSC; (f) X.-S. Wu, M.-G. Zhou, Y. Chen and S.-K. Tian, Asian J. Org. Chem., 2014, 3, 711 CrossRef CAS.
- (a) B. M. Trost and E. Keinan, J. Org. Chem., 1980, 45, 2741 CrossRef CAS; (b) R. V. Kunakova, R. L. Gaisin, M. M. Sirazova and U. M. Dzhemilev, Izv. Akad. Nauk. SSSR, Ser. Khim., 1983, 32, 157 Search PubMed; R. V. Kunakova, R. L. Gaisin, M. M. Sirazova and U. M. Dzhemilev, Bull. Acad. Sci. USSR Div. Chem. Sci. (Engl. Transl.), 1983, 32, 133 Search PubMed; (c) S.-I. Murahashi, Y. Makabe and K. Kunita, J. Org. Chem., 1988, 53, 4489 CrossRef CAS; (d) F. Garro-Helion, A. Merzouk and F. Guibé, J. Org. Chem., 1993, 58, 6109 CrossRef CAS; (e) B. M. Trost and M. D. Spagnol, J. Chem. Soc., Perkin Trans. 1, 1995, 2083 RSC; (f) H. Bricout, J.-F. Carpentier and A. Mortreux, Chem. Commun., 1997, 1393 RSC; (g) J. Pawlas, Y. Nakao, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 3669 CrossRef CAS PubMed; (h) I. D. G. Watson and A. K. Yudin, J. Am. Chem. Soc., 2005, 127, 17516 CrossRef CAS PubMed; (i) S. Mukherjee and B. List, J. Am. Chem. Soc., 2007, 129, 11336 CrossRef CAS PubMed; (j) B. M. Trost, M. Osipov and G. Dong, J. Am. Chem. Soc., 2010, 132, 15800 CrossRef CAS PubMed; (k) I. Dubovyk, D. Pichugin and A. K. Yudin, Angew. Chem., Int. Ed., 2011, 50, 5924 CrossRef CAS PubMed; (l) X. Zhao, D. Liu, H. Guo, Y. Liu and W. Zhang, J. Am. Chem. Soc., 2011, 133, 19354 CrossRef CAS PubMed.
- For the use of other alkylamines as sp3 carbon electrophiles, see: (a) W. Geng, W.-X. Zhang, W. Hao and Z. Xi, J. Am. Chem. Soc., 2012, 134, 20230 CrossRef CAS PubMed; (b) Y. Xie, J. Hu, Y. Wang, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 20613 CrossRef CAS PubMed; (c) H. Huang, X. Ji, W. Wu, L. Huang and H. Jiang, J. Org. Chem., 2013, 78, 3774 CrossRef CAS PubMed; (d) Y. Xie, J. Hu, P. Xie, B. Qian and H. Huang, J. Am. Chem. Soc., 2013, 135, 18327 CrossRef CAS PubMed.
- T. G. Schenck and B. Bosnich, J. Am. Chem. Soc., 1985, 107, 2058 CrossRef CAS.
- For a review, see: Y. Gu and S.-K. Tian, Synlett, 2013, 1170 CAS.
- (a) F.-L. Yang, X.-T. Ma and S.-K. Tian, Chem. – Eur. J., 2012, 18, 1582 CrossRef CAS PubMed; (b) F.-L. Yang and S.-K. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929 CrossRef CAS PubMed; (c) Y.-H. Su, Z. Wu and S.-K. Tian, Chem. Commun., 2013, 49, 6528 RSC; (d) Y.-G. Zhang, X.-L. Liu, Z.-Y. He, X.-M. Li, H.-J. Kang and S.-K. Tian, Chem. – Eur. J., 2014, 20, 2765 CrossRef CAS PubMed; (e) F.-L. Yang, F.-X. Wang, T.-T. Wang, Y.-J. Wang and S.-K. Tian, Chem. Commun., 2014, 50, 2111 RSC.
- The absolute configuration of compound 3i was assigned to be S by single-crystal X-ray analysis (CCDC 997538) and that of compounds 3a–e, 3g, 3h, 3j, and 3k was assigned by correlation with compound 3i. The absolute configuration of the rest of products was assigned by analogy.
- The reaction with sulfonyl-protected hydrazines (sulfonyl hydrazides) gave allylic sulfones instead of sulfonyl-protected N-allylhydrazines, which are incompatible with air. See: ref. 9e.
- For the synthetic applications of enantioenriched N-allylhydrazines, see: (a) B. F. Strick, D. A. Mundal and R. J. Thomson, J. Am. Chem. Soc., 2011, 133, 14252 CrossRef CAS PubMed; (b) O. Gutierrez, B. F. Strick, R. J. Thomson and D. J. Tantillo, Chem. Sci., 2013, 4, 3997 RSC; (c) J. C. T. Reddel, K. E. Lutz, A. B. Diagne and R. J. Thomson, Angew. Chem., Int. Ed., 2014, 53, 1395 CrossRef CAS PubMed.
- The corresponding reaction with racemic 3j was reported to give racemic 6a as an inseparable 82:14 mixture of diastereomers under identical conditions. See: M. Tiecco, L. Testaferri and F. Marini, Tetrahedron, 1996, 52, 11841 CrossRef CAS.
- Electrospray ionization (ESI) mass spectrometric analysis of the reaction mixture of amine 1a with carbazate 2a allowed us to assign π-allylpalladium intermediate 8a (R1 = Ph, R2 = H, R3 = Me, Ln = BINAP) according to the high resolution mass data [HRMS (ESI) calcd for C54H43P2Pd+ 859.1869, found 859.1889].
Footnote |
| † Electronic supplementary information (ESI) available: General information, experimental procedures, characterization data, copies of 1H NMR and 13C NMR spectra and HPLC traces, and crystal data of compound 3i. CCDC 997538. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00155a |
| This journal is © the Partner Organisations 2014 |