
Synthesis of 2-deoxy-C-glycosides via Lewis acid-mediated rearrangement of 2,3-anhydro-1-thiopyranosides†
De-Cai
Xiong
,
Chao
Gao
,
Wenming
Li
,
Yuan
Wang
,
Qin
Li
and
Xin-Shan
Ye
*
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Xue Yuan Road 38, Beijing, 100191, China. E-mail: xinshan@bjmu.edu.cn; Fax: +86 10 8280 2724
First published on 9th July 2014
Abstract
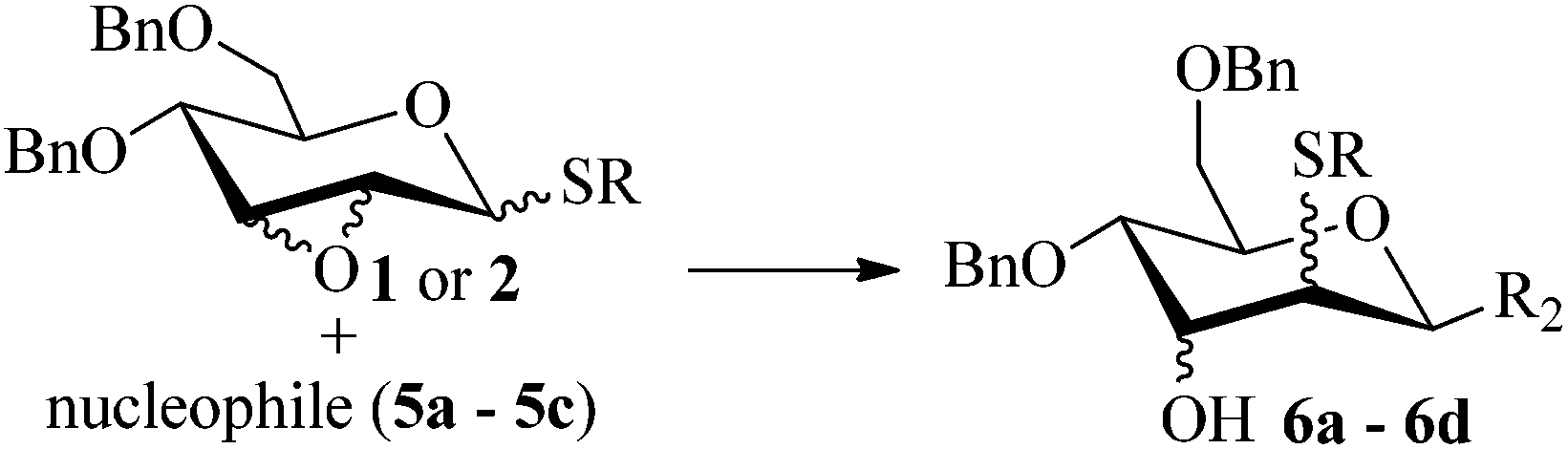
An approach to the regio- and stereo-selective construction of 2-deoxy-C-glycosides via Lewis acid-mediated rearrangement of 2,3-anhydro-1-thiopyranosides is disclosed. Treatment of 2,3-anhydro-1-thiopyranosides with phenols in the presence of TMSOTf, the migration–O-glycosylation and Fries-like O to C rearrangement took place in succession, providing aryl 2-thio-2-deoxy-C-glycosides with the single thermodynamically favourable configuration (1C4 or 4C1) in good to excellent yields. The coupling reaction of 2,3-anhydro-1-thiopyranosides with trimethylsilylated or tributylstanylated nucleophiles in the presence of TMSOTf or Sc(OTf)3, via the migration–C-glycosylation process, afforded 2-thio-2-deoxy-C-glycosides in a stereospecific manner in moderate to good yields and with the C-1 and C-2 substituents opposite. The 2-thio-functionality was further removed to produce the corresponding 2-deoxy-C-glycosides. This method may provide a convenient route for the preparation of 2-deoxy-C-glycosides.
Introduction
C-Glycosides are a class of carbohydrate analogues in which the glycosidic oxygen is replaced by a carbon atom. This novel C–C glycosidic bond is unique and it confers remarkable stability to both enzymatic and/or chemical hydrolysis. In addition, C-glycosides exist in natural products and exhibit a diverse range of biological activities.1 In particular, the 2-deoxy-C-glycoside structure is embodied in a variety of therapeutically important natural products such as pluramycins, galtamycin, and aquayamycin.2 Therefore, the synthesis of C-glycosides has been one of the main topics in carbohydrate chemistry. The stereoselective assembly of 2-deoxy-C-glycosyl linkages is more challenging than that of other C-glycosyl linkages due to the non-existence of participating groups at the 2-position. So far, a series of chemical transformations have been reported for the synthesis of 2-deoxy-C-glycosides,3 these transformations mainly include the electrophilic reaction,4 cross-coupling reaction,5 free radical reaction,6 cyclization,7 intramolecular O–C rearrangement,8 umpolung,9 and other miscellaneous reactions. These reactions involve various glycosyl donors such as glycosyl halides,10 glycosyl acetates,11 and glycals.12 Although advances have been made, stereospecific C-glycosylation remains a difficult task. Therefore, it would be highly desirable to develop new approaches to synthesize diverse 2-deoxy-C-glycosides.2,3-Anhydro-1-thioglycosides13 are a type of intriguing donor. They can undergo stereoselectivity-controllable glycosylation reactions generating 2-thio-2-deoxyglycosides. These coupling reactions occurred both regio- and stereo-selectively: the arylthio groups migrated to the position 2 and the nucleophile attacked at the anomeric position; the groups at C-1 and C-2 adopt the trans-configuration and the substituents at C-1 and C-3 adopt the cis-configuration. Inspired by these results, we reasoned that C-glycosylation might highly benefit from 2,3-anhydro-1-thioglycosides. Herein we report C-glycosylations of 2,3-anhydro-1-thioglycosides with phenols via O–C rearrangement or with trimethylsilylated (tributylstanylated) nucleophiles.
Results and discussion
Firstly, p-tolyl 2,3-anhydro-4,6-di-O-benzyl-1-thio-β-D-allopyranoside (1) was prepared (Scheme 1). Treatment of thioglycoside 1a13a,j with CSA in MeOH provided the de-benzylidene product 1b in 54% yield. By using Ag2O, O-benzylation of 1b proceeded very smoothly and compound 1 was obtained in 91% yield. Next, the reaction of 2,3-anhydro-1-thio-β-glycoside 1 with m-methoxyphenol (3a) was carried out to explore the feasibility of C-glycosylation of 1 (Table 1). As expected, the reaction of 1 and 3a in the presence of BF3·Et2O afforded the desired 2-tolylthio-C-glycoside 4a in 9% yield with high β-selectivity (entry 1). Encouraged by this result, a series of other Lewis acids including SnCl4, TMSOTf, Sc(OTf)3, and Cu(OTf)2 were screened in order to find out the optimal conditions (entries 2–5). The results indicated that the reaction displayed the highest efficiency when TMSOTf was used (entry 3, 65% yield), whereas Sc(OTf)3 and SnCl4 took the second place in around 50% yield (entries 2 and 4). Notably, the use of Cu(OTf)2 resulted in no detectable product 4a (entry 5). Therefore, TMSOTf was chosen as the preferred Lewis acid in the following experiments.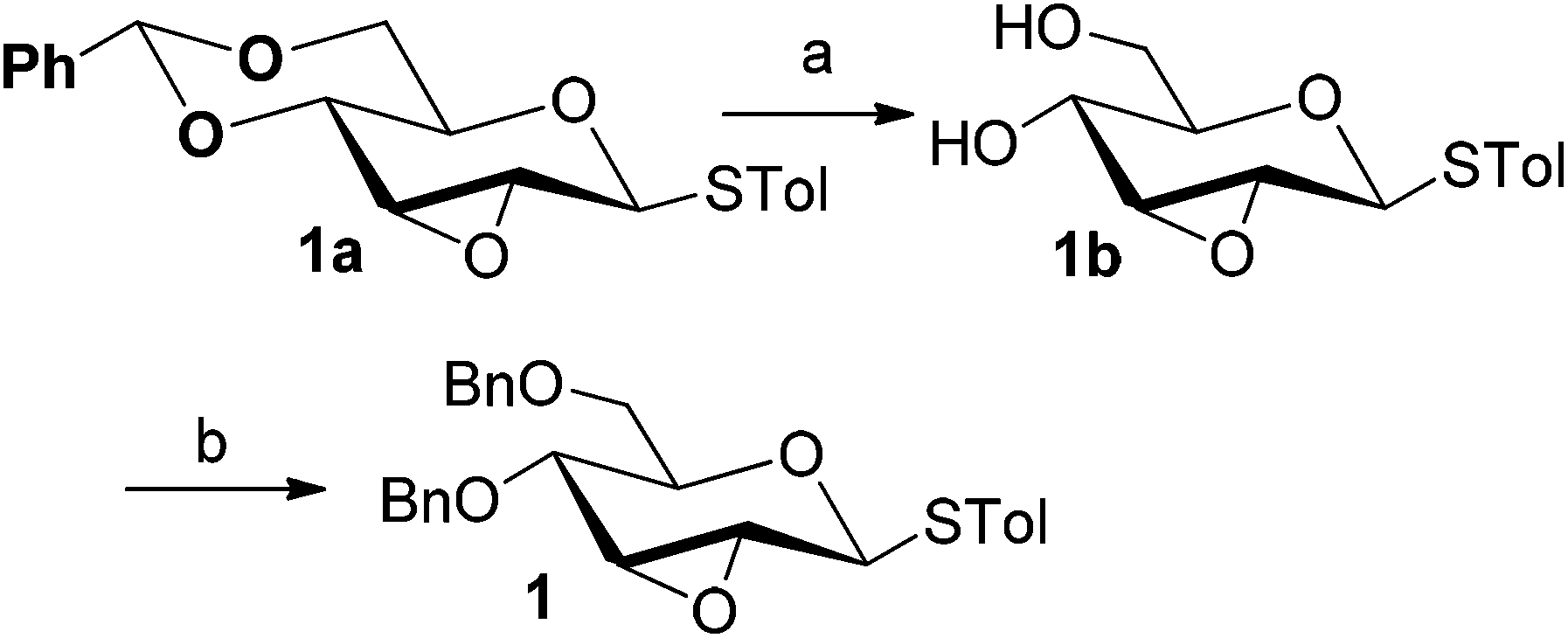 | ||
| Scheme 1 Synthesis of 2,3-anhydro-1-thioglycoside 1. (a) CSA, MeOH, 54%; (b) 1b (1.0 equiv.), BnBr (7.5 equiv.), Ag2O (8.0 equiv.), KI (8.0 equiv.), 91%. | ||
|
|
|||
|---|---|---|---|
| Entry | Lewis acid | Yieldb (%) | β/α ratioc |
| a Conditions: 1 (0.122 mmol), 3a (0.146 mmol) and Lewis acid (0.122 mmol) in CH2Cl2 (3 mL) from −78 °C to rt. b Isolated yield after chromatography. c Ratio determined by 1H NMR spectra after chromatography. | |||
| 1 | BF3·Et2O | 9 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 :0 |
| 2 | SnCl4 | 46 | 1:0 |
| 3 | TMSOTf | 65 | 1:0 |
| 4 | Sc(OTf)3 | 55 | 1:0 |
| 5 | Cu(OTf)2 | 0 | — |
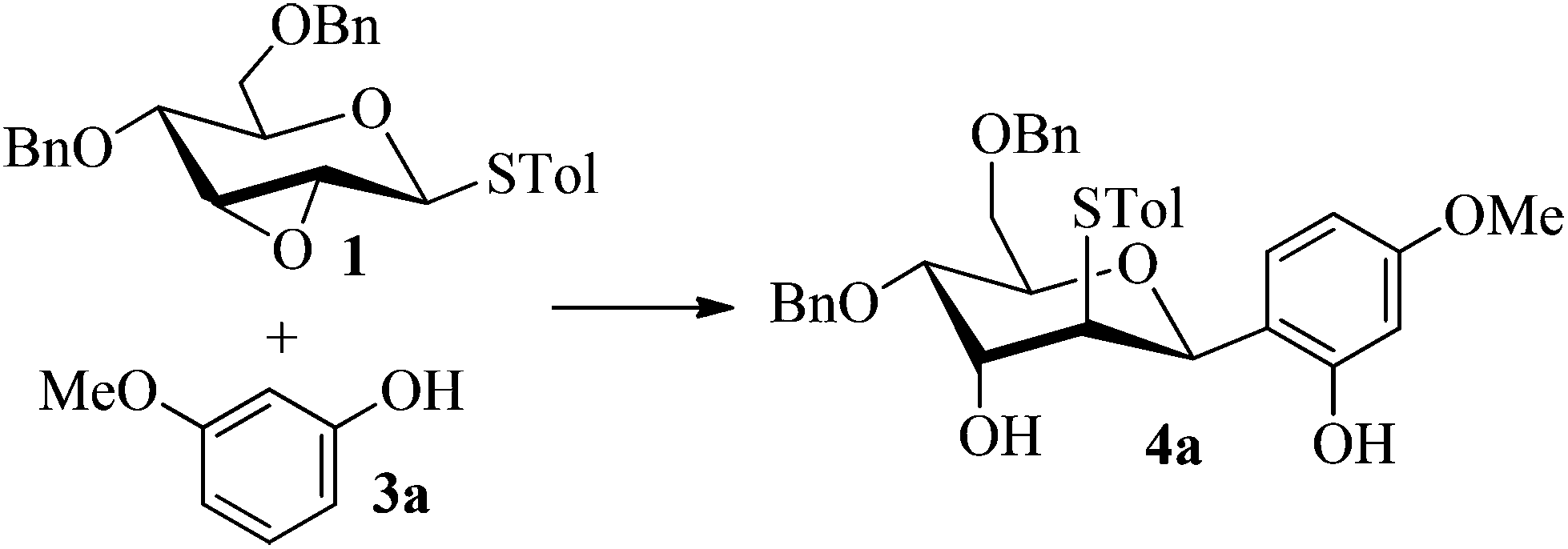
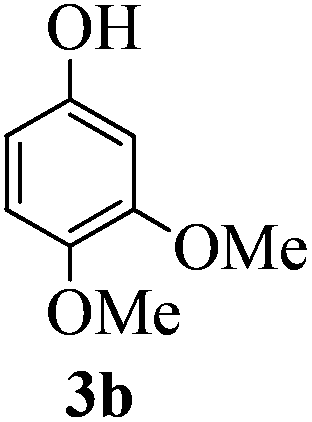
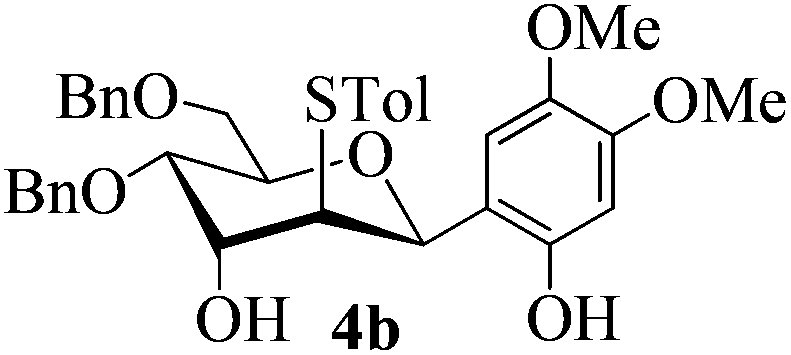
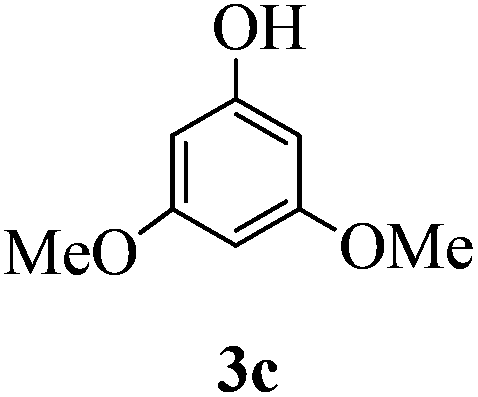
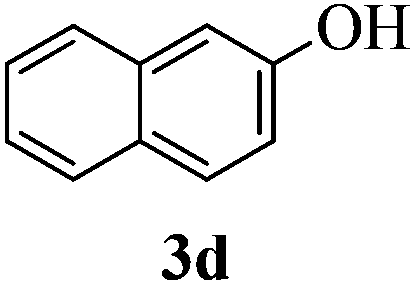
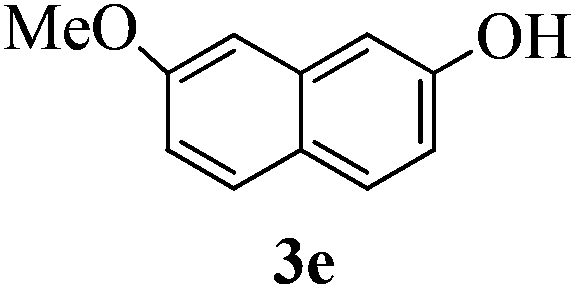
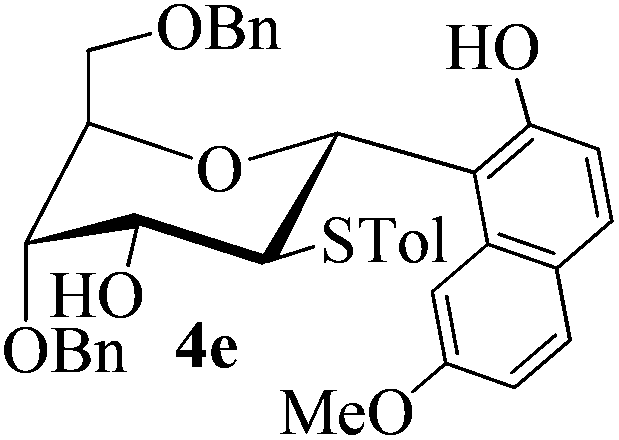
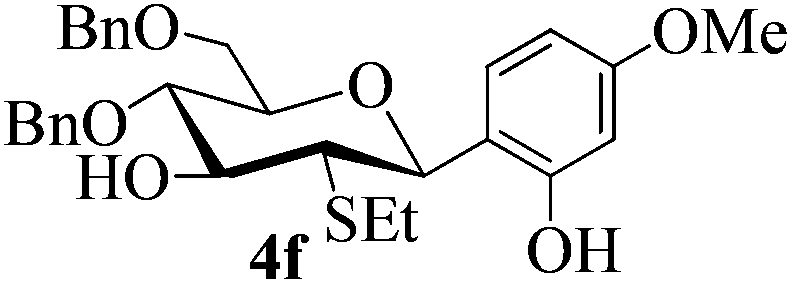
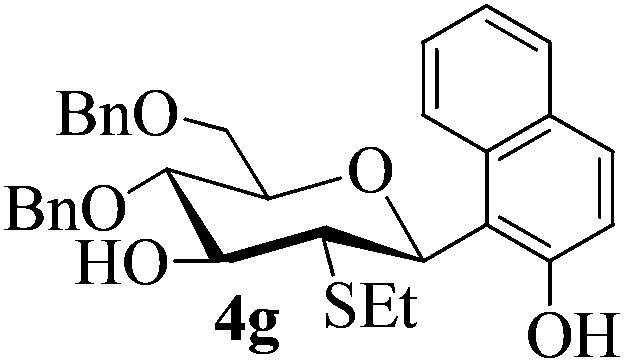
With the above optimized conditions in hand, we next turned our attention to exploration of the scope of phenols for C-glycosylation with 1 in the presence of TMSOTf (Table 2, entries 1–4). The results showed that electron-rich phenols 3b–3e were suitable substrates for this reaction. Reaction of 3,4-dimethoxyphenol (3b) with 1 afforded product 4b with β-configuration in 90% yield (entry 1), and reaction of 3,5-dimethoxyphenol (3c) with 1 furnished product 4c with α-configuration in 65% yield (entry 2). Gratifyingly, reactions of 2-naphthol (3d) and 7-methoxy-2-naphthol (3e) provided products 4d and 4e with α-configuration in 71% and 73% yield, respectively (entries 3 and 4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Product | Yieldb [%] | β/α ratioc |
| a Conditions: 1 or 2 (0.122 mmol), acceptor (0.146 mmol), TMSOTf (0.122 mmol) and 4 Å MS in CH2Cl2 (3 mL) from −78 °C to rt. b Isolated yield after chromatography. c Ratio determined by 1H NMR spectra after chromatography. | |||||
| 1 | 1 |
|
|
90 | 0:1 |
| 2 | 1 |
|
|
65 | 1:0 |
| 3 | 1 |
|

|
71 | 1:0 |
| 4 | 1 |
|
|
73 | 1:0 |
| 5 | 2 | 3a |
|
81 | 0:1 |
| 6 | 2 | 3d |
|
79 | 0:1 |
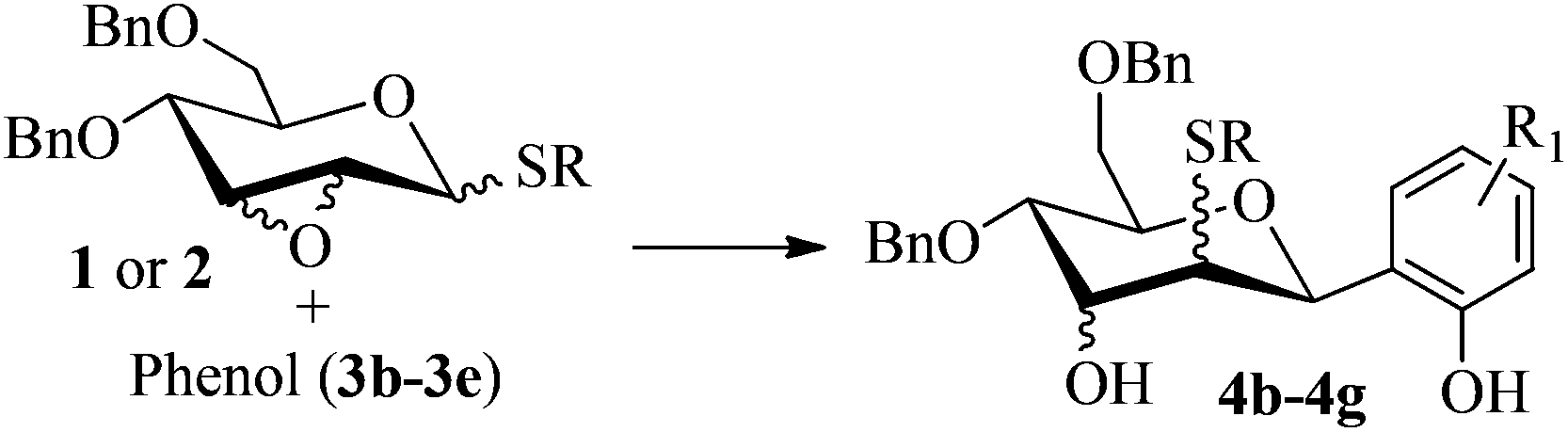
To investigate the influence of the anomeric configuration of thioglycosides, ethyl 2,3-anhydro-1-thio-α-glycoside 2 was prepared (Scheme 2). Initially, ethyl 4,6-O-benzylidene-1-thio-α-D-glucopyranoside (2a)14a was selectively sulfonated at the 2-position to obtain the mono-sulfonate 2b in 85% yield.14b Then, 2b was treated with sodium methoxide in methanol providing anhydro-glycoside 2c in 81% yield. Next, debenzylidenation of 2c, which was followed by benzylation, gave the desired ethyl 2,3-anhydro-1-thio-α-glycoside 2 smoothly. Subsequently, compound 2 was employed to check the C-glycosylation reaction (Table 2, entries 5 and 6). As shown, the C-glycosylations of 2 with phenols 3a and 3d proceeded with high β-anomeric selectivity in 81% and 79% yield, respectively. It seems that the substituent at C-1 is not always opposite to the thio-group at C-2 when phenols are used as substrates. The aryl group at C-1 always occupies the equatorial position.
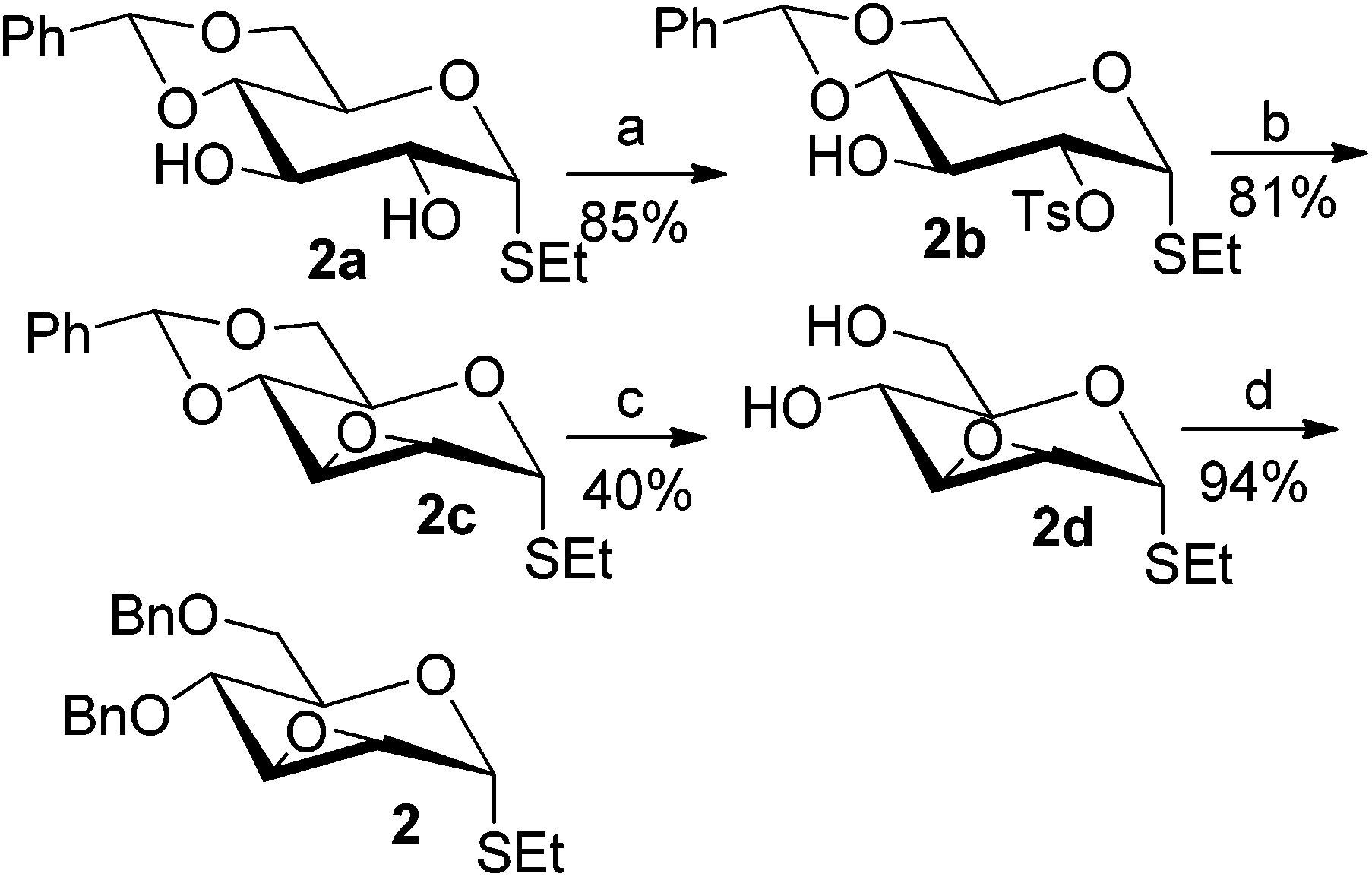 | ||
| Scheme 2 Synthesis of anhydro-thio-α-glycoside 2. (a) 2a (1.0 equiv.), TsCl (2.0 equiv.), Ag2O (2.0 equiv.), KI (0.4 equiv.), CH2Cl2, 12 h; (b) 2b (1.0 equiv.), NaOMe (3.0 equiv.), MeOH; (c) 2c (1.0 equiv.), CSA (0.3 equiv.), MeOH; (d) 2d (1.0 equiv.), BnBr (7.5 equiv.), Ag2O (8.0 equiv.), KI (1.0 equiv.), DMF. | ||
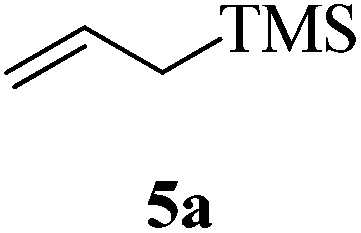
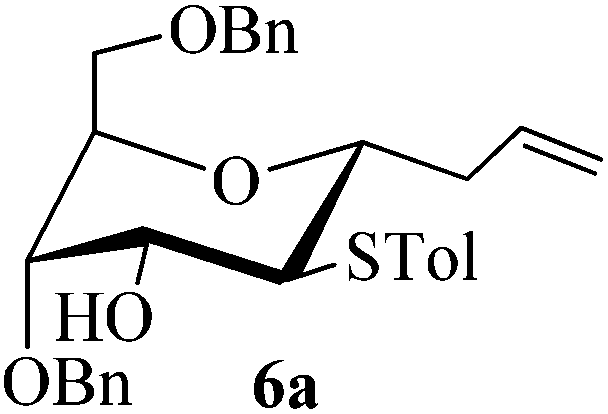
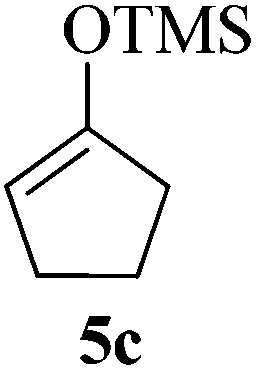
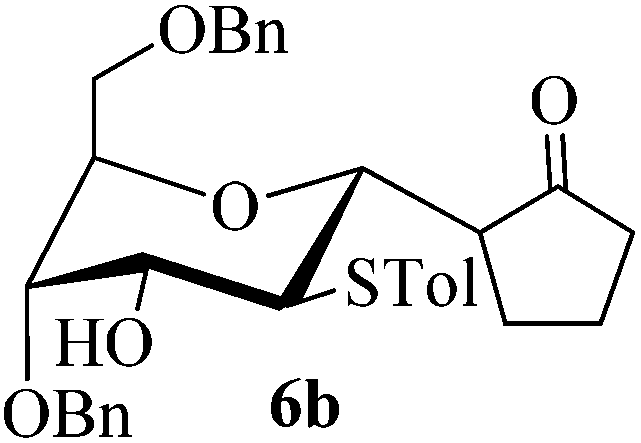
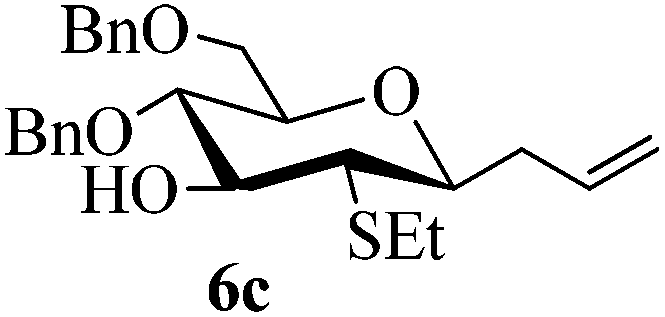
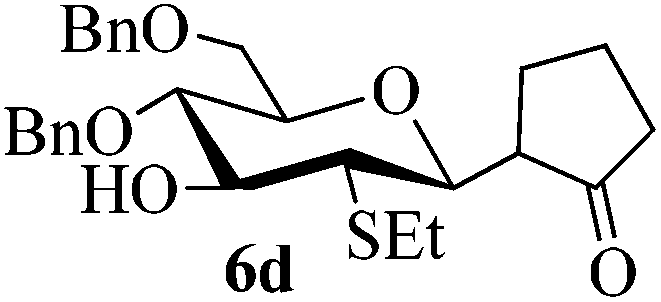
Given the successful results from phenols as substrates, we next want to test the reaction of 2,3-anhydro-1-thioglycosides with trimethylsilylated (tributylstanylated) nucleophiles, which failed in the previous report.15 Thus, upon treatment with TMSOTf at −78 °C, the reaction of anhydro-donor 1 with allyltrimethylsilane (5a) proceeded smoothly, providing C-glycoside 6a with α-anomeric selectivity in 50% isolated yield (Table 3, entry 1). When tributylallylstanane (5b) was used as the substrate, coupling product 6a was also obtained with α-stereoselectivity in 64% yield (Table 3, entry 2). However, under the same conditions, when 1-(trimethylsiloxy)cyclopentene (5c) was used for the reaction, only a small amount of C-glycoside 6b was isolated (10% yield) with α-stereochemistry. Fortunately, compound 6b was prepared from 5c and 1 in an improved yield (51% yield) when Sc(OTf)3 was used as the promoter (Table 3, entry 3). Similarly, when donor 2 was reacted with 5b in the presence of TMSOTf at −78 °C, coupling product 6c was obtained in 41% yield and with β-stereoselectivity (Table 3, entry 5). Again, using Sc(OTf)3 as the Lewis acid, the C-glycosylations of 2 with 5a and 5c were greatly improved, producing C-glycosides 6c and 6d with β-stereochemistry in 46% and 70% yield, respectively (Table 3, entries 4 and 6). It is apparent that the group at C-1 is opposite to the thio-group at C-2 when using trimethylsilylated or tributylstanylated nucleophiles as substrates.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Product | Yieldb [%] | β/α ratioc |
| a Conditions: donor (0.122 mmol), acceptor (0.146 mmol), TMSOTf (0.122 mmol) and 4 Å MS in CH2Cl2 (3 mL) from −78 °C to rt. b Isolated yield after chromatography. c Ratio determined by 1H NMR spectra after chromatography. d Sc(OTf)3 as a Lewis acid. | |||||
| 1 | 1 |
|
|
50 | 1:0 |
| 2 | 1 |
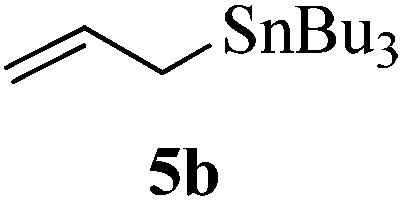
|
6a | 64 | 1:0 |
| 3 | 1 |
|
|
10 (51d) | 1:0 |
| 4 | 2 | 5a |
|
Trace (46d) | 0:1 |
| 5 | 2 | 5b | 6c | 41 | 0:1 |
| 6 | 2 | 5c |
|
Trace (70d) | 0:1 |
The anomeric configurations of the coupling products were unambiguously identified by their NMR analyses (Table 4).16 In the NMR spectra of 4a, the coupling constants of hydrogen signals 3JH1,H2 = 2.8 Hz (axial–equatorial relationship), 3JH2,H3 = 2.8 Hz, 3JH3,H4 = 2.8 Hz, and 3JH4,H5 = 10.0 Hz (axial–axial relationship), suggest that the sugar ring exists in a 4C1 conformation; the nuclear Overhauser effect (NOE) between H-1 and H-5 indicates that the H-1 is in the axial position. Therefore, the anomeric configuration of 4a is β. By the similar analyses, the NMR data of 4b are consistent with a β-anomeric configuration and a 4C1 conformation. For compound 4c, the values of 3JH1,H2 = 11.0 Hz (axial–axial relationship), 3JH3,H4 = 2.0 Hz and 3JH4,H5 = 3.0 Hz (axial–equatorial relationship) suggest the 1C4 conformation and α-anomeric configuration. The structure of 4c was further confirmed by the observations of NOEs between H-1 and H-3, H-1 and H-6. In the same way, the structures of compounds 4d, 4e, 6a, and 6b were identified with α-anomeric configuration and in the 1C4 conformation. Compounds 4f, 4g, 6c, and 6d with β-anomeric configuration and in 4C1 conformation were analyzed from their 1H NMR by a large 3JH1,H2 coupling constant (>10 Hz, axial–axial relationship).17
| Compound | NOE | 3 J H,H [Hz] | Conformation | Configuration |
|---|---|---|---|---|
| 4a | H1↔H5 | 3 J H1,H2 = 2.8 | 4C1 | β |
| 3 J H2,H3 = 2.8 | ||||
| 3 J H3,H4 = 2.8 | ||||
| 3 J H4,H5 = 10.0 | ||||
| 3 J H1,H2 = 2.5 | ||||
| 4b | H1↔H5 | 3 J H2,H3 = 3.5 | 4C1 | β |
| 3 J H3,H4 = 2.5 | ||||
| 3 J H4,H5 = 10.0 | ||||
| 4c | H1↔H3 | 3 J H1,H2 = 11.0 | 1C4 | α |
| H1↔H6a | 3 J H3,H4 = 2.0 | |||
| H1↔H6b | 3 J H4,H5 = 3.0 | |||
| 4d | 3 J H1,H2 = 10.8 | 1C4 | α | |
| 4e | H1↔H3 | 3 J H1,H2 = 10.5 | 1C4 | α |
| H1↔H6a | 3 J H3,H4 = 2.5 | |||
| H1↔H6b | 3 J H4,H5 = 2.5 | |||
| 4f | 3 J H1,H2 = 11.0 | 4C1 | β | |
| 4g | 3 J H1,H2 = 10.5 | 4C1 | β | |
| 6a | H1↔H3 | 3 J H1,H2 = 6.5 | 1C4 | α |
| H1↔H6a | 3 J H2,H3 = 6.5 | |||
| H1↔H6b | ||||
| 6b | H1↔H3 | 3 J H1,H2 = 9.0 | 1C4 | α |
| H1↔H6a | 3 J H2,H3 = 9.0 | |||
| H1↔H6b | ||||
| 6c | 3 J H1,H2 = 10.5 | 4C1 | β | |
| 6d | 3 J H1,H2 = 11.0 | 4C1 | β |
After having checked the feasibility of C-glycosylations of 2,3-anhydro-1-thiopyranosides with phenols and trimethylsilylated (tributylstanylated) nucleophiles, the desulfurization of 2-tolylthio-C-glycoside 4b was carried out (Scheme 3). After treatment of 4b with tri-n-butyltin hydride and AIBN in toluene at 105 °C, the 2-deoxy-C-glycoside 7 was obtained in 72% isolated yield. The anomeric configuration of 7 is completely consistent with that identified in 4b.
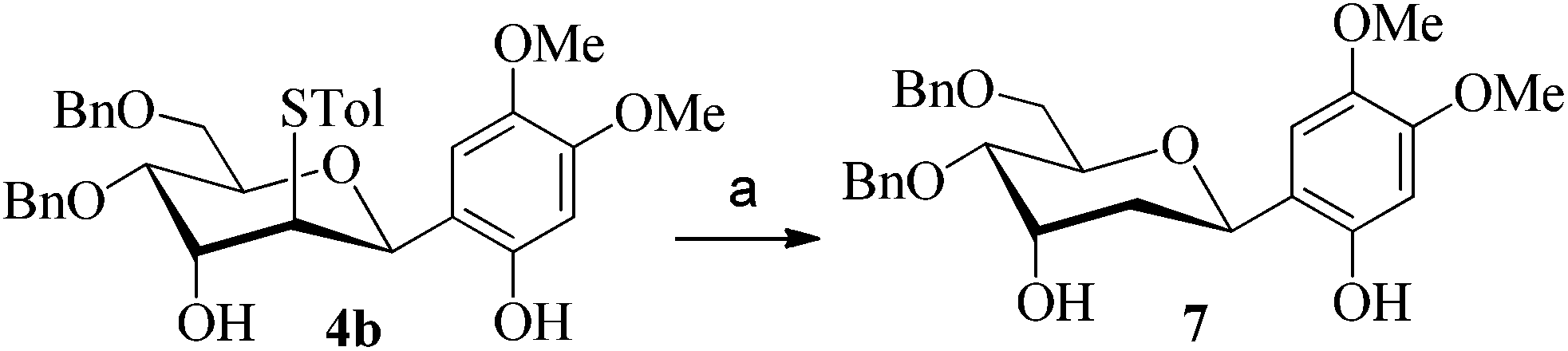 | ||
| Scheme 3 Desulfurization of compound 4b. (a) AIBN, n-Bu3SnH, 105 °C, 72%. | ||
Although the details are not yet known, a proposed mechanism for C-glycosylation is shown in Fig. 1. The reaction may proceed through an oxocarbenium ion A or intermediate B.13a When phenol as a nucleophile attacks the anomeric carbon at low temperature, a phenolic glycoside C would be generated. During raising the reaction temperature to room temperature, C would rearrange to the thermodynamically favourable C-glycoside G or Hvia the Fries-like O to C rearrangement.18 The exclusive anomeric configuration may be attributed to the only stable conformation (1C4 or 4C1), arising from the severe repulsion between the tolylthio group at C-2 and the aryl group at C-1 with other atoms or groups. Different substituents on phenol may result in different conformations. The aryl group at C-1 is always in the equatorial position, but not always opposite to the thio-group at C-2, as in the case of coupling products 4a and 4b. When the nucleophilic attack is performed by a trimethylsilylated (tributylstanylated) nucleophile, it would form a C-glycoside I with the C-1 and C-2 substituents opposite.
 | ||
| Fig. 1 A proposed pathway leading to C-glycosides. | ||
Conclusions
We have described a new method for the synthesis of 2-deoxy-C-glycosides which is both facile and highly stereoselective. By utilizing 2,3-anhydro-1-thiopyranosides and a variety of nucleophiles, this method affords C-glycosides via the Lewis acid-mediated rearrangement. Promoted by TMSOTf, a reaction of 2,3-anhydro-1-thiopyranosides with phenols gives aryl 2-thio-2-deoxy-C-glycosides, resulting from the successive migration–O-glycosylation and Fries-like O to C rearrangement. These reactions provide C-glycosides with a single anomeric configuration in good to excellent yields. The formed configuration depends on the thermodynamically favourable chair conformation (1C4 or 4C1) of the C-glycoside, and the aryl group at C-1 always takes the equatorial position. By treatment of 2,3-anhydro-1-thiopyranosides with trimethylsilylated (tributylstanylated) nucleophiles in the presence of TMSOTf or Sc(OTf)3, the migration–C-glycosylation reaction proceeds, producing 2-thio-2-deoxy-C-glycosides in moderate to good yields and with the C-1 and C-2 substituents opposite. Finally, treatment of the resulting 2-thio-2-deoxy-C-glycosides with tri-n-butyltin hydride and AIBN can afford the corresponding 2-deoxy-C-glycosides. This method may find applications in the preparation of 2-deoxy-C-glycosides with biological importance.Experimental section
General methods
All chemicals purchased were of reagent grade and used without further purification, unless noted otherwise. Dichloromethane (CH2Cl2) was distilled over calcium hydride (CaH2). Methanol was distilled from magnesium. DMF was stirred with CaH2 and distilled under reduced pressure. Toluene was distilled over sodium. All reactions were carried out under anhydrous conditions with freshly distilled solvents, unless stated otherwise. Reactions were monitored by analytical thin-layer chromatography on silica gel 60 F254 precoated on aluminium plates. Spots were detected under UV light and/or by staining with acidic ceric ammonium molybdate. Column chromatography was performed on silica gel (200–300 mesh). 1H-NMR spectra were recorded on a JEOL AL-300, Varian INOVA-500 or Advance DRX Bruker-500 spectrometer at 25 °C. Chemical shifts (in ppm) were referenced to tetramethylsilane (δ = 0 ppm) in deuterated chloroform. 13C-NMR spectra were obtained using the same NMR spectrometers and were calibrated with CDCl3 (δ = 77.00 ppm). Mass spectra were recorded using a PE SCLEX QSTAR spectrometer.General procedures for C-glycosylations
To a mixture of acceptors (0.146 mmol), donor 1 or 2 (0.122 mmol) and 4 Å molecular sieves was added CH2Cl2 (3 mL). The mixture was stirred for 10 min at room temperature and then cooled down to −72 °C. Lewis acid (0.122 mmol) was added and the temperature was raised until TLC showed that the donor was consumed. After neutralization by triethylamine, the reaction mixture was concentrated, and the residue was purified by column chromatography on silica gel to afford product.:1) to provide 1b (269 mg, 54% yield) as a colorless oil, and unreacted 1a was recovered (90.0 mg, 18%); 1H NMR (500 MHz, CDCl3) δ 2.24 (brs, 1H), 2.35 (s, 3H), 2.52 (brs, 1H), 3.37 (d, 1H, J = 4.0 Hz), 3.44–3.48 (m, 1H), 3.66 (d, 1H, J = 4.0 Hz), 3.74 (dd, 1H, J = 5.0 Hz, 12.0 Hz), 3.82–3.85 (m, 2H), 5.16 (d, 1H, J = 8.5 Hz), 7.14 (d, 2H, J = 7.5 Hz), 7.43 (d, 1H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 21.16, 54.82, 59.16, 62.54, 65.64, 74.58, 79.95, 127.66, 129.84, 133.55, 138.82; HRMS (ESI) Anal. calcd for C13H16O4S [M + NH4]+: 286.1108, found: 286.1108.
:1) to yield 1 (308 mg, 91% yield) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 2.30 (s, 3H), 3.28 (d, 1H, J = 4.5 Hz), 3.56 (d, 1H, J = 4.0 Hz), 3.64 (dd, 1H, J = 4.5 Hz, 11.0 Hz), 3.68–3.72 (m, 2H), 3.77 (dd, 1H, J = 1.5 Hz, 9.5 Hz), 4.54–4.63 (m, 3H), 5.13 (s, 1H), 7.03 (d, 2H, J = 8.0 Hz), 7.24–7.35 (m, 10H), 7.44–7.47 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 21.07, 52.39, 58.46, 69.10, 71.55, 71.67, 72.82, 73.22, 80.09, 127.42, 127.57, 127.81, 127.84, 128.21, 128.32, 129.52, 133.37, 137.67, 138.17, 138.32; HRMS (ESI) Anal. calcd for C27H28O4S [M + NH4]+: 466.2047, found: 466.2049.
:1, containing 1% triethylamine), providing compound 2b (2.55 g, 85% yield) as a white solid.
To a solution of 2b (1.95 g, 4.18 mmol) in MeOH (25 mL) was added NaOMe (30% in MeOH, 3 mL, 15.99 mmol). After stirring for 1 d at room temperature, the reaction mixture was concentrated and the residue was dissolved in ethyl acetate. Then the solution was washed with brine and water. The organic layer was dried, filtered, and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 15:1, containing 1% triethylamine) to yield 2c (0.99 g, 81% yield) as a white solid; 1H NMR (500 MHz, CDCl3) δ 1.33 (t, 3H, J = 7.5 Hz), 2.67 (qd, 1H, J = 7.5 Hz, 13.0 Hz), 2.77 (qd, 1H, J = 7.5 Hz, 13.0 Hz), 3.28 (d, 1H, J = 4.0 Hz), 3.50 (d, 1H, J = 3.5 Hz), 3.72 (d, 1H, J = 10.5 Hz), 3.76 (d, 1H, J = 10.0 Hz), 3.93 (td, 1H, J = 4.5 Hz, 10.0 Hz), 4.22 (dd, 1H, J = 4.5 Hz, 10.5 Hz), 5.51 (s, 1H), 5.57 (s, 1H), 7.35–7.41 (m, 3H), 7.49–7.52 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 15.09, 25.20, 51.90, 53.96, 62.05, 69.33, 75.28, 80.15, 102.44, 126.17, 128.36, 129.27, 137.02; HRMS (ESI) Anal. calcd for C15H18O4S [M + Na]+: 317.0818, found: 317.0821.
:1) to yield 2d (70.1 mg, 40% yield) as a colorless oil and recovered the unreacted 2c (70.0 mg, 28%); 1H NMR (500 MHz, CDCl3) δ 1.33 (t, 3H, J = 7.5 Hz), 1.62 (s, 1H), 1.92 (t, 1H, J = 6.0 Hz), 2.55 (d, 1H), 2.63–2.78 (m, 2H), 3.27 (d, 1H, J = 3.5 Hz), 3.33 (d, 1H, J = 3.5 Hz), 3.77–3.84 (m, 3H), 3.97 (dd, 1H, J = 5.0 Hz, 9.0 Hz), 5.47 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 15.07, 25.21, 51.52, 55.46, 63.20, 63.28, 69.17, 79.40; HRMS (ESI) Anal. calcd for C8H14O4S [M + Na]+: 229.0505, found: 229.0498.
:1) to yield 2 (370 mg, 94%) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 1.30 (t, 3H, J = 7.5 Hz), 2.64 (qd, 1H, J = 7.5 Hz, 13.0 Hz), 2.76 (qd, 1H, J = 7.5 Hz, 13.0 Hz), 3.21 (d, 1H, J = 3.5 Hz), 3.36 (d, 1H, J = 4.0 Hz), 3.58 (d, 1H, J = 3.0 Hz), 3.71 (d, 1H, J = 9.5 Hz), 4.00 (dt, 1H, J = 3.5 Hz, 9.5 Hz), 4.44 (d, 1H, J = 12.5 Hz), 4.46 (d, 1H, J = 12.0 Hz), 4.58 (d, 1H, J = 12.5 Hz), 4.70 (d, 1H, J = 11.5 Hz), 5.51 (s, 1H), 7.25–7.34 (m, 10H); 13C NMR (125 MHz, CDCl3) δ 14.96, 24.84, 51.30, 53.45, 67.48, 68.94, 69.27, 72.19, 73.14, 79.25, 127.54, 127.72, 127.99, 128.26, 128.46, 137.29, 138.12; HRMS (ESI) Anal. calcd for C22H26O4S [M + NH4]+: 404.1890, found: 404.1892.
:1) as a colorless oil (65% yield): Rf 0.30 (petroleum ether–ethyl acetate, 2:1); 1H NMR (400 MHz, CDCl3) δ 2.28 (s, 3H, tolyl CH3), 2.63 (s, 1H, OH), 3.53 (t, 1H, J = 2.8 Hz, H-2), 3.69 (dd, 1H, J = 4.4 Hz, 10.4 Hz, H-6a), 3.75 (dd, 1H, J = 1.6 Hz, 10.4 Hz, H-6b), 3.77 (s, 3H, OCH3), 3.98 (ddd, 1H, J = 2.0 Hz, 4.4 Hz, 10.0 Hz, H-5), 4.18 (dd, 1H, J = 2.8 Hz, 10.0 Hz, H-4), 4.33 (t, 1H, J = 2.8 Hz, H-3), 4.50 (d, 1H, J = 11.6 Hz, PhCH2), 4.56 (d, 1H, J = 12.0 Hz, PhCH2), 4.57 (d, 1H, J = 11.6 Hz, PhCH2), 4.65 (d, 1H, J = 12.0 Hz, PhCH2), 5.47 (d, 1H, J = 2.0 Hz, H-1), 6.38 (dd, 1H, J = 2.4 Hz, 8.4 Hz), 6.46 (d, 1H, J = 2.4 Hz), 6.83 (d, 1H, J = 8.8 Hz), 6.98 (d, 2H, J = 8.4 Hz), 7.01 (d, 2H, J = 8.4 Hz), 7.23–7.38 (m, 10H), 8.36 (s, 1H, ArOH); 13C NMR (125 MHz, CDCl3) δ 21.03, 55.22, 57.22 (C-2), 68.07 (C-3), 68.93 (C-6), 71.31 (C-4), 72.02, 73.48, 75.23 (C-5), 77.25 (C-1), 102.35, 105.97, 114.56, 127.31, 127.66, 127.75, 128.15, 128.24, 128.45, 128.60, 129.70, 131.30, 132.27, 137.21, 137.38, 138.02, 157.80, 160.29; HRMS (ESI) Anal. calcd for C34H36O6S [M + NH4]+: 466.2047, found: 466.2049.
:1) as a colorless oil (90% yield): Rf 0.30 (petroleum ether–ethyl acetate, 1:1); 1H NMR (500 MHz, CDCl3) δ 2.28 (s, 3H, tolyl CH3), 2.67 (d, 1H, J = 1.0 Hz, OH), 3.55 (dd, 1H, J = 2.5 Hz, 3.5 Hz, H-2), 3.69 (s, 3H, OCH3), 3.70 (dd, 1H, J = 4.5 Hz, 11.0 Hz, H-6a), 3.75 (dd, 2H, J = 2.5 Hz, 11.0 Hz, H-6b), 3.84 (s, 3H, OCH3), 3.98 (ddd, 1H, J = 2.0 Hz, 4.5 Hz, 10.0 Hz, H-5), 4.16 (dd, 1H, J = 2.5 Hz, 10.0 Hz, H-4), 4.38 (t, 1H, J = 3.0 Hz, H-3), 4.51 (d, 1H, J = 11.5 Hz, PhCH2), 4.55 (d, 1H, J = 12.0 Hz, PhCH2), 4.58 (d, 1H, J = 11.5 Hz, PhCH2), 4.65 (d, 1H, J = 12.5 Hz, PhCH2), 5.43 (d, 1H, J = 2.0 Hz, H-1), 6.37 (s, 1H), 6.47 (s, 1H), 6.97–7.02 (m, 4H), 7.24–7.38 (m, 10H), 8.07 (s, 1H, ArOH); 13C NMR (125 MHz, CDCl3) δ 20.99, 55.77, 56.58, 57.37 (C-2), 68.22 (C-3), 68.90 (C-6), 71.31 (C-4), 72.03, 73.48, 75.20 (C-5), 77.39 (C-1), 101.78, 109.97, 112.36, 127.66, 127.72, 128.14, 128.25, 128.44, 128.60, 129.66, 131.38, 132.26, 137.17, 137.44, 138.00, 142.06, 149.43, 150.91; HRMS (ESI) Anal. calcd for C35H38O7S [M + Na]+: 625.2231, found: 625.2238.
:1) as a colorless oil (65% yield): Rf 0.35 (petroleum ether–ethyl acetate, 2:1); [α]20D −84.4 (c 0.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 2.26 (s, 3H, tolyl CH3), 2.63 (d, 1H, J = 6.5 Hz, OH), 3.52 (dd, 1H, J = 5.5 Hz, 10.5 Hz, H-6a), 3.62 (dd, 1H, J = 5.5 Hz, 10.5 Hz, H-6b), 3.67 (t, 1H, J = 10.5 Hz, H-2), 3.74 (s, 3H, OCH3), 3.75–3.79 (m, 1H, H-3), 3.81 (s, 3H, OCH3), 3.87 (t, 1H, J = 2.0 Hz, 3.0 Hz, H-4), 4.26–4.32 (m, 1H, H-5), 4.47 (s, 2H, PhCH2), 4.73 (d, 1H, J = 12.0 Hz, PhCH2), 4.76 (d, 1H, J = 12.0 Hz, PhCH2), 5.33 (d, 1H, J = 11.0 Hz, H-1), 6.00 (d, 1H, J = 2.0 Hz), 6.02 (d, 1H, J = 2.5 Hz), 6.94 (d, 1H, J = 8.0 Hz), 7.14 (dd, 2H, J = 2.5 Hz, 6.0 Hz), 7.25–7.37 (m, 10H), 7.73 (s, 1H, ArOH); 13C NMR (125 MHz, CDCl3) δ 21.05, 53.48 (C-2), 55.26, 55.55, 67.46 (C-6), 68.71 (C-3), 70.43 (C-1), 72.92, 73.39, 74.96 (C-5), 76.09 (C-4), 91.11, 94.72, 105.95, 127.54, 127.80, 127.91, 128.48, 128.58, 129.22, 129.43, 133.76, 137.60, 158.04, 158.25, 161.35; HRMS (ESI) Anal. calcd for C35H38O7S [M + Na]+: 625.2231, found: 625.2238.
:1) as a colorless oil (71% yield): Rf 0.20 (petroleum ether–ethyl acetate, 3:1); [α]20D −40.0 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.13 (s, 3H), 2.81 (d, 1H, J = 6.0 Hz), 3.76 (dd, 1H, J = 5.2 Hz, 10.4 Hz), 3.80 (dd, 1H, J = 5.2 Hz, 10.4 Hz), 3.96 (t, 1H, J = 10.8 Hz), 4.04 (t, 1H, J = 2.8 Hz), 4.07–4.12 (m, 1H), 4.34–4.37 (m, 1H), 4.55 (d, 1H, J = 12.0 Hz), 4.59 (d, 1H, J = 12.0 Hz), 4.83 (s, 2H), 5.79 (d, 1H, J = 10.8 Hz), 6.45 (d, 2H, J = 8.0 Hz), 6.63 (d, 2H, J = 8.0 Hz), 6.93 (d, 1H, J = 8.8 Hz), 7.26–7.45 (m, 12H), 7.56 (d, 1H, J = 8.8 Hz), 7.66 (d, 1H, J = 7.6 Hz), 7.96 (d, 1H, J = 8.8 Hz), 8.05 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ 20.87, 54.57, 68.43, 69.61, 73.07, 73.14, 73.62, 75.43, 76.13, 119.29, 122.73, 122.90, 126.00, 127.67, 127.90, 127.99, 128.00, 128.23, 128.64, 128.80, 129.00, 129.70, 130.19, 131.96, 132.74, 136.87, 137.46, 137.69; HRMS (ESI) Anal. calcd for C35H38O7S [M + NH4]+: 610.2622, found: 610.2645.
:1) as a colorless oil (73% yield): Rf 0.35 (petroleum ether–ethyl acetate, 2:1); [α]20D −133.2 (c 0.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 2.12 (s, 3H, tolyl CH3), 2.81 (d, 1H, J = 6.5 Hz, OH), 3.73 (s, 1H, OCH3), 3.80 (dd, 1H, J = 4.5 Hz, 10.5 Hz, H-6a), 3.85 (dd, 1H, J = 4.5 Hz, 10.5 Hz, H-6b), 3.98 (t, 1H, J = 10.5 Hz, H-2), 4.07 (t, 1H, J = 2.5 Hz, H-4), 4.20 (ddd, 1H, J = 3.0 Hz, 6.0 Hz, 13.5 Hz, H-3), 4.32–4.34 (m, 1H, H-5), 4.56 (d, 1H, J = 12.0 Hz, PhCH2), 4.60 (d, 1H, J = 12.0 Hz, PhCH2), 4.82 (s, 2H, PhCH2), 5.79 (d, 1H, J = 11.0 Hz), 6.52 (d, 2H, J = 8.0 Hz), 6.64 (d, 2H, J = 8.0 Hz), 6.80 (d, 1H, J = 8.5 Hz), 6.95 (dd, 1H, J = 2.0 Hz, 8.0 Hz), 7.26–7.43 (m, 11H), 7.48 (d, 1H, J = 8.5 Hz), 7.55 (d, 1H, J = 8.5 Hz), 8.08 (brs, 1H, ArOH); 13C NMR (125 MHz, CDCl3) δ 20.87, 54.60 (C-2), 55.11, 69.37 (C-6), 69.74 (C-3), 73.14, 73.61 (C-1), 75.33 (C-5), 76.27 (C-4), 102.88, 114.60, 116.81, 127.27, 127.93, 128.04, 128.62, 128.65, 128.99, 129.68, 129.94, 131.81, 133.96, 136.82, 137.46, 137.64, 157.89; HRMS (ESI) Anal. calcd for C38H38O6S [M + NH4]+: 640.2727, found: 640.2714.
:1) as a colorless oil (81% yield): Rf 0.40 (petroleum ether–acetone, 2:1); [α]20D +78.4 (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.98 (t, 3H, J = 7.5 Hz, ethyl CH3), 1.80–1.85 (m, 2H, ethyl CH2), 2.87 (t, 1H, J = 10.5 Hz, H-2), 3.13 (d, 1H, J = 1.0 Hz, OH), 3.51–3.59 (m, 2H, H-3 and H-5), 3.68 (dd, 1H, J = 2.0 Hz, 10.0 Hz, H-6a), 3.76–3.84 (m, 5H, H-6b, OCH3 and H-4), 4.36 (d, 1H, J = 11.0 Hz, H-1), 4.46 (d, 1H, J = 12.5 Hz, PhCH2), 4.58 (d, 1H, J = 12.0 Hz, PhCH2), 4.59 (d, 1H, J = 11.0 Hz, PhCH2), 4.97 (d, 1H, J = 11.0 Hz, PhCH2), 6.42 (dd, 1H, J = 2.5 Hz, 8.5 Hz), 6.47 (d, 1H, J = 2.5 Hz), 7.08 (d, 1H, J = 8.5 Hz), 7.25–7.34 (m, 10H), 7.86 (s, 1H, ArOH); 13C NMR (100 MHz, CDCl3) δ 14.71, 26.28, 53.77 (C-2), 55.29, 68.04 (C-6), 73.46, 74.85, 75.57 (C-3), 77.39 (C-4), 78.01 (C-5), 83.48 (C-1), 102.82, 105.71, 115.56, 127.72, 127.76, 127.79, 128.10, 128.40, 128.43, 130.38, 137.92, 138.37, 156.97, 160.96; HRMS (ESI) Anal. calcd for C29H34O6S [M + NH4]+: 528.2414, found: 528.2430.
:1) as a colorless oil (79% yield): Rf 0.35 (petroleum ether–acetone, 3:1); [α]20D +90.4 (c 0.3, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.67 (t, 3H, J = 7.5 Hz, ethyl CH3), 1.41–1.59 (m, 2H, ethyl CH2), 3.19 (d, 1H, J = 1.5 Hz, OH), 3.25 (t, 1H, J = 11.5 Hz, H-2), 3.66–3.73 (m, 3H, H-6a, H-3 and H-5), 3.82 (dd, 1H, J = 2.5 Hz, 10.0 Hz, H-6b), 3.94 (t, 1H, J = 9.0 Hz, H-4), 4.47 (d, 1H, J = 12.0 Hz, PhCH2), 4.60 (d, 1H, J = 12.5 Hz, PhCH2), 4.63 (d, 1H, J = 11.5 Hz, PhCH2), 5.01 (d, 1H, J = 11.0 Hz, PhCH2), 5.44 (d, 1H, J = 10.5 Hz, H-1), 7.16 (d, 1H, J = 9.0 Hz), 7.26–7.35 (m, 11H), 7.44–7.47 (m, 1H), 7.72–7.75 (m, 2H), 8.07 (d, 1H, J = 8.5 Hz), 8.50 (s, 1H, ArOH); 13C NMR (125 MHz, CDCl3) δ 14.34, 26.23, 52.80 (C-2), 67.89 (C-6), 73.41, 74.91, 75.81 (C-3), 77.36 (C-4), 77.60 (C-1), 78.37 (C-5), 115.27, 119.66, 123.12, 123.16, 125.98, 127.73, 127.80, 128.13, 128.22, 128.42, 128.73, 130.49, 132.38, 137.86, 138.33, 154.69; HRMS (ESI) Anal. calcd for C32H34O5S [M + NH4]+: 548.2465, found: 548.2454.
:1) as a colorless oil (50% or 64% yield): Rf 0.35 (petroleum ether–ethyl acetate, 6:1); [α]20D −17.2 (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ 2.32 (s, 3H, tolyl CH3), 2.57–2.63 (m, 1H, CH2CHCH2), 2.65 (d, 1H, J = 4.5 Hz, OH), 2.75–2.80 (m, 1H, CH2CHCH2), 3.19 (t, 1H, J = 6.5 Hz, H-2), 3.50 (dd, 1H, J = 4.5 Hz, 10.5 Hz, H-6a), 3.55 (dd, 1H, J = 5.5 Hz, 10.5 Hz, H-6b), 3.76 (ddd, 1H, J = 4.5 Hz, 6.5 Hz, 8.0 Hz, H-1), 3.86–3.91 (m, 2H, H-3 and H-4), 4.09 (dd, 1H, J = 5.0 Hz, 9.5 Hz, H-5), 4.45 (d, 1H, J = 12.0 Hz, PhCH2), 4.51 (d, 1H, J = 12.0 Hz, PhCH2), 4.56 (d, 1H, J = 11.5 Hz, PhCH2), 4.62 (d, 1H, J = 11.5 Hz, PhCH2), 5.05–5.11 (m, 2H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.80–5.89 (m, 1H, –CH), 7.09 (d, 2H, J = 8.0 Hz), 7.27–7.35 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 21.11, 37.14, 51.97 (C-2), 68.19 (C-3), 69.02 (C-6), 71.18 (C-5), 72.03, 73.32, 74.36 (C-4), 74.73 (C-1), 117.05, 127.65, 127.90, 127.95, 128.35, 128.47, 129.16, 129.81, 133.53, 134.92, 137.80, 137.93, 138.04; HRMS (ESI) Anal. calcd for C30H34O4S [M + NH4]+: 508.2516, found: 508.2518.
:1) as a colorless oil (51% yield): Rf 0.30 (petroleum ether–acetone, 5:1); [α]20D −42.0 (c 0.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.67–1.71 (m, 1H), 1.83–1.88 (m, 1H), 1.96–2.05 (m, 2H), 2.08–2.15 (m, 1H), 2.28 (t, 1H, J = 8.5 Hz), 2.32 (s, 3H), 2.72 (d, 1H, J = 6.0 Hz), 3.11 (td, 1H, J = 3.5 Hz, 9.0 Hz), 3.26 (t, 1H, J = 9.0 Hz), 3.47 (d, 2H, J = 6.0 Hz), 3.79 (qd, 1H, J = 3.5 Hz, 6.0 Hz, 9.5 Hz), 3.86 (t, 1H, J = 3.5 Hz), 4.01 (dd, 1H, J = 4.0 Hz, 9.0 Hz), 4.05 (td, 1H, J = 3.5 Hz, 5.5 Hz), 4.41 (d, 1H, J = 12.0 Hz, PhCH2), 4.45 (d, 1H, J = 12.0 Hz, PhCH2), 4.57 (d, 1H, J = 12.0 Hz, PhCH2), 4.65 (d, 1H, J = 12.0 Hz, PhCH2), 7.10 (d, 2H, J = 8.0 Hz), 7.26–7.36 (m, 10H), 7.38 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 20.59, 21.15, 23.35, 39.19, 50.52, 51.33, 68.01, 68.78, 71.90, 72.47, 73.40, 73.63, 75.40, 127.58, 127.62, 127.77, 127.83, 128.07, 128.38, 128.46, 129.86, 134.36, 137.93, 138.33, 219.69; HRMS (ESI) Anal. calcd for C32H36O5S [M + NH4]+: 550.2622, found: 550.2627.
:1) as a colorless oil (46% or 41% yield): Rf 0.35 (petroleum ether–acetone, 5:1); 1H NMR (500 MHz, CDCl3) δ 1.28 (t, 3H, J = 7.5 Hz, ethyl CH3), 2.37–2.42 (m, 2H, H-2 and CH2CHCH2), 2.55–2.67 (m, 2H, ethyl CH2), 2.78–2.83 (m, 1H, CH2CHCH2), 3.02 (d, 1H, J = 1.5 Hz, OH), 3.33 (ddd, 1H, J = 3.0 Hz, 8.0 Hz, 10.5 Hz, H-1), 3.39 (ddd, 1H, J = 2.0 Hz, 4.5 Hz, 9.5 Hz, H-5), 3.49 (t, 1H, J = 8.5 Hz, 9.5 Hz, H-4), 3.54–3.58 (m, 1H, H-3), 3.69 (dd, 1H, J = 4.5 Hz, 11 Hz, H-6b), 3.74 (dd, 1H, J = 2.0 Hz, 10.5 Hz, H-6a), 4.57 (d, 1H, J = 12.5 Hz, PhCH2), 4.61 (d, 1H, J = 11.0 Hz, PhCH2), 4.63 (d, 1H, J = 12.5 Hz, PhCH2), 4.91 (d, 1H, J = 11.5 Hz, PhCH2), 5.09–5.15 (m, 2H, CH2), 5.91–5.99 (m, 1H, –CH), 7.26–7.35 (m, 10H); 13C NMR (125 MHz, CDCl3) δ 15.29, 25.03, 36.91, 53.75 (C-2), 69.17 (C-6), 73.40, 74.60, 76.15 (C-3), 78.55 (C-4, C-5), 78.96 (C-1), 117.10, 127.51, 127.71, 127.99, 128.31, 128.38, 134.78, 138.47; HRMS (ESI) Anal. calcd for C25H32O4S [M + NH4]+: 446.2359, found: 446.2357.
:1) as a colorless oil (70% yield): Rf 0.30 (petroleum ether–acetone, 5:1); [α]20D −23.2 (c 1.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.26 (t, 3H, J = 7.5 Hz, ethyl CH3), 1.73–1.84 (m, 1H), 2.00–2.15 (m, 4H), 2.28–2.34 (m, 1H), 2.42 (t, 1H, J = 10.5 Hz, H-2), 2.55–2.66 (m, 2H, ethyl CH2), 2.86 (dd, J = 9.0 Hz, 1H), 2.97 (d, 1H, J = 1.5 Hz, OH), 3.39 (ddd, 1H, J = 2.0 Hz, 4.0 Hz, 9.5 Hz, H-5), 3.54 (dd, 1H, J = 9.0 Hz, 10.0 Hz, H-4), 3.61–3.67 (m, 2H, H-3, H-6a), 3.72 (dd, 1H, J = 4.0 Hz, 11.5 Hz, H-6b), 3.87 (dd, 1H, J = 2.0 Hz, 11.0 Hz, H-1), 4.50 (d, 1H, J = 12.5 Hz, PhCH2), 4.54 (d, 1H, J = 12.0 Hz, PhCH2), 4.66 (d, 1H, J = 11.0 Hz, PhCH2), 4.89 (d, 1H, J = 11.0 Hz, PhCH2), 7.27–7.33 (m, 10H); 13C NMR (125 MHz, CDCl3) δ 15.09, 20.84, 22.76, 23.46, 39.07, 50.19, 51.42 (C-2), 68.94 (C-6), 73.16, 74.61, 75.56 (C-3), 77.19 (C-1), 78.35 (C-5), 78.47 (C-4), 127.47, 127.70, 127.98, 128.26, 128.39, 138.47, 219.58; HRMS (ESI) Anal. calcd for C27H34O5S [M + NH4]+: 488.2465, found: 488.2474.
:1) as a colorless oil (72% yield): Rf 0.40 (toluene–CH3CN, 4:1); 1H NMR (400 MHz, CDCl3) δ 1.99–2.06 (m, 1H), 2.18 (dd, 1H, J = 3.2 Hz, 12.0 Hz), 2.49 (s, 1H), 3.71 (d, 2H, OCH3, J = 2.4 Hz), 3.75 (d, 1H, J = 2.4 Hz), 3.78 (s, 1H), 3.83 (s, 1H), 3.98 (d, 1H, J = 10.0 Hz), 4.31 (d, 1H, J = 2.0 Hz), 4.47–4.51 (m, 2H), 4.59–4.62 (m, 2H), 5.00 (dd, 1H, J = 2.0 Hz, 11.6 Hz, H-1), 6.49 (s, 1H), 6.50 (s, 1H), 7.24–7.31 (m, 10H), 7.93 (s, 1H, OH); 13C NMR (125 MHz, CDCl3) δ 36.97, 55.85, 56.61, 64.30, 68.70, 71.79, 73.44, 73.69, 74.01, 74.49, 102.13, 110.36, 115.70, 127.70, 127.78, 127.99, 128.21, 128.42, 128.60, 137.35, 137.98, 142.14, 149.40, 149.97; HRMS (ESI) Anal. calcd for C28H32O7 [M + H]+: 481.2221, found: 481.2232.
CH2), 5.80–5.89 (m, 1H, –CH), 7.09 (d, 2H, J = 8.0 Hz), 7.27–7.35 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 21.11, 37.14, 51.97 (C-2), 68.19 (C-3), 69.02 (C-6), 71.18 (C-5), 72.03, 73.32, 74.36 (C-4), 74.73 (C-1), 117.05, 127.65, 127.90, 127.95, 128.35, 128.47, 129.16, 129.81, 133.53, 134.92, 137.80, 137.93, 138.04; HRMS (ESI) Anal. calcd for C30H34O4S [M + NH4]+: 508.2516, found: 508.2518.
:1) as a colorless oil (51% yield): Rf 0.30 (petroleum ether–acetone, 5:1); [α]20D −42.0 (c 0.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.67–1.71 (m, 1H), 1.83–1.88 (m, 1H), 1.96–2.05 (m, 2H), 2.08–2.15 (m, 1H), 2.28 (t, 1H, J = 8.5 Hz), 2.32 (s, 3H), 2.72 (d, 1H, J = 6.0 Hz), 3.11 (td, 1H, J = 3.5 Hz, 9.0 Hz), 3.26 (t, 1H, J = 9.0 Hz), 3.47 (d, 2H, J = 6.0 Hz), 3.79 (qd, 1H, J = 3.5 Hz, 6.0 Hz, 9.5 Hz), 3.86 (t, 1H, J = 3.5 Hz), 4.01 (dd, 1H, J = 4.0 Hz, 9.0 Hz), 4.05 (td, 1H, J = 3.5 Hz, 5.5 Hz), 4.41 (d, 1H, J = 12.0 Hz, PhCH2), 4.45 (d, 1H, J = 12.0 Hz, PhCH2), 4.57 (d, 1H, J = 12.0 Hz, PhCH2), 4.65 (d, 1H, J = 12.0 Hz, PhCH2), 7.10 (d, 2H, J = 8.0 Hz), 7.26–7.36 (m, 10H), 7.38 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 20.59, 21.15, 23.35, 39.19, 50.52, 51.33, 68.01, 68.78, 71.90, 72.47, 73.40, 73.63, 75.40, 127.58, 127.62, 127.77, 127.83, 128.07, 128.38, 128.46, 129.86, 134.36, 137.93, 138.33, 219.69; HRMS (ESI) Anal. calcd for C32H36O5S [M + NH4]+: 550.2622, found: 550.2627.
:1) as a colorless oil (46% or 41% yield): Rf 0.35 (petroleum ether–acetone, 5:1); 1H NMR (500 MHz, CDCl3) δ 1.28 (t, 3H, J = 7.5 Hz, ethyl CH3), 2.37–2.42 (m, 2H, H-2 and CH2CHCH2), 2.55–2.67 (m, 2H, ethyl CH2), 2.78–2.83 (m, 1H, CH2CHCH2), 3.02 (d, 1H, J = 1.5 Hz, OH), 3.33 (ddd, 1H, J = 3.0 Hz, 8.0 Hz, 10.5 Hz, H-1), 3.39 (ddd, 1H, J = 2.0 Hz, 4.5 Hz, 9.5 Hz, H-5), 3.49 (t, 1H, J = 8.5 Hz, 9.5 Hz, H-4), 3.54–3.58 (m, 1H, H-3), 3.69 (dd, 1H, J = 4.5 Hz, 11 Hz, H-6b), 3.74 (dd, 1H, J = 2.0 Hz, 10.5 Hz, H-6a), 4.57 (d, 1H, J = 12.5 Hz, PhCH2), 4.61 (d, 1H, J = 11.0 Hz, PhCH2), 4.63 (d, 1H, J = 12.5 Hz, PhCH2), 4.91 (d, 1H, J = 11.5 Hz, PhCH2), 5.09–5.15 (m, 2H, CH2), 5.91–5.99 (m, 1H, –CH), 7.26–7.35 (m, 10H); 13C NMR (125 MHz, CDCl3) δ 15.29, 25.03, 36.91, 53.75 (C-2), 69.17 (C-6), 73.40, 74.60, 76.15 (C-3), 78.55 (C-4, C-5), 78.96 (C-1), 117.10, 127.51, 127.71, 127.99, 128.31, 128.38, 134.78, 138.47; HRMS (ESI) Anal. calcd for C25H32O4S [M + NH4]+: 446.2359, found: 446.2357.
:1) as a colorless oil (70% yield): Rf 0.30 (petroleum ether–acetone, 5:1); [α]20D −23.2 (c 1.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.26 (t, 3H, J = 7.5 Hz, ethyl CH3), 1.73–1.84 (m, 1H), 2.00–2.15 (m, 4H), 2.28–2.34 (m, 1H), 2.42 (t, 1H, J = 10.5 Hz, H-2), 2.55–2.66 (m, 2H, ethyl CH2), 2.86 (dd, J = 9.0 Hz, 1H), 2.97 (d, 1H, J = 1.5 Hz, OH), 3.39 (ddd, 1H, J = 2.0 Hz, 4.0 Hz, 9.5 Hz, H-5), 3.54 (dd, 1H, J = 9.0 Hz, 10.0 Hz, H-4), 3.61–3.67 (m, 2H, H-3, H-6a), 3.72 (dd, 1H, J = 4.0 Hz, 11.5 Hz, H-6b), 3.87 (dd, 1H, J = 2.0 Hz, 11.0 Hz, H-1), 4.50 (d, 1H, J = 12.5 Hz, PhCH2), 4.54 (d, 1H, J = 12.0 Hz, PhCH2), 4.66 (d, 1H, J = 11.0 Hz, PhCH2), 4.89 (d, 1H, J = 11.0 Hz, PhCH2), 7.27–7.33 (m, 10H); 13C NMR (125 MHz, CDCl3) δ 15.09, 20.84, 22.76, 23.46, 39.07, 50.19, 51.42 (C-2), 68.94 (C-6), 73.16, 74.61, 75.56 (C-3), 77.19 (C-1), 78.35 (C-5), 78.47 (C-4), 127.47, 127.70, 127.98, 128.26, 128.39, 138.47, 219.58; HRMS (ESI) Anal. calcd for C27H34O5S [M + NH4]+: 488.2465, found: 488.2474.
:1) as a colorless oil (72% yield): Rf 0.40 (toluene–CH3CN, 4:1); 1H NMR (400 MHz, CDCl3) δ 1.99–2.06 (m, 1H), 2.18 (dd, 1H, J = 3.2 Hz, 12.0 Hz), 2.49 (s, 1H), 3.71 (d, 2H, OCH3, J = 2.4 Hz), 3.75 (d, 1H, J = 2.4 Hz), 3.78 (s, 1H), 3.83 (s, 1H), 3.98 (d, 1H, J = 10.0 Hz), 4.31 (d, 1H, J = 2.0 Hz), 4.47–4.51 (m, 2H), 4.59–4.62 (m, 2H), 5.00 (dd, 1H, J = 2.0 Hz, 11.6 Hz, H-1), 6.49 (s, 1H), 6.50 (s, 1H), 7.24–7.31 (m, 10H), 7.93 (s, 1H, OH); 13C NMR (125 MHz, CDCl3) δ 36.97, 55.85, 56.61, 64.30, 68.70, 71.79, 73.44, 73.69, 74.01, 74.49, 102.13, 110.36, 115.70, 127.70, 127.78, 127.99, 128.21, 128.42, 128.60, 137.35, 137.98, 142.14, 149.40, 149.97; HRMS (ESI) Anal. calcd for C28H32O7 [M + H]+: 481.2221, found: 481.2232.
Acknowledgements
This work was financially supported by the grants (2012ZX09502001-001, 2012CB822100) from the Ministry of Science and Technology of China, the National Natural Science Foundation of China (grant no. 21232002, 21102008), Beijing Natural Science Foundation (2142015) and Beijing Higher Education Young Elite Teacher Project (YETP0063).Notes and references
- (a) X. Yuan and R. J. Linhardt, Curr. Top. Med. Chem., 2005, 5, 1393 CrossRef CAS; (b) Z. Wei, Curr. Top. Med. Chem., 2005, 5, 1363 CrossRef; (c) J.-M. Beau and T. Gallagher, Top. Curr. Chem., 1997, 187, 1 CrossRef CAS; (d) P. Sinay, Pure Appl. Chem., 1997, 69, 459 CrossRef CAS; (e) D. E. Levy and C. Tang, The Chemistry of C-Glycosides, Pergamon, Oxford, 1995 Search PubMed; (f) K. Suzuki and T. Matsumoto, in Recent Progress in the Chemical Synthesis of Antibiotics and Related Microbial Products, ed. G. Lukacs, Springer, Berlin, 1993, vol. 2, p. 353 Search PubMed; (g) U. Hacksell and G. D. Daves Jr., Prog. Med. Chem., 1985, 22, 1 CrossRef CAS.
- T. Bililign, B. R. Griffith and J. S. Thorson, Nat. Prod. Rep., 2005, 22, 742 RSC , and references therein.
- (a) J. Stambasky, M. Hocek and P. Kocovsky, Chem. Rev., 2009, 109, 6729 CrossRef CAS PubMed; (b) D. Y. W. Lee and M. He, Curr. Top. Med. Chem., 2005, 5, 1333 CrossRef CAS; (c) P. G. Hultin, Curr. Top. Med. Chem., 2005, 5, 1299 CrossRef CAS; (d) Y. Du, R. J. Linhardt and I. R. Vlahov, Tetrahedron, 1998, 54, 9913 CrossRef CAS; (e) W. Kevin, K. W. Wellington and S. A. Benner, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 1309 CrossRef PubMed.
- B. Apsel, J. A. Bender, M. Escobar, D. E. Kaelin Jr., O. D. Lopez and S. F. Martin, Tetrahedron Lett., 2003, 44, 1075 CrossRef CAS.
- G. D. Daves Jr., Acc. Chem. Res., 1990, 23, 201 CrossRef.
- G. Li, D.-C. Xiong and X.-S. Ye, Synlett, 2010, 1577 Search PubMed.
- G. V. M. Sharma, K. R. Kumar, P. Sreenivas, P. R. Krishna and M. S. Chorghade, Tetrahedron: Asymmetry, 2002, 13, 687 CrossRef CAS.
- (a) R. Santos, A. R. Jesus, J. M. Caio and A. P. Rauter, Curr. Org. Chem., 2011, 15, 128 CrossRef; (b) A. Ben, T. Yamauchi, T. Matsumoto and K. Suzuki, Synlett, 2004, 225 CAS; (c) T. Yamauchi, Y. Watanabe, K. Suzuki and T. Matsumoto, Synlett, 2010, 1577 Search PubMed; (d) T. Matsumoto, T. Hosoya and K. Suzuki, Tetrahedron Lett., 1990, 31, 4629 CrossRef CAS; (e) T. Matsumoto, M. Katsuki and K. Suzuki, Tetrahedron Lett., 1988, 29, 6935 CrossRef CAS; (f) J. R. Mahling and R. R. Schmidt, Synthesis, 1993, 325 CrossRef CAS; (g) K. Kitamura, Y. Ando, T. Matsumoto and K. Suzuki, Angew. Chem., Int. Ed., 2014, 53, 1258 CrossRef CAS PubMed; (h) K. Kitamura, Y. Maezawa, Y. Ando, T. Kusumi, T. Matsumoto and K. Suzuki, Angew. Chem., Int. Ed., 2014, 53, 1262 CrossRef CAS PubMed.
- S. Vijayasaradhi and I. S. Aidhen, Org. Lett., 2002, 4, 1739 CrossRef CAS PubMed.
- P. de Pouilly, A. Chenede, J.-M. Mallet and P. Sinai, Bull. Soc. Chim. Fr., 1993, 130, 256 CAS.
- T. Mukaiyama, H. Uchiro, N. Hirano and T. Ishikawa, Chem. Lett., 1996, 629 CrossRef CAS.
- (a) D.-C. Xiong, X.-S. Ye and L.-H. Zhang, Org. Lett., 2009, 11, 1709 CrossRef CAS PubMed; (b) H.-H. Li and X.-S. Ye, Org. Biomol. Chem., 2009, 7, 3855 RSC; (c) D. Pan and N. Jiao, Synlett, 2010, 1577 CAS.
- (a) D. Hou, H. A. Taha and T. L. Lowary, J. Am. Chem. Soc., 2009, 131, 12937 CrossRef CAS PubMed; (b) D. Hou and T. L. Lowary, J. Org. Chem., 2009, 74, 2278 CrossRef CAS PubMed; (c) C. Gao, D.-C. Xiong, Y. Wang and X.-S. Ye, J. Chin. Pharm. Sci., 2011, 20, 139 CAS; (d) D. Hou and T. L. Lowary, Org. Lett., 2007, 9, 4487 CrossRef CAS PubMed; (e) Y. Bai and T. L. Lowary, J. Org. Chem., 2006, 71, 9658 CrossRef CAS PubMed; (f) O. M. Cociorva and T. L. Lowary, Tetrahedron, 2004, 60, 1481 CrossRef CAS PubMed; (g) C. S. Callam, R. R. Gadikota, D. M. Krein and T. L. Lowary, J. Am. Chem. Soc., 2003, 125, 13112 CrossRef CAS PubMed; (h) R. R. Gadikota, C. S. Callam, T. Wagner, B. Del Fraino and T. L. Lowary, J. Am. Chem. Soc., 2003, 125, 4155 CrossRef CAS PubMed; (i) Y. Wang, Q. Li, S. Cheng, Y. Wu, D. Guo, Q.-H. Fan, X. Wang, L.-H. Zhang and X.-S. Ye, Org. Lett., 2005, 7, 5577 CrossRef CAS PubMed; (j) B. Yu and Z. Yang, Org. Lett., 2001, 3, 377 CrossRef CAS.
- (a) K. Takeo, K. Maki, Y. Wada and S. Kitamura, Carbohydr. Res., 1993, 245, 81 CrossRef CAS; (b) H. Wang, J. She, L.-H. Zhang and X.-S. Ye, J. Org. Chem., 2004, 69, 5774 CrossRef CAS PubMed.
- D. Hou and T. L. Lowary, Tetrahedron, 2009, 65, 5916 CrossRef CAS PubMed.
- R. Benhaddou, S. Czernecki and G. Ville, J. Org. Chem., 1992, 57, 4612 CrossRef CAS.
- V. Bellmta, C. Chaseagnard and S. Czemecki, Carbohydr. Res., 1991, 219, 1 CrossRef.
- (a) T. Hosoya, Y. Ohashi, T. Matsumoto and K. Suzuki, Tetrahedron Lett., 1996, 37, 663 CrossRef CAS; (b) T. Hosoya, E. Takashiro, T. Matsumoto and K. Suzuki, J. Am. Chem. Soc., 1994, 116, 1004 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra of 1, 1b, 2c, 2d, 2, 4a–4g, 6a–6d, 7. See DOI: 10.1039/c4qo00113c |
| This journal is © the Partner Organisations 2014 |