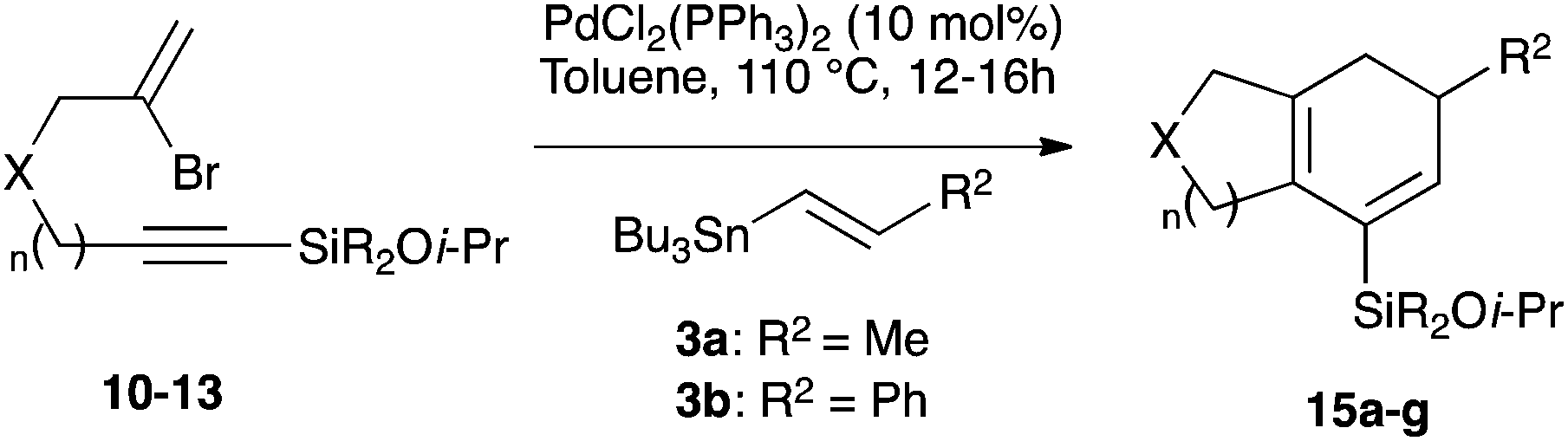DOI:
10.1039/C4QO00123K
(Research Article)
Org. Chem. Front., 2014,
1, 661-673
Carbopalladation of bromoene-alkynylsilanes: mechanistic insights and synthesis of fused-ring bicyclic silanes and phenols†
Received
25th April 2014
, Accepted 20th May 2014
First published on 28th May 2014
Abstract
The palladium-catalyzed cascade cyclization of silylated bromoenynes and alkenylstannanes provides a straightforward route to a range of bicyclic silylated cyclohexadienes. Mechanistic insights into aspects of carbopalladation and unusual palladium-mediated isomerizations have been obtained through the detection of reaction intermediates, the isolation of byproducts, and reaction monitoring by VT NMR spectroscopy. The utility of the bicyclic products is illustrated through oxidation to bicyclic enones and phenols.
Introduction
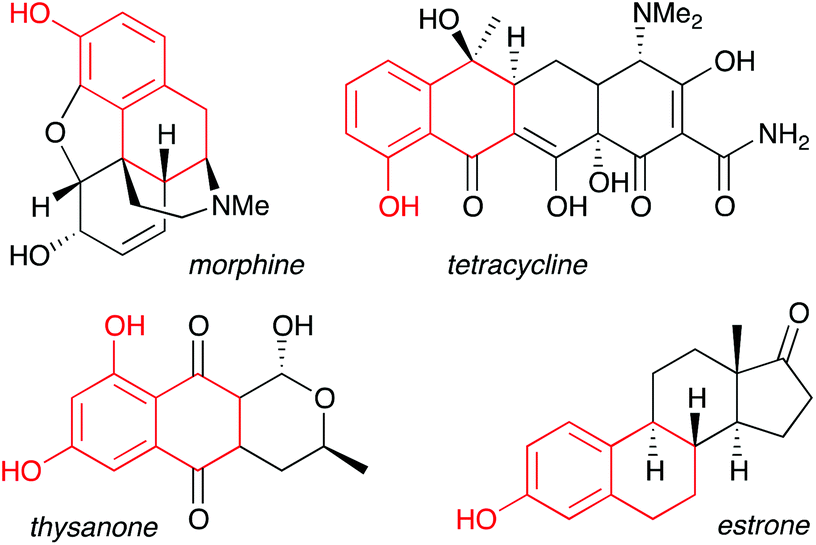
Phenols represent one of the most important aromatic functional groups, featuring in numerous natural products and drugs.1 Polycyclic phenols are of particular interest, as illustrated by highly bioactive compounds such as the anthracycline antibiotics, estrone, and morphine (Fig. 1).2 As traditional methods for phenol preparation require harsh conditions that may not be compatible with sensitive functional groups,3 phenols are usually incorporated at an early stage of a synthesis, thus restricting synthetic planning.
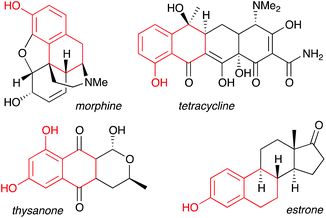 |
| | Fig. 1 Fused-ring phenols in bioactive natural products. | |
Late-stage phenolation is therefore an attractive prospect that has driven the development of transition metal-catalyzed aryl halide hydroxylation and aryl C–H activation/oxidation methods.4 An alternative that avoids the prolonged heating often required by these processes is the use of a phenol surrogate, which could be revealed at a late stage of a synthetic route, but is stable to intermediate transformations. Here, arylboron derivatives have met with some success, particularly due to Molander's elegant work on the oxidation of aryl trifluoroborate salts,5 although the synthetic processability of these remains to be proven. In contrast, organosilanes – stalwarts of the protecting group field – display unrivalled tolerance towards multistep synthesis, and in recent work we have reported the use of arylsilanes as a source of phenols through Tamao-type oxidation.6,7
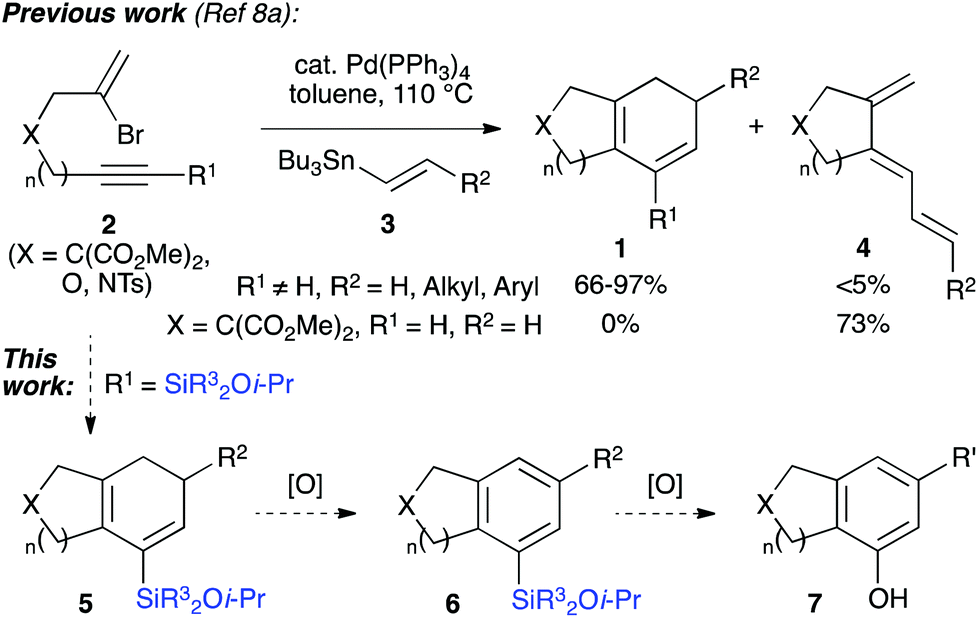
We have also disclosed a palladium-catalyzed cascade cyclization which prepares bi- or tricyclic products (1, Scheme 1) from the coupling of bromoenynes 2 with alkenyl- and dienylstannanes 3,8 a reaction pioneered and impressively explored by Suffert et al. for the synthesis of various polycycles.9 A feature of this work is the requirement for an internal alkyne (i.e. R1 ≠ H, Scheme 1) to avoid the formation of undesired triene isomers 4, which cannot undergo electrocyclization. One solution is to employ trimethylsilyl alkynes,10 and although this functionality indeed enables the desired cascade, it was clear that the resultant TMS-substituted cyclohexadienes would be of limited synthetic utility.
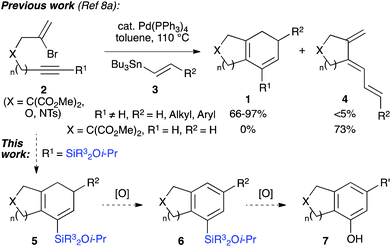 |
| | Scheme 1 Proposed synthesis of bicyclic phenols. | |
We realized that the use of a more functional silane would lead to a silicon-containing product capable of undergoing a range of further transformations, such as the aforementioned Tamao oxidation or Hiyama coupling.11 Indeed, we have illustrated this principle in approaches to the CDE rings of rubriflordilactone A, where arylsilanes formed from the cyclizations of bromoenynes could be oxidized to give specific tricyclic phenols.8b,12 In this paper we wish to report methods for the synthesis of a range of silylated bromoenynes, and their intermolecular coupling with alkenylstannanes to provide bicyclic silylcyclohexadienes 5. The oxidation of these dienes and silane substituents leads to bicyclic arylsilanes (6) and phenols (7) that would be challenging to prepare by other routes, and offers a de novo approach to ring systems of this type.13 Alongside this synthetic work, we expand on the mechanistic pathways operative in the cascade cyclization, including two unusual palladium-mediated isomerization processes. The unexpected observation of an unprecedented formal 4-endo-trig cyclization to afford fused ring cyclobutenes is also described.
Results and discussion
Synthesis of cascade cyclization substrates
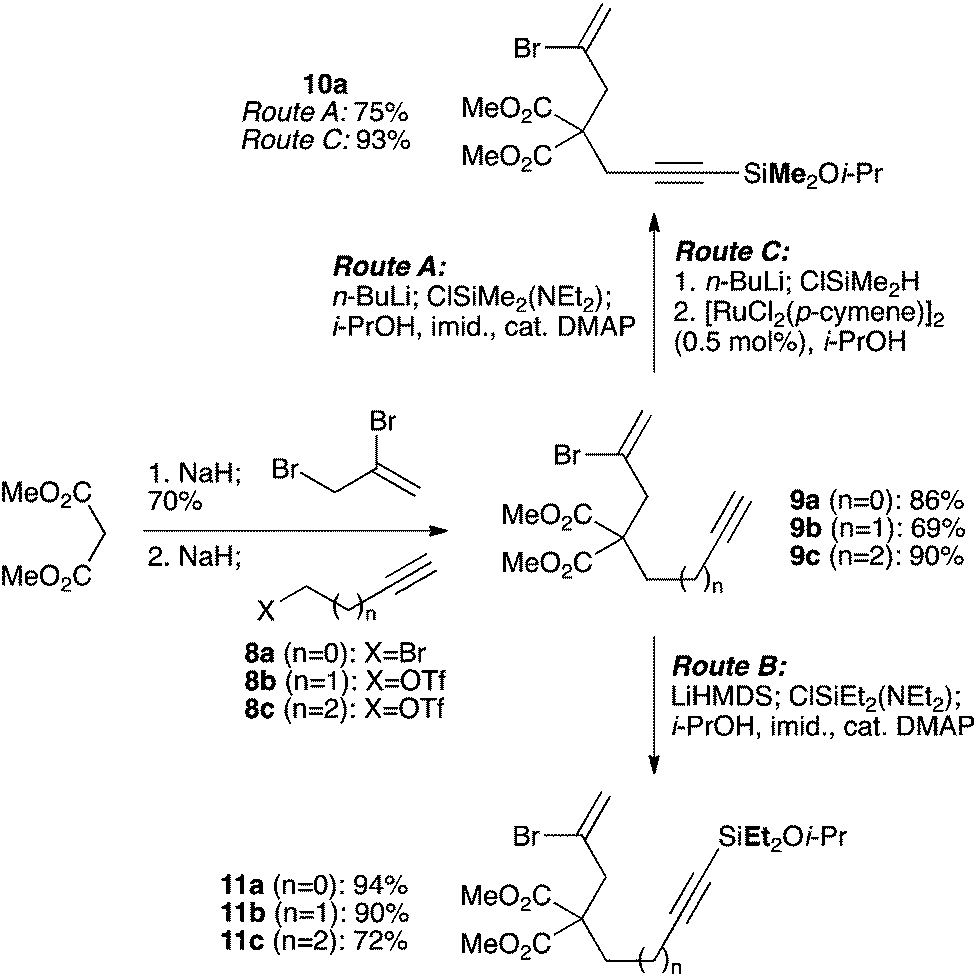
We selected a range of linkers to connect the bromoalkene and silylalkyne, such that bicyclic products containing 5- to 7-membered rings, and heterocycles, could be obtained. Synthesis of carbon-tethered bromoenynes began with dimethylmalonate (Scheme 2), which following monoallylation with 2,3-dibromopropene (70%) was alkylated with alkynyl electrophiles 8a–c to give bromoenynes 9a–c (69–90%). The silylation of these alkynes was initially attempted using conditions employed in our arylsilane work.6a Thus, deprotonation of 9a with n-butyllithium followed by trapping of the resultant alkynyllithium species with the moisture-sensitive (diethylamino)chlorodimethylsilane (Et2NMe2SiCl) gave an intermediate aminosilane which was not isolated, but immediately converted to the dimethylisopropoxysilane 10a (Route A, Scheme 2).14 Perhaps unsurprisingly, this silane proved to be rather unstable towards chromatography, however through rapid chromatographic purification using a solvent system buffered with triethylamine, 10a could be isolated in 75% yield. In order to address the chromatographic instability of this dimethylisopropoxysilane, we employed the analogous diethylsilane reagent (Et2N)SiEt2SiCl,15 which gave the alkynyl isopropoxydiethylsilanes 11a–c in much improved yields following chromatographic purification (Route B). An alternative solution to the synthesis of 10a proved to be the use of dimethylchlorosilane (HMe2SiCl) as the acetylide trapping agent (Route C); the ensuing hydrolytically stable intermediate alkynylhydrosilane could be easily purified, and then smoothly oxidized to 10a using Lee and Chang's elegant ruthenium-catalyzed dehydrogenative silylation methodology,16 which improved the yield of 10a to 93%.
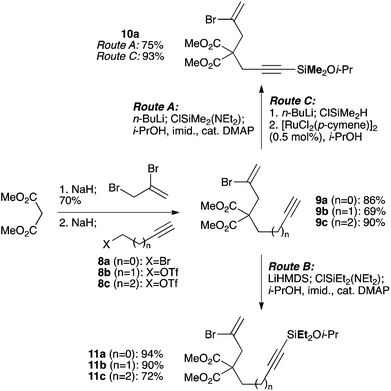 |
| | Scheme 2 Synthesis of malonate-derived silylbromoenynes. | |
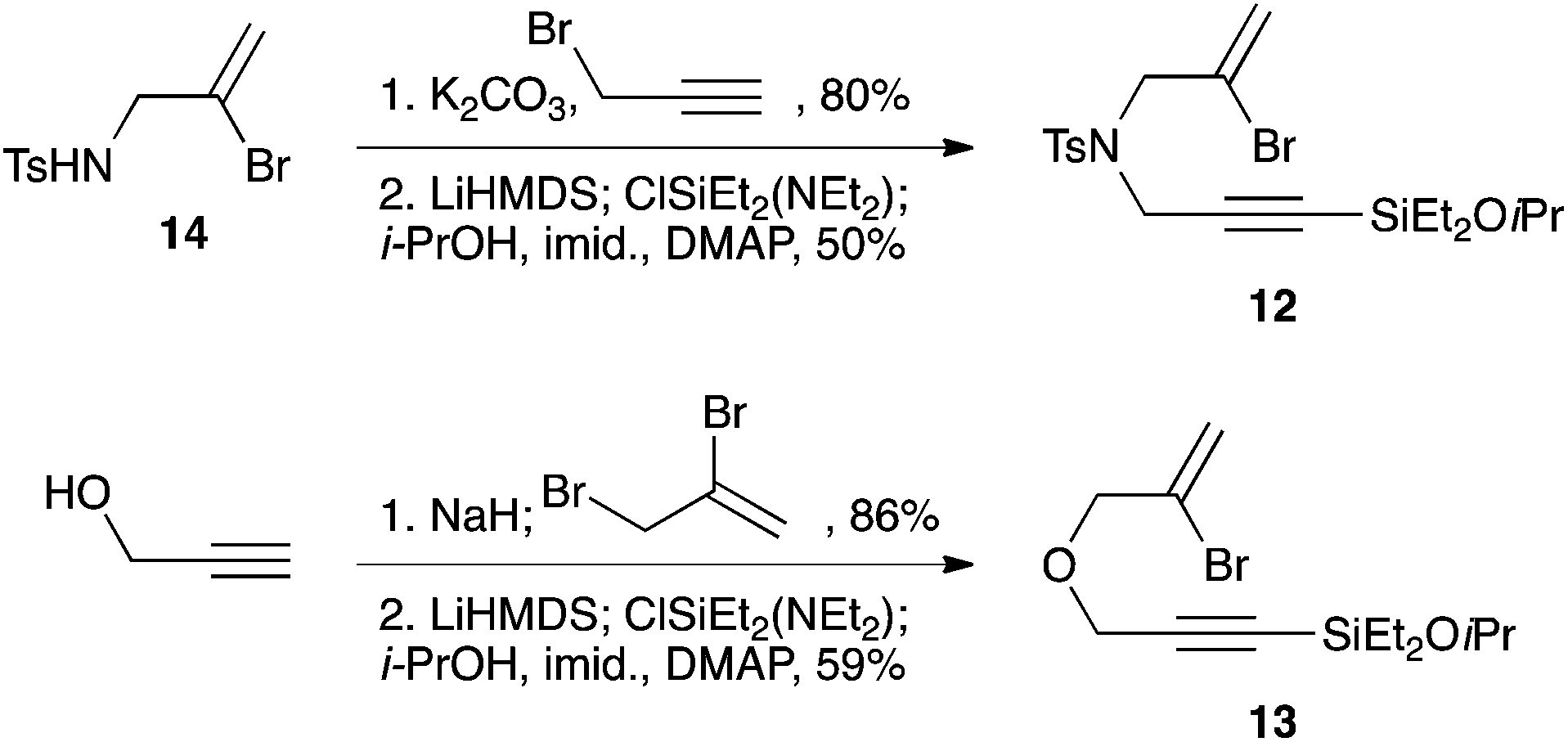
The syntheses of heteroatom-tethered silylbromoenynes could be achieved uneventfully using equivalent chemistry, with sulfonamide-tethered enyne 12 and ether-linked enyne 13 being prepared in reasonable yields over two steps from known sulfonamide 14 and propargyl alcohol, respectively (Scheme 3).
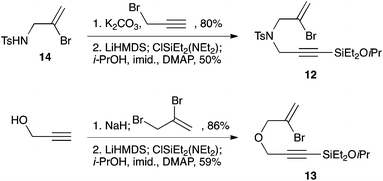 |
| | Scheme 3 Synthesis of heteroatom-tethered silylbromoenynes. | |
Cascade cyclizations of silylated bromoenynes
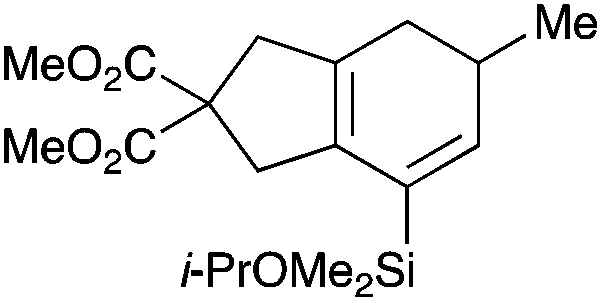
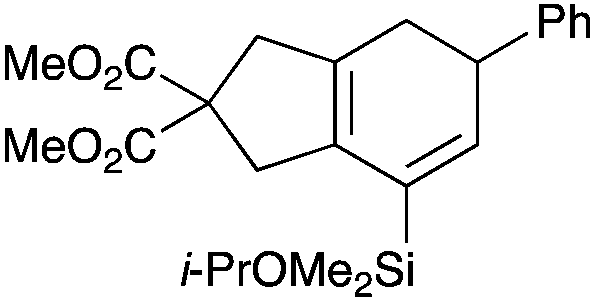
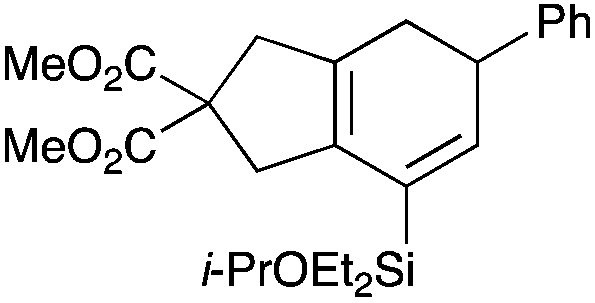
With a selection of silylated alkynes in hand, their palladium-catalyzed cascade cyclizations were examined. These reactions were carried out according to our previously optimized conditions,8a using PdCl2(PPh3)2 as precatalyst in refluxing toluene (Table 1). For the purposes of this work, the propenyl and styrenyl stannanes 3a and 3b were selected as generic cross-coupling partners. The reaction of dimethylsilane 10a was first investigated, which in spite of the relative instability of the alkynylsilane cyclized in excellent yields with both stannanes (92–97%, entries 1, 2). Notably, the hydrolytic stability of the silane increases markedly upon cyclization owing to the increased steric bulk of the cyclohexadienyl substituent in 15a compared to the alkyne substituent of the starting material. The cyclizations of the (more robust) diethylsilane substrates proved more challenging (entries 3–5), with these reactions requiring longer reaction times, likely due to steric hindrance of the key transmetallation step following carbopalladation. This resulted in lower isolated yields, as exemplified by the malonate and sulfonamide tethered enynes 11a and 12 which gave the corresponding bicycles 15c and 15d in 77 and 63% yields respectively. The oxygen-tethered substrate 13 proved particularly troublesome, delivering the dihydrofuran product 15e in low yield, possibly due to instability of the allyl propargyl ether. Reaction of substrates featuring a longer tether also proved effective, giving the 6,6- and 7,6-bicyclic products 15f and 15g in good yields (entries 6, 7).
Table 1 Cascade cyclizations of silylated bromoenynes 10–13
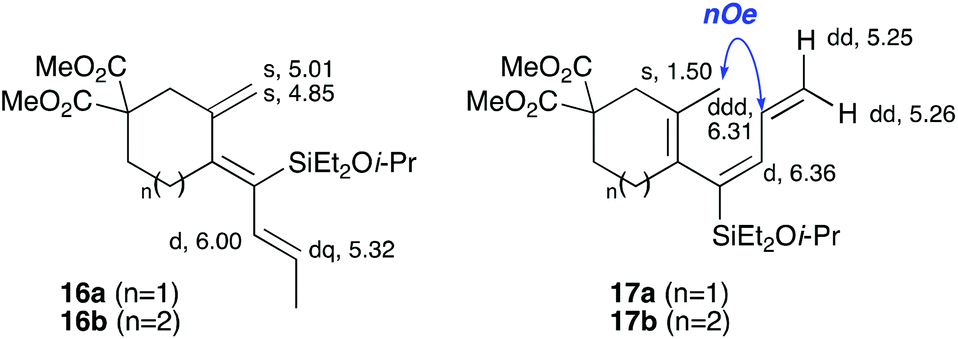
In the latter two cases, the desired bicyclic products were contaminated with significant quantities of partially separable isomeric byproducts 16a, b and 17a, b (Fig. 2), which were identified by careful analysis of 1H–1H COSY, and 1H–13C HSQC and HMBC 2D NMR spectra;17 the assignment of one of these sets of byproducts (16a and 17a) is discussed here. Byproduct 16a contains an exo-methylene unit (δH 5.01 and 4.85 ppm), and two vicinal alkene protons (δH 6.00 ppm, dd, J = 15.5, 1.5 Hz; and 5.32 ppm, dq, J = 15.5 and 6.5 Hz), the latter being characteristic of the trans-alkene of the propenyl unit. These data, together with key HMBC correlations, are strongly suggestive of the formal anti-carbopalladation/cross-coupling product 16a, the formation of which is entirely consistent with our earlier results.8a The identification of the second byproduct proved less straightforward. Key signals in the 1H NMR spectrum at δH 6.36 (d, J = 10.6 Hz), 6.31 (ddd, J = 16.5, 10.6 and 9.7 Hz), 5.26 (dd, J = 16.5 and 2.0 Hz) and 5.25 ppm (dd, J = 9.8 and 2.0 Hz) revealed a connectivity between four alkene protons and thus the surprising presence of a 1,3-butadienyl unit. A further significant piece of evidence was the detection of a methyl singlet at 1.50 ppm, to which the proton at 6.33 ppm showed an nOe enhancement. These combined observations, and 1H–13C correlations, led us to propose structure 17a, which features an (E)-alkenylsilane as part of a conjugated triene.
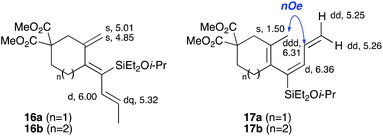 |
| | Fig. 2 Cyclization byproducts 16a/b and 17a/b. Selected key 1H NMR chemical shifts/multiplicities (in CDCl3) for 16a and 17a are shown. | |
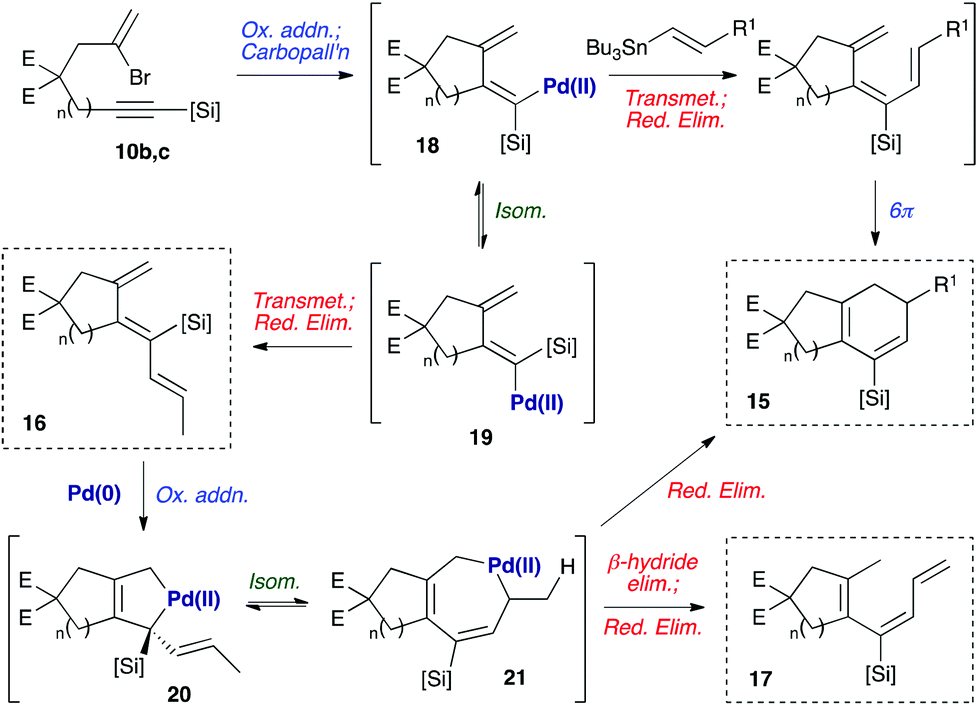
A mechanistic hypothesis for the formation of these byproducts is depicted in Scheme 4. The formation of byproduct 16 is consistent with our earlier observation8a that such undesired anti-trienes form in the course of the cyclization reaction, potentially via isomerization of the intermediate dienylpalladium complex 18 to its isomer 19.18 The rate of transmetallation of complex 19 is likely to exceed that of 18, so even small amounts of 19 may lead to significant quantities of anti-triene 16 (i.e., a Curtin–Hammett situation). The formation of increased quantities of this triene for substrates 11b and 11c may reflect the increased flexibility of the larger tethering ring, which can distort to alleviate 1,3-allylic strain between the silyl substituent and exo-methylene in 19 – and therefore reduce the steric cost of placing a silane in this more hindered position. The formation of 17 may be explained by our second previous observation8a that anti-trienes can isomerize to bicyclic products on prolonged exposure to the reaction conditions. We suggest that this process may proceed via oxidative addition of Pd(0) with diene 16 to give a palladacyclopentene 20.19 This could undergo a 1,3-allylic migration of the palladium atom to the 7-membered palladacycle 21, reductive elimination from which would afford the cyclohexadiene product 15.20 The formation of byproduct 17 could be rationalised by β-hydride elimination from this common palladacycle intermediate 21, followed by reductive elimination of the resultant palladium(II) hydride species. We presume that the steric hindrance imposed by the methyl group in 17 prevents a 6π-electrocyclization of this compound.
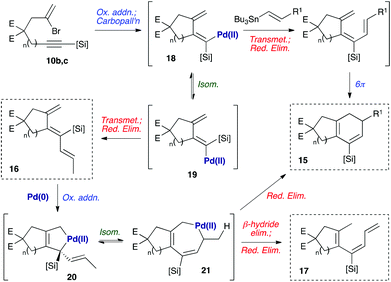 |
| | Scheme 4 Mechanistic proposal for byproduct formation. E = CO2Me, [Si] = SiEt2Oi-Pr. | |
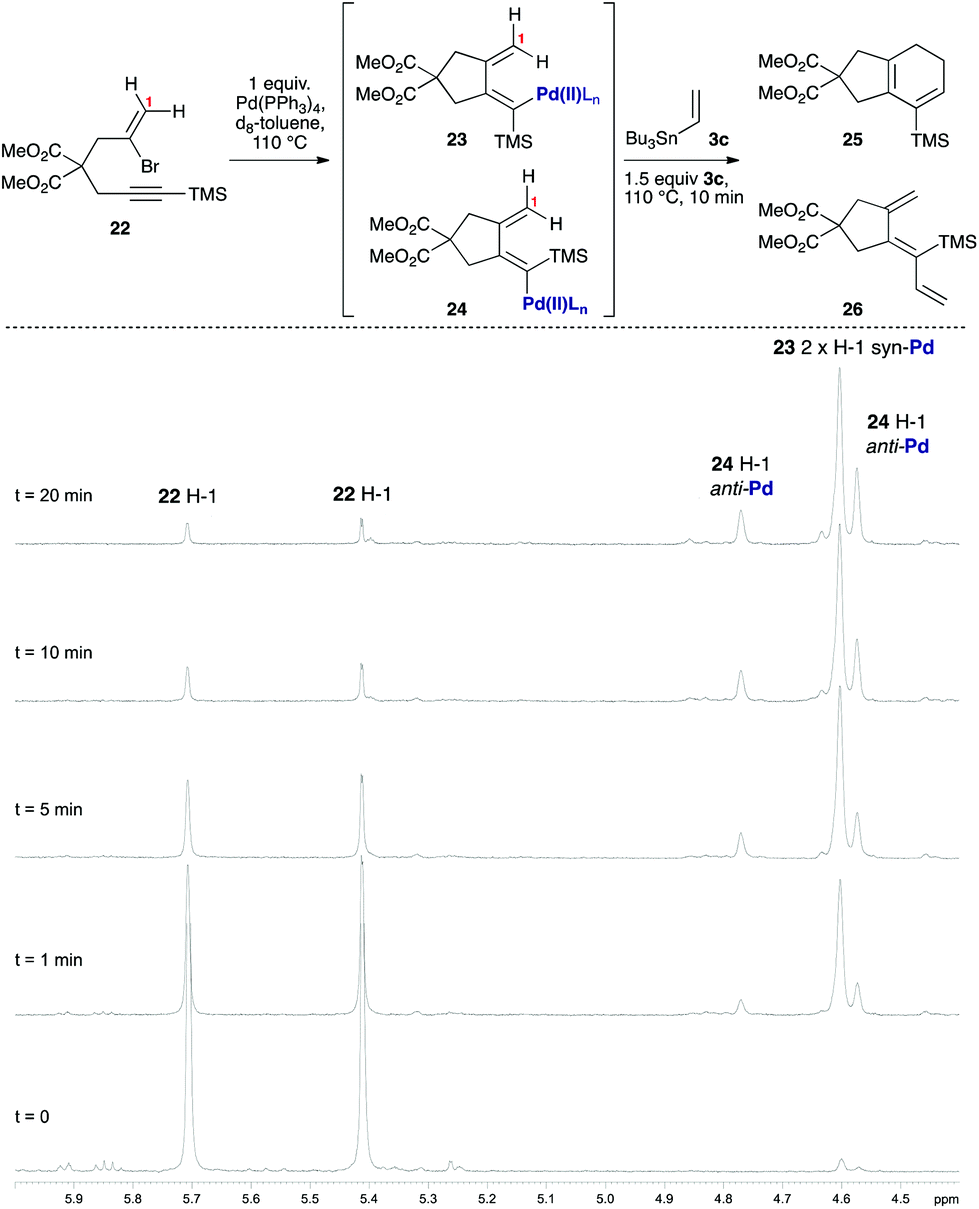
As [4 + 1] oxidative additions of Pd(0) to dienes are rare,19 more concrete evidence to support these pathways was sought. Firstly, the formation of 16via the intermediacy of dienylpalladium complex 19 was probed through the exposure of alkynylsilane 22 to one equivalent of Pd(PPh3)4 in d8-toluene at 110 °C (Fig. 3). The characteristic methylene signals of 22 at δH 5.41 and 5.71 ppm were rapidly converted (10 min) to two new sets of peaks: a prominent (apparent) singlet at 4.60 ppm, and two smaller singlets at 4.58 and 4.78 ppm. These were tentatively assigned as the exo-methylene peaks of the syn- and anti-dienylpalladium complexes 23 and 24 respectively, based on analysis by COSY and HSQC experiments. Support for this assignment was gained through the addition of 1.5 equivalents of tributylvinyltin to the NMR tube; further heating for 10 minutes led to exclusive formation of cyclohexadiene 25 and triene 26 in a ratio mirroring that of these intermediate species (84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16).
:16).
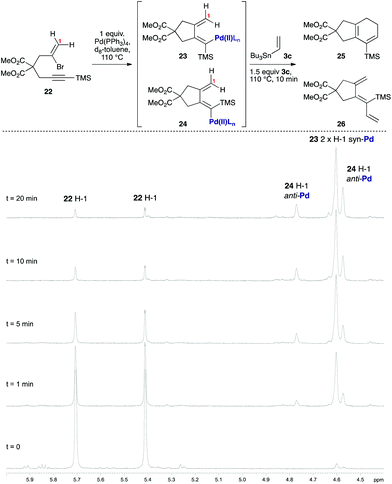 |
| | Fig. 3 Use of stoichiometric Pd(PPh3)4/VT 1H NMR spectroscopy to probe the putative dienylpalladium(II) intermediates. | |
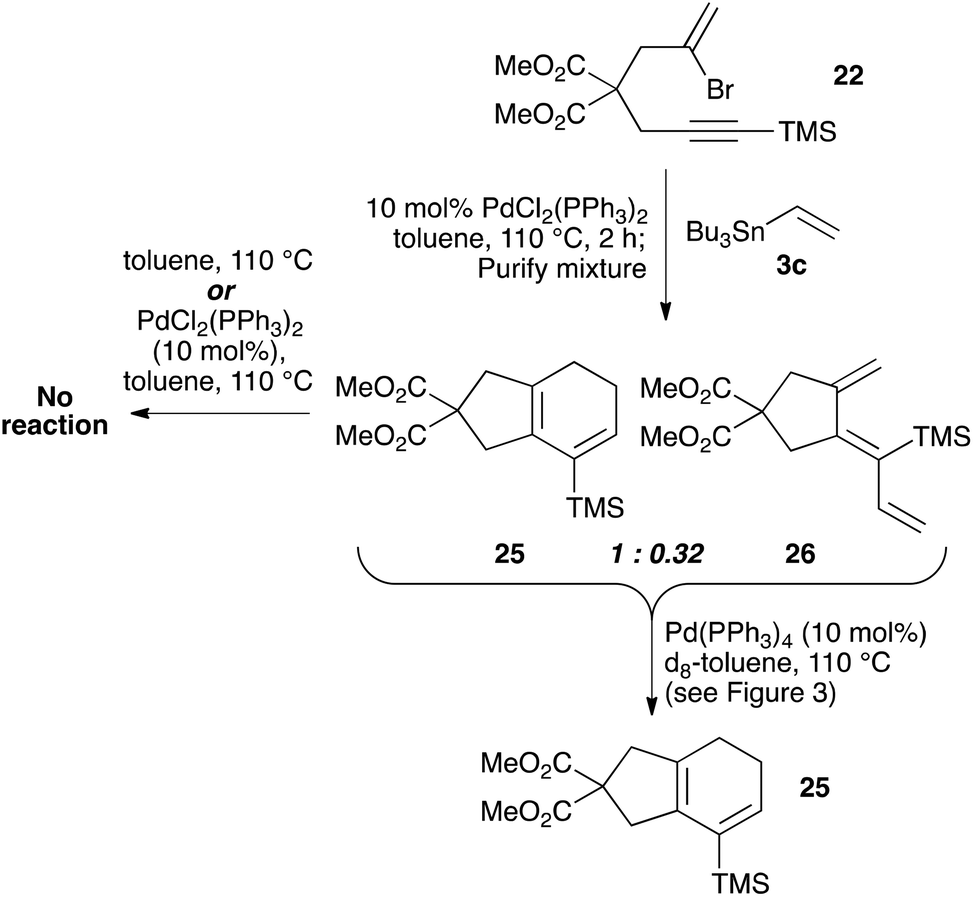
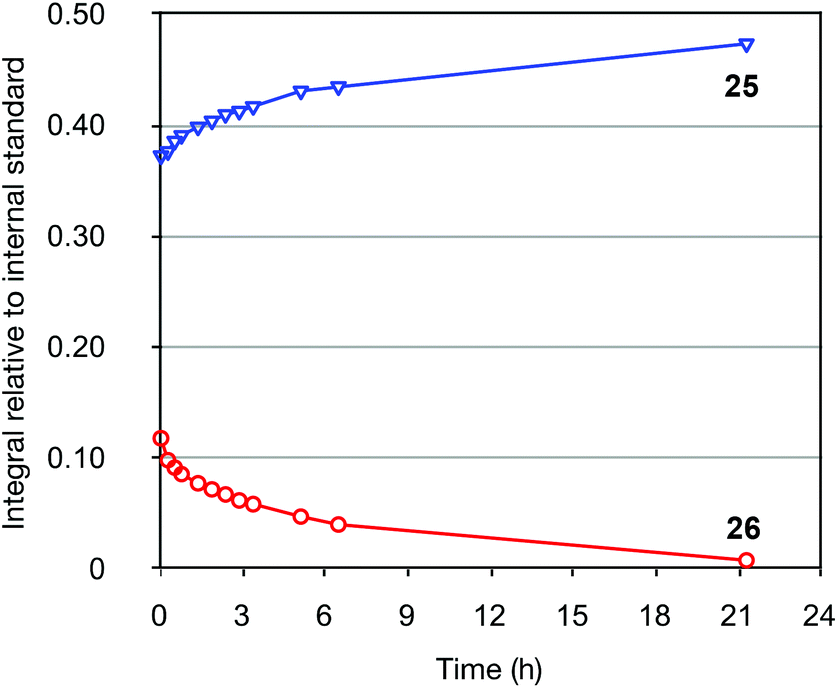
To explore the isomerization of 26 to 25, a 1:0.32 mixture of these compounds, formed from reaction of silane 22 with vinyltributyltin for two hours, was purified by silica gel chromatography (Scheme 5); the triene 26 in this mixture would be expected to isomerize to give the 25 upon resubmission to the reaction conditions. Heating the mixture in toluene overnight at 110 °C in the absence of catalyst led to no conversion of 26 to the bicyclic product 25, with both compounds recovered unchanged after this period (thus ruling out a purely thermal process). Palladium(II) salts have previously been shown to promote alkene isomerisation,21 but subjection of the anti-triene to PdCl2(PPh3)2 at 110 °C also led to no reaction. However, heating the mixture in the presence of Pd(PPh3)4 for 20 h led to complete consumption of anti-triene, and by performing this isomerization in d8-toluene with monitoring by 1H NMR spectroscopy in the presence of an internal standard (1,4-dimethoxybenzene), a smooth conversion of 26 to 25 was observed (Fig. 4). This clearly demonstrates that the isomerization of anti-triene to product is not a thermal process, and in fact requires a Pd(0) catalyst, thus offering some support to our proposed mechanism. At no point do we detect the formation of syn-triene, which lends some weight to our mechanistic hypothesis for the direct conversion of anti-trienes to bicyclic products (Scheme 4, although we recognise that any syn-triene formed could electrocyclize rapidly).
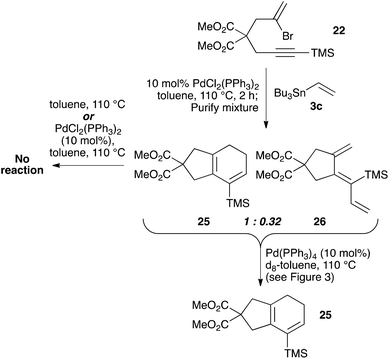 |
| | Scheme 5 Exploration of conditions to effect the isomerization of anti-triene 26 to bicycle 25. | |
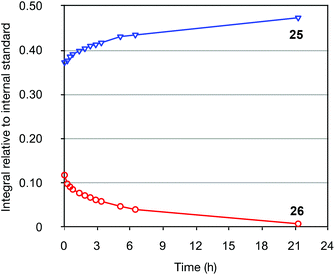 |
| | Fig. 4
1H NMR spectroscopic monitoring of the conversion of 26 to 25 catalyzed by Pd(PPh3)4. 1,4-Dimethoxybenzene was used as internal standard. | |
Cyclization to 7,4-fused ring cyclobutenes
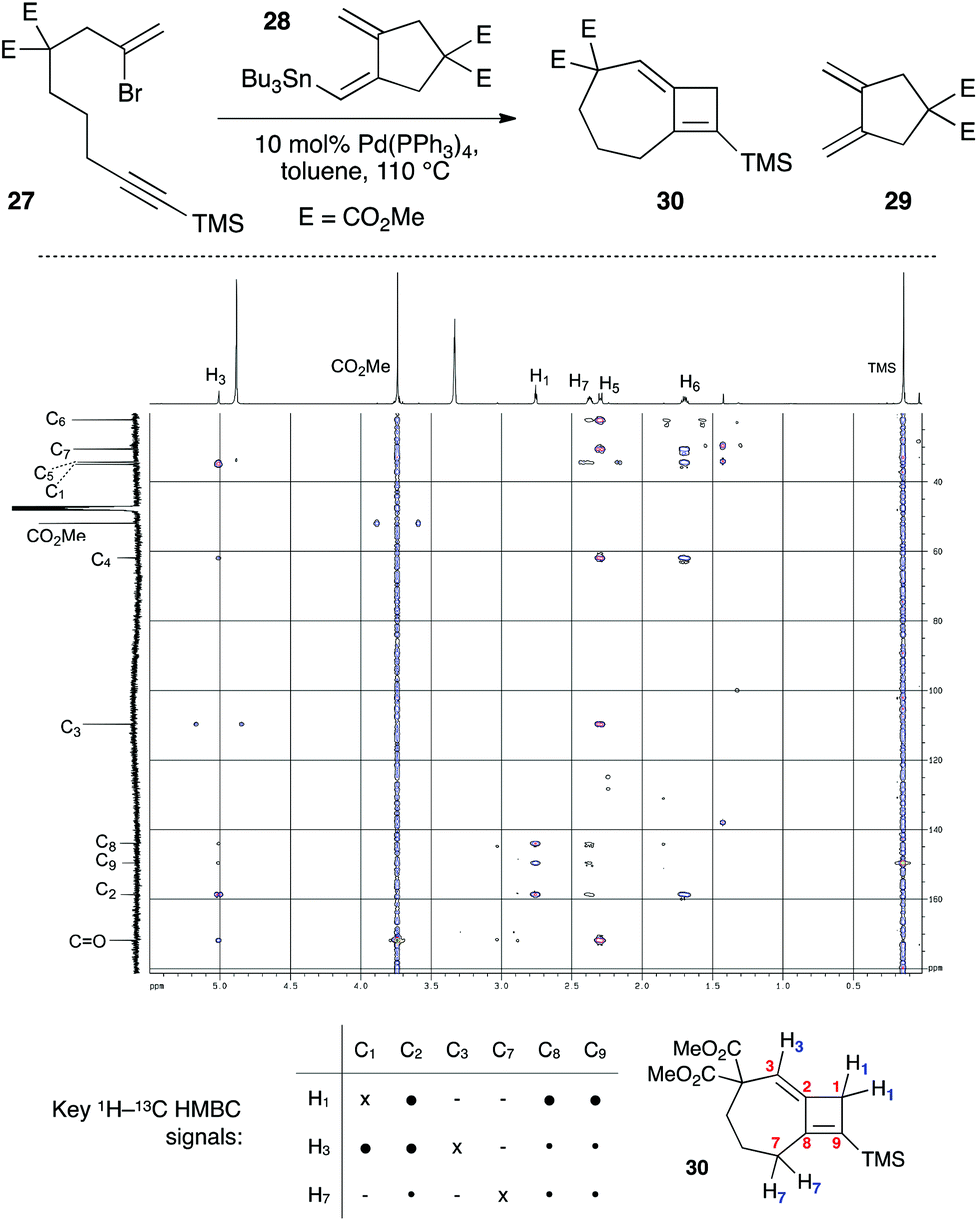
In the course of cyclization reactions to form seven-membered rings (including 11c), we had noticed the occasional formation of a different byproduct to those discussed thus far, the production of which seemed highly dependent on the reaction concentration, and quantity of stannane coupling partner. Although this byproduct was not observed under our optimized conditions, the use of <1.5 equivalents of stannane, or more dilute reaction conditions (i.e. such that transmetallation would be slowed) increased its formation. In fact, we had first observed such a species in the attempted 8π-electrocyclic coupling of bromoenyne 27 with stannane 28 (Fig. 5), which resulted in a surprising degree of apparent protodestannylation of 28 (leading to the known diene 29).22 We assigned the product formed in this reaction as the 7,4-fused cyclobutene 30 based on detailed analysis by 2D NMR experiments. Specifically, a complete set of HMBC correlations (Fig. 5) was observed between protons H1, H3 and H7, and carbons C2, C8 and C9, together with long-range coupling between H1 and H7 (5J = 2.7 Hz; this coupling was also observed in a 1H–1H COSY spectrum).
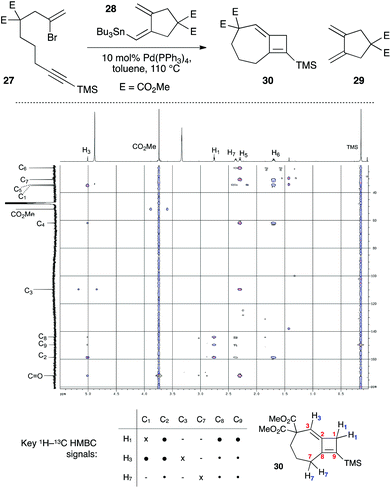 |
| | Fig. 5 Initial synthesis of 7,4-fused cyclobutene 30, and key HMBC spectra correlations. E = CO2Me. ● = strong correlation; • = weak correlation. | |
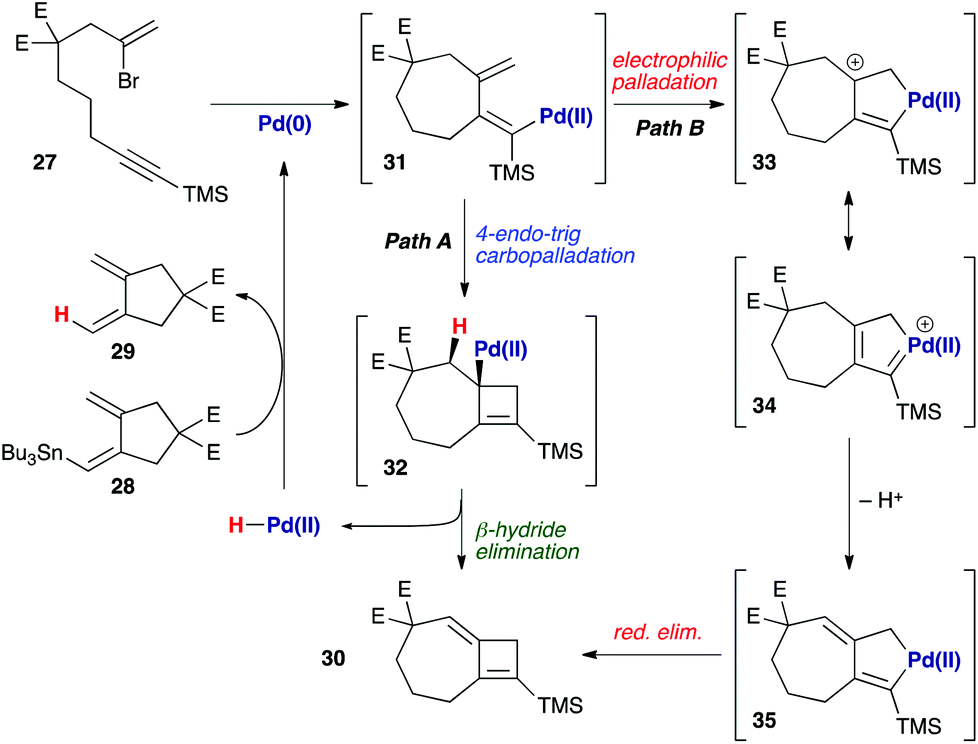
The formation of this product is not unreasonable if potential mechanisms for its formation are considered (Scheme 6). Following carbopalladation (31), one possibility would involve a 4-endo-trig carbopalladation (Path A), leading to cyclobutene 32 – a pathway that might be favoured, in spite of ring strain, due to the formation of an allylpalladium intermediate. β-Hydride elimination would afford the observed product 30, and liberate a palladium(II) hydride species that could be reduced to palladium(0) by transmetallation with 28, thus leading to the ‘protodestannylated’ product 29. However, due to a lack of precedent for this mode of carbopalladation, we also consider an electrophilic palladation route feasible (Path B), in which attack by the exo-methylene on the proximal palladium(II) atom leads to palladacycle 33 – which may again be rendered possible by the formation of an allyl cation in this process. Now, loss of a proton generates palladacyclopentene 35, reductive elimination from which leads to 30.23 The resemblance of intermediate 35 to those proposed in enyne cycloisomerization processes is clear;24 the proton lost from this pathway could then effect protodestannylation of the coupling partner 28 to afford 29.
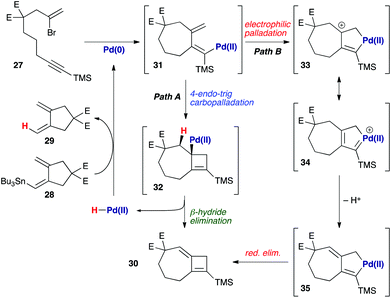 |
| | Scheme 6 Possible mechanisms for the formation of 7,4-fused cyclobutene 30. E = CO2Me. | |
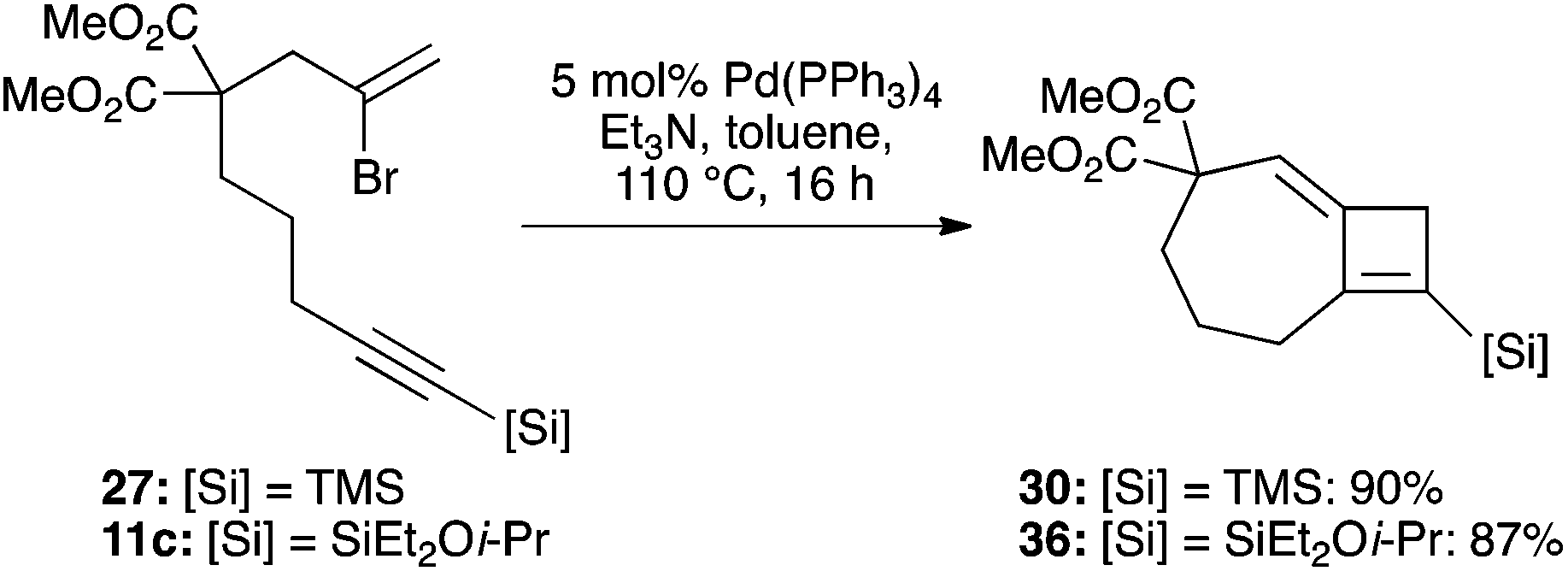
Whatever mechanism is operational, it was clear to us that this process overall corresponds to a Heck reaction in which regeneration of the palladium(0) catalyst is mediated by the stannane reagent. This suggested that an amine base might perform a similar role, and to our delight, the use of common Heck conditions (toluene, triethylamine) at just 5 mol% catalyst loading indeed led to a high-yielding cyclization of 27 to 30 (90%, Scheme 7). The more functional bromoenyne 11c was also tested in this chemistry, which gave the corresponding 7,4-fused silylcyclobutene 36 in excellent yield (86%). This efficient process offers an alternative entry to this type of fused cyclobutene ring system.25
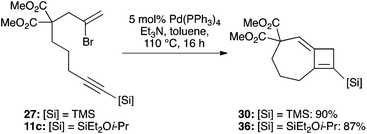 |
| | Scheme 7 Heck conditions for formation of 7,4-fused cyclobutenes. | |
Oxidations of silylcyclohexadienes
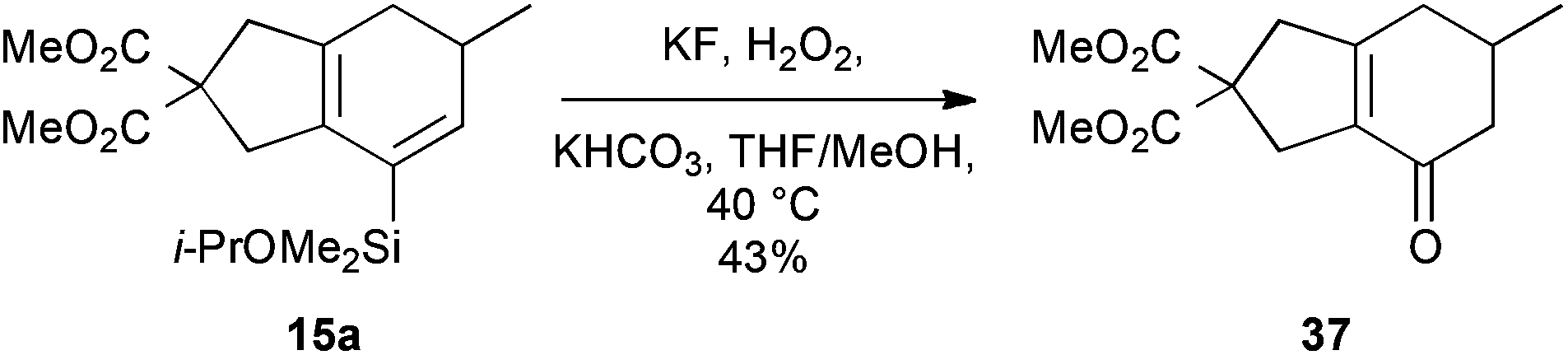
To illustrate the potential utility of this chemistry, we subjected the product dienylsilanes to a selection of oxidative transformations. Firstly, a direct Tamao oxidation of the dienylsilane was carried out,26 which we hoped would deliver a bicyclic enone. In the case of the isopropoxydimethylsilane 15a, this met with some success (Scheme 8), delivering 37 in moderate yield (43%). The successful formation of this product, which lacks any olefinic protons, could be confirmed by 13C NMR (δC 194.9, 158.7, 134.8 ppm for the enone region) and IR spectroscopy (νmax 1667 cm−1). Attempted oxidation of the equivalent diethylisopropoxysilanes proved unsuccessful, potentially due to competing nucleophilic epoxidation of the product enone, which highlights a reactivity benefit of the less-hindered dimethylalkoxysilane group.
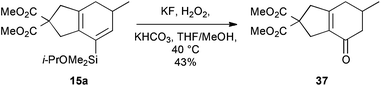 |
| | Scheme 8 Bicyclic enone formation. | |
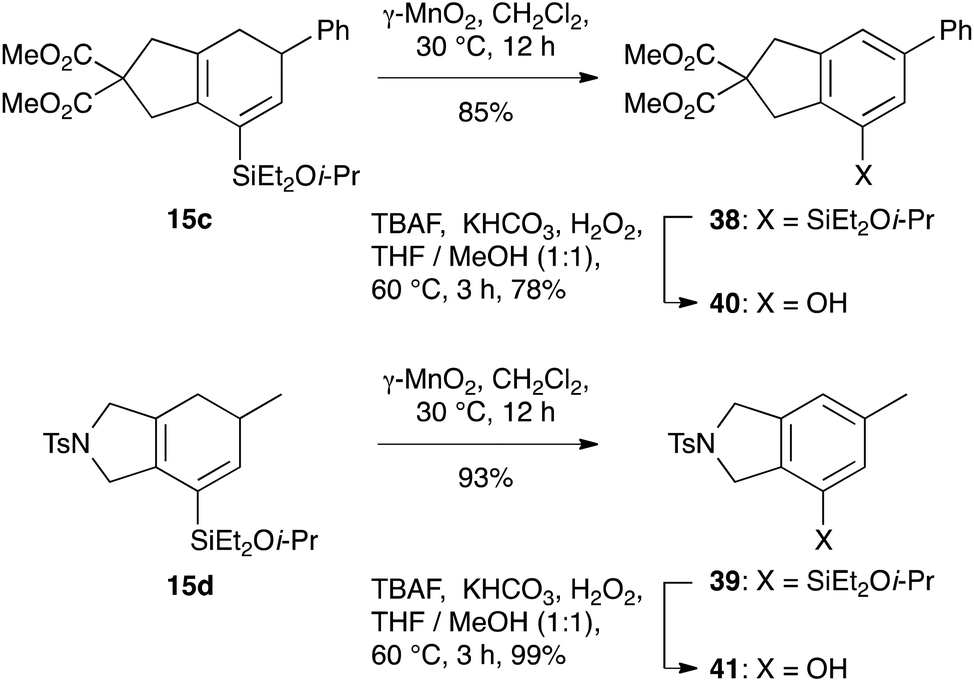
The silylcyclohexadiene frameworks could also be readily oxidized to the corresponding arylsilanes using manganese dioxide, conditions that we had successfully employed in other work27 and which proved superior to the use of other oxidants such as DDQ. The resultant arylsilanes could generally be isolated in excellent yield; two examples are shown in Scheme 9 (38: 88%; 39: 93%). These arylsilanes show potential for a range of transformations – but here, in keeping with our interests in the synthesis of phenols from arylsilanes,6 we chose to investigate Tamao oxidation. Under Tamao conditions (TBAF, KHCO3, H2O2, 60 °C),28 good yields of the corresponding phenols 40 and 41 were obtained, thus validating this approach to the synthesis of bi/polycyclic phenols.
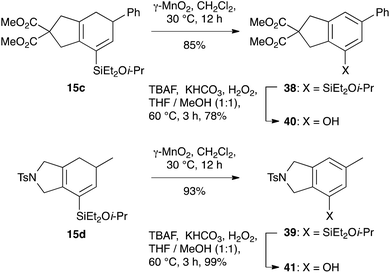 |
| | Scheme 9 Oxidation of the silylcyclohexadiene framework. | |
Conclusions
In summary, we have developed efficient routes for the synthesis of functional alkynylsilanes and demonstrated their application in bicyclization cascade reactions. The resultant silylated bicyclic cyclohexadienes are substrates for oxidation to bicyclic enones, arylsilanes, and phenols; the latter process thus affords bicyclic phenols from acyclic precursors in just three steps. Investigations into the mechanism of the cascade cyclization have revealed some unusual palladium-mediated isomerization pathways. Finally, 7,4-fused cyclobutene ring systems, arising from cyclization of a 7-membered exocyclic dienylpalladium complex, could be formed under standard ‘Heck’ type conditions. Together, these processes underline the rich reactivity – both expected and unexpected – that can be harvested from carbopalladation chemistry.
Experimental
General
Reagents were used as purchased, or purified by standard laboratory techniques. Reaction solvents were purified using an alumina column drying system, and reactions were performed under inert atmosphere unless otherwise stated.
Dimethyl 2-(2-bromoallyl)malonate.
To a suspension of NaH (60% dispersion in mineral oil, 2.32 g, 58.0 mmol) in THF (50 mL) at 0 °C was added dropwise dimethyl malonate (5.52 mL, 48.4 mmol), and the mixture was stirred for 1 h. 2,3-dibromopropene (80% tech. grade, 5.52 mL, 48.4 mmol) was added to the milky yellow mixture, and stirring was continued for 3 h until completion of the reaction as monitored by TLC. The reaction was quenched with NH4Cl (sat., aq.). The aqueous phase was extracted with Et2O and the combined organic extracts washed with brine and dried (MgSO4). The solvent was evaporated in vacuo and the crude product purified by distillation at (101–106 °C, 1 mmHg) to afford the product as a colourless oil (8.50 g, 33.9 mmol, 70%). Rf 0.22 (5:1 pet. ether–ether); 1H NMR (500 MHz, CDCl3) δH 5.69 (1H, dt, J = 2.0, 1.0 Hz), 5.48 (1H, d, J = 2.0 Hz), 3.89 (1H, t, J = 7.5 Hz), 3.76 (6H, s), 3.03 (2H, dd, J = 7.5, 1.0 Hz); 13C NMR (125 MHz, CDCl3) δC 168.7, 129.2, 119.9, 52.7, 50.4, 40.5. Data in accordance with the literature.29
General procedure 1: alkylation of dimethyl 2-(2-bromoallyl)malonate
A solution of dimethyl 2-(2-bromoallyl)malonate (1 equiv.) in THF (2 mL mmol−1) was added to a stirred suspension of NaH (60% dispersion in mineral oil, 1.2 equiv.) and the alkyne electrophile (1.2 equiv.) in THF (2 mL mmol−1). The resulting mixture was stirred at room temperature under argon until completion of the reaction as monitored by TLC. The reaction was quenched by addition of NH4Cl (sat., aq.). The aqueous layer was extracted with Et2O and the combined organic extracts washed with brine, and dried (MgSO4). The solvent was evaporated in vacuo and the crude product purified by flash chromatography to afford the bromoenyne product.
Dimethyl 2-(2-bromoallyl)-2-(prop-2-ynyl)malonate (9a).
Synthesised from dimethyl 2-(2-bromoallyl)malonate (2.90 g, 11.7 mmol) and propargyl bromide (1.60 mL, 14.0 mmol) using general procedure 1. The crude product was purified by flash chromatography (5:1 pet. ether–ether) to afford 9a as a colourless oil (2.91 g, 10.1 mmol, 86%). Rf 0.31 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 3289, 2954, 1737; 1H NMR (500 MHz, CDCl3) δH 5.83 (1H, t, J = 0.6 Hz), 5.63 (1H, d, J = 1.6 Hz), 3.77 (6H, s), 3.31 (2H, br s), 2.93 (2H, d, J = 2.7 Hz), 2.05 (1H, t, J = 2.7 Hz); 13C NMR (125 MHz, CDCl3) δC 169.5, 126.1, 122.9, 78.6, 72.1, 56.0, 53.0, 42.9, 22.2; HRMS (ES+) calcd for C11H13BrO4 [M + H]+ 289.0074, found 289.0070. Data in accordance with the literature.8a
Dimethyl 2-(2-bromoallyl)-2-(but-3-ynyl)malonate (9b).
Synthesised from dimethyl 2-(2-bromoallyl)malonate (876 mg, 3.49 mmol) and but-3-ynyl trifluoromethanesulfonate (847 mg, 4.19 mmol) using general procedure 1. The crude product was purified by flash chromatography (5:1 pet. ether–ether) to afford 9b as a colourless oil (730 mg, 2.41 mmol, 69%). Rf 0.26 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 3285, 2954, 1730, 1624, 1433, 1201, 1150, 959, 649; 1H NMR (500 MHz, CDCl3) δH 5.70–5.68 (1H, m), 5.61 (1H, d, J = 1.8 Hz), 3.75 (6H, s), 3.19 (2H, d, J = 0.5 Hz), 2.30–2.28 (2H, m), 2.21–2.19 (2H, m), 1.97 (1H, t, J = 2.8 Hz); 13C NMR (125 MHz, CDCl3) δC 170.4, 126.7, 122.2, 82.8, 68.9, 56.4, 52.8, 43.3, 30.7, 14.0; HRMS (ES+) calcd for C12H15BrNaO4 [M + Na]+ 325.0070, found 325.0062.
Dimethyl 2-(2-bromoallyl)-2-(pent-4-ynyl)malonate (9c).
Synthesised from dimethyl 2-(2-bromoallyl)malonate (200 mg, 0.796 mmol) and pent-4-ynyl trifluoromethanesulfonate (207 mg, 0.957 mmol) using general procedure 1. The crude product was purified by flash chromatography (5:1 pet. ether–ether) to afford 9c as a colourless oil (227 mg, 0.716 mmol, 90%). Rf 0.26 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 3291, 2952, 1731, 1625, 1434, 1278, 1150, 901, 640; 1H NMR (500 MHz, CDCl3) δH 5.69–5.67 (1H, m), 5.59 (1H, d, J = 1.7 Hz), 3.75 (6H, s), 3.17 (2H, s), 2.21 (2H, dt, J = 7.0, 2.6 Hz), 2.12–2.10 (2H, m), 1.96 (1H, t, J = 2.6 Hz), 1.44–1.42 (2H, m); 13C NMR (125 MHz, CDCl3) δC 170.8, 126.9, 121.9, 83.5, 68.8, 56.8, 52.7, 43.1, 30.8, 23.4, 18.5; HRMS (ES+) calcd for C13H17BrNaO4 [M + Na]+ 339.0202, found 339.0200.
Dimethyl 2-(2-bromoallyl)-2-(3-(isopropoxydimethylsilyl)prop-2-ynyl)malonate (10a).
Method A. To a solution of 9a (150 mg, 0.519 mmol) in dry THF (5 mL) at −78 °C was added n-BuLi (1.6 M in hexanes, 405 μL, 0.649 mmol, 1.25 equiv.), and the mixture was stirred for 20 min. Chloro(diethylamino)dimethylsilane (130 μL, 0.649 mmol, 1.25 equiv.) was added, and the reaction mixture was allowed to warm to rt. When complete as judged by TLC, the reaction was concentrated, diluted with dry pentane, and stirred for 15 min, then filtered through oven-dried Celite. The clear filtrate was evaporated to give the intermediate aminosilane. This was dissolved in CH2Cl2 (3 mL) and cooled to 0 °C, then imidazole (71 mg, 1.04 mmol, 2 equiv.), isopropanol (80 μL, 1.04 mmol, 2 equiv.) and DMAP (cat.) were added to the mixture, which was stirred at rt for 2 h. The reaction was quenched with NaHCO3 (aq., sat.), the aqueous layer was extracted with CH2Cl2, and the combined organic extracts were washed with brine, and dried (MgSO4). The solvent was evaporated in vacuo and the product was purified by flash chromatography (10:1 pet. ether–ether with 1% Et3N) to afford the 10a as a colourless oil (158 mg, 0.39 mmol, 75%). Rf 0.38 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2360, 1658, 1568, 1382, 1369, 1290, 1030; 1H NMR (400 MHz, CDCl3) δH 5.83–5.81 (1H, m), 5.63 (1H, d, J = 1.5 Hz), 4.09 (1H, septet, J = 6.1 Hz), 3.76 (6H, s), 3.30 (2H, s), 2.97 (2H, s), 1.18 (6H, d, J = 6.1 Hz), 0.22 (6H, s); 13C NMR (125 MHz, CDCl3) δC 169.4, 126.2, 122.7, 100.8, 87.4, 65.9, 56.0, 53.0, 43.0, 25.4, 23.4, 0.6; HRMS (ES+) calcd for C16H25BrNaO5Si [M + Na]+ 427.0533, found 427.0547.
Dimethyl 2-(2-bromoallyl)-2-(3-(isopropoxydimethylsilyl)prop-2-ynyl)malonate (10a).
Method B. To a solution of 9a (200 mg, 0.691 mmol) in THF (2 mL) at −78 °C was added LiHMDS (1 M in hexanes, 830 μL, 0.83 mmol) and the mixture was stirred for 30 min. ClSiMe2H (116 μL, 1.038 mmol) was then added, and the reaction was allowed to warm to rt, then stirred until completion as monitored by TLC. The reaction was quenched with H2O, and the aqueous layer was extracted with ether. The combined organic extracts washed with brine and dried (MgSO4). The solvent was evaporated in vacuo and the crude product was purified by flash chromatography (10:1 pet. ether–ether), to afford the intermediate alkynyl hydrosilane as a colourless oil (224 mg, 0.645 mmol, 93%). 1H NMR (400 MHz, CDCl3) δH 5.65–5.58 (1H, m), 5.43 (1H, d, J = 1.5 Hz), 3.87 (1H, septet, J = 3.7 Hz), 3.56 (6H, s), 3.10 (2H, s), 2.76 (2H, s), 0.01 (6H, d, J = 3.7 Hz); 13C NMR (100 MHz, CDCl3) δC 169.4, 126.1, 122.8, 102.5, 86.0, 56.0, 53.0, 42.9, 23.6, −3.0; HRMS (ES+) calcd for C13H19BrNaO4Si [M + Na]+ 369.0128, found 369.0128. To a solution of this hydrosilane (30 mg, 0.086 mmol) in i-PrOH (100 μL) was added [RuCl2(p-cymene)]2 (0.3 mg, 0.0004 mmol). The reaction was stirred at rt for 10 min, then concentrated. Pentane (1 mL) was added, and the crude product was filtered through Celite (ether eluent). Concentration of the filtrate gave 10a as a pale pink oil (35 mg, 0.086 mmol, 99%).
(Diethylamino)diethylchlorosilane.
To a solution of dichlorodiethylsilane (20 mL, 134 mmol) and anhydrous triethylamine (20.6 mL, 147 mmol) in THF at 0 °C was added a solution of anhydrous diethylamine (13.8 mL, 137 mmol) in THF (6 mL) over 2 h. The resulting beige suspension was warmed to rt overnight with vigorous stirring maintained. The mixture was then filtered through Celite under N2, the filter cake washed with anhydrous pentane, and the filtrate concentrated in vacuo to give an orange oil. This residue was purified by vacuum distillation (bp 92–94 °C, 36 mbar) to afford (diethylamino)diethylchlorosilane as a colourless oil (17.6 g, 90.8 mmol, 68%). 1H NMR (250 MHz, CDCl3) δH 2.88 (4H, q, J = 7.0 Hz), 1.07–0.99 (12H, m), 0.90 (4H, q, J = 6.5 Hz). Data in accordance with literature.30
General procedure 2: alkyne silylation with (diethylamino)diethylchlorosilane
To a solution of bromoenyne (1 equiv.) in THF (4.5 mL mmol−1) at −78 °C under Ar was added LiHMDS (1 M in THF, 1.5 equiv.). The pale yellow solution was stirred at −78 °C for 20 min and (diethylamino)diethylchlorosilane (1.5 equiv.) was added dropwise to the reaction mixture. After stirring for 30 min, after which time the starting material had been consumed (as monitored by TLC), i-PrOH (4 equiv.), DMAP (cat.) and imidazole (4 equiv.) were added and the reaction was allowed to warm to rt. The reaction was quenched by addition of water, and the aqueous layer was extracted with ether. The combined organic extracts were washed with brine, dried (MgSO4), and concentrated. The residue was purified by flash chromatography to afford the silylated bromoenyne.
Dimethyl 2-(2-bromoallyl)-2-(3-(diethyl(isopropoxy)silyl)prop-2-ynyl)malonate (11a).
Synthesised from 9a (500 mg, 1.73 mmol), LiHMDS (2.60 mL, 2.60 mmol), ClSiEt2(NEt2) (606 μL, 2.60 mmol), i-PrOH (530 μL, 6.90 mmol) and imidazole (471 mg, 6.92 mmol) using general procedure 2. The product was purified by flash chromatography (5:1 pet. ether–ether) to afford 11a as a colourless oil (701 mg, 1.62 mmol, 94%). Rf 0.26 (5:1 pet. ether–ether); Anal. calcd for C18H29BrO5Si: C, 49.88; H, 6.74, found: C, 49.96; H, 6.72; IR (thin film) νmax/cm−1 2956, 2178, 1742, 1626, 1025, 733; 1H NMR (500 MHz, CDCl3) δH 5.84–5.82 (1H, m), 5.63 (1H, d, J = 1.7 Hz), 4.10 (1H, septet, J = 6.1 Hz), 3.76 (6H, s), 3.32 (2H, s), 3.00 (2H, s), 1.18 (6H, d, J = 6.1 Hz), 0.98 (6H, t, J = 7.9 Hz), 0.62 (4H, app. qd, J = 7.9 Hz, 2.2 Hz); 13C NMR (125 MHz, CDCl3) δC 169.3, 126.2, 122.7, 101.6, 85.5, 66.0, 56.1, 53.0, 42.9, 25.4, 23.5, 6.8, 6.6; HRMS (ES+) calcd for C18H29NaBrO5Si [M + Na]+ 455.0860, found 455.0862.
Dimethyl 2-(2-bromoallyl)-2-(4-(diethyl(isopropoxy)silyl)but-3-ynyl)malonate (11b).
Synthesised from 9b (545 mg, 1.80 mmol), LiHMDS (2.70 mL, 2.70 mmol), ClSiEt2(NEt2) (634 μL, 2.70 mmol), i-PrOH (550 μL, 7.20 mmol) and imidazole (490 mg, 7.20 mmol) using general procedure 2. The product was purified by flash chromatography (5:1 pet. ether–ether) to afford 11b as a colourless oil (723 mg, 1.62 mmol, 90%); Rf 0.26 (5:1 Pet. ether–ether); Anal. calcd for C19H31BrO5Si: C, 51.00; H, 6.98, found: C, 50.92, H; 7.02; IR (thin film) νmax/cm−1 2956, 2173, 1735, 1625, 1435, 1088, 1027, 732; 1H NMR (500 MHz, CDCl3) δH 5.84–5.82 (1H, m), 5.63 (1H, d, J = 1.7 Hz), 4.10 (1H, septet, J = 6.1 Hz), 3.76 (6H, s), 3.18 (2H, s), 2.31–2.29 (2H, m), 2.23–2.24 (2H, m), 1.18 (6H, d, J = 6.1 Hz), 0.98 (6H, t, J = 7.9 Hz), 0.62 (4H, app. qd, J = 7.9 Hz, 2.2 Hz); 13C NMR (125 MHz, CDCl3) δC 170.4, 126.6, 122.2, 106.0, 81.9, 65.9, 56.5, 52.8, 43.4, 30.9, 25.5, 15.3, 6.8, 6.5; HRMS (ES+) calcd for C19H31BrNaO5Si [M + Na]+ 469.1016, found 469.1015.
Dimethyl 2-(2-bromoallyl)-2-(5-(diethyl(isopropoxy)silyl)pent-4-ynyl)malonate 11c).
Synthesised from 9c (320 mg, 1.01 mmol), LiHMDS (1.50 mL, 1.50 mmol), ClSiEt2(NEt2) (355 μL, 1.50 mmol), i-PrOH (306 μL, 4.00 mmol) and imidazole (272 mg, 4.00 mmol) using general procedure 2. The product was purified by flash chromatography (5:1 pet. ether–ether) to afford 11c as a colourless oil (337 mg, 0.730 mmol, 72%); Rf 0.26 (5:1 pet. ether–ether); Anal. calcd for C20H33BrO5Si: C, 52.05; H, 7.21, found: C, 51.94; H, 7.30; IR (thin film) νmax/cm−1 2955, 2172, 1735, 1625, 1434, 1278, 1026, 732; 1H NMR (500 MHz, CDCl3) δH 5.68–5.66 (1H, m), 5.59 (1H, d, J = 1.5 Hz), 4.14 (1H, septet, J = 6.4 Hz), 3.75 (6H, s), 3.18 (2H, s), 2.29 (2H, t, J = 7.3 Hz), 2.14–2.12 (2H, m), 1.49–1.47 (2H, m), 1.20 (6H, d, J = 3.4 Hz), 1.00 (6H, t, J = 6.4 Hz), 0.65–0.62 (4H, m); 13C NMR (125 MHz, CDCl3) δC 170.8, 127.0, 121.9, 106.8, 81.8, 65.9, 56.9, 52.9, 43.2, 30.9, 25.5, 23.4, 19.9, 6.9, 6.6; HRMS (ES+) calcd for C20H33BrNaO5Si [M + Na]+ 483.1173, found 483.1179.
N-(2-Bromoallyl)-4-methyl-N-(prop-2-ynyl)benzene sulfonamide.
To a mixture of N-(2-bromoallyl)-4-methylbenzenesulfonamide (1.00 g, 3.45 mmol) and K2CO3 (953 mg, 6.89 mmol) in acetone (10 mL) was added propargyl bromide (80% in toluene, 460 μL, 4.14 mmol). The reaction mixture was stirred at 60 °C for 12 h, then it was cooled to rt and quenched with water. The aqueous layer was extracted with Et2O, then the combined organic layers were dried (MgSO4) and concentrated in vacuo. The crude product was purified by flash chromatography (2:1 pet. ether–ether) to afford the title compound as a colourless oil (903 mg, 2.75 mmol, 80%); Rf 0.35 (2:1 pet. ether–ether); IR (thin film) νmax/cm−1 3280, 1629, 1344, 1181, 1150, 892, 679; 1H NMR (500 MHz, CDCl3) δH 7.75 (2H, d, J = 8.1 Hz), 7.31 (2H, d, J = 8.1 Hz), 5.97–5.95 (1H, m), 5.69–5.67 (1H, m), 4.13 (2H, d, J = 2.3 Hz), 4.08 (2H, s), 2.44 (3H, s), 2.05 (1H, t, J = 2.3 Hz); 13C NMR (125 MHz, CDCl3) δC 143.9, 135.8, 129.6, 127.7, 126.9, 120.5, 76.0, 74.2, 53.8, 36.1, 21.6; HRMS (ES+) calcd for C14H18BrNNaO3S [M + MeOH + Na]+ 382.0083, found 382.0077.
N-(2-Bromoallyl)-N-(3-(diethyl(isopropoxy)silyl)prop-2-ynyl)-4-methylbenzene sulfonamide (12).
Synthesised from N-(2-bromoallyl)-4-methyl-N-(prop-2-ynyl)benzene sulfonamide (150 mg, 0.457 mmol), LiHMDS (686 μL, 0.686 mmol), ClSiEt2(NEt2) (160 μL, 0.686 mmol), i-PrOH (140 μL, 1.80 mmol) and imidazole (125 mg, 1.84 mmol) using general procedure 2. The product was purified by flash chromatography (5:1 pet. ether–ether) to afford 12 as a colourless oil (107 mg, 0.226 mmol, 50%); Rf 0.26 (5:1 pet. ether–ether); Anal. calcd for C20H30BrNO3SSi: C, 50.84; H, 6.40; N, 2.96, found: C, 50.90; H, 6.32; N, 2.95. IR (thin film) νmax/cm−1 2960, 1353, 1163, 1119, 1092, 1002, 899, 734, 661; 1H NMR (400 MHz, CDCl3) δH 7.77 (2H, d, J = 8.1 Hz), 7.33 (2H, d, J = 8.1 Hz), 5.98 (1H, d, J = 1.8 Hz), 5.72 (1H, d, J = 1.8 Hz), 4.23 (2H, s), 4.11 (2H, s), 3.95 (1H, sept, J = 6.0 Hz), 2.45 (3H, s), 1.14 (6H, d, J = 6.0 Hz), 0.89 (6H, t, J = 7.8 Hz), 0.51 (4H, q, J = 7.8 Hz); 13C NMR (125 MHz, CDCl3) δC 144.0, 136.3, 130.4, 130.1, 128.1, 119.7, 98.9, 66.5, 45.4, 41.0, 38.6, 26.2, 25.8, 7.8, 7.0; HRMS (ES+) calcd for C20H30BrNNaO3SSi [M + Na]+ 494.0791, found 494.0789.
2-Bromo-3-(prop-2-ynyloxy)prop-1-ene.
To a solution of NaH (60% in mineral oil, 1.50 g, 35.8 mmol) in THF (10 mL) at 0 °C was added propargyl alcohol (1.00 mL, 17.0 mmol) and the reaction mixture was stirred for 30 min. 2,3-Dibromopropene (1.50 mL, 14.3 mmol) was slowly added, then the reaction mixture was allowed to warm to rt and stirred for 12 h. The reaction was quenched by addition of water and the aqueous layer was extracted with Et2O; the combined organic extracts were washed with brine and dried (MgSO4). The solvent was evaporated in vacuo and the crude product purified by flash chromatography (5:1 pet. ether–ether) to afford the 2-bromo-3-(prop-2-ynyloxy)prop-1-ene as a colourless oil (2.15 g, 12.3 mmol, 86%); Rf 0.26 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 3300, 2923, 2130, 1639, 1443, 1l62, 1086, 899, 668; 1H NMR (400 MHz, CDCl3) δH 5.97–5.95 (1H, m), 5.67–5.65 (1H, m), 4.23–4.19 (4H, m), 2.47 (1H, t, J = 2.4 Hz); 13C NMR (125 MHz, CDCl3) δC 128.4, 118.7, 78.8, 75.1, 73.3, 57.0; HRMS (ES+) calcd for C6H7BrO [M + H]+ 173.9681, found 173.9679. Data in accordance with the literature.31
(3-(2-Bromoallyloxy)prop-1-ynyl)diethyl(isopropoxy)silane (13).
Synthesised from 2-bromo-3-(prop-2-ynyloxy) prop-1-ene (500 mg, 2.86 mmol), LiHMDS (4.30 mL, 4.30 mmol), ClSiEt2(NEt2) (1.00 mL, 4.30 mmol), i-PrOH (875 μL, 11.4 mmol) and imidazole (778 mg, 11.4 mmol) using general procedure 2. The product was purified by flash chromatography (5:1 pet. ether–ether) to afford 13 as a colourless oil (537 mg, 1.68 mmol, 59%); Rf 0.26 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2969, 1461, 1380, 1240, 1122, 1088, 878, 734; 1H NMR (400 MHz, CDCl3) δH 5.93 (1H, d, J = 1.7 Hz), 5.64–5.62 (1H, m), 4.23 (2H, s), 4.21 (2H, s), 4.12 (1H, septet, J = 6.1 Hz), 1.17 (6H, d, J = 6.1 Hz), 0.96 (6H, t, J = 7.8 Hz), 0.58 (4H, q, J = 7.9 Hz); 13C NMR (125 MHz, CDCl3) δC 128.7, 118.8, 101.3, 89.1, 73.3, 66.3, 57.9, 25.7, 6.8, 6.7; HRMS (ES+) calcd for C13H23BrNaO2Si [M + Na]+ 341.0543, found 341.0531.
General procedure 3: cyclisation of bromoenynes to silylcyclohexyldienes
PdCl2(PPh3)2 (0.1 equiv.) was added to a degassed solution of bromoenyne (1 equiv.) and vinyl stannane (1.5–1.7 equiv.) in toluene (20 mL mmol−1) under argon. The reaction mixture was heated to reflux (110 °C) until completion as monitored by TLC and/or 1H NMR spectroscopic analysis of an aliquot. After concentration, the crude mixture was purified by flash chromatography to obtain the bicyclic diene.
Dimethyl 7-(isopropoxydimethylsilyl)-5-methyl-4,5-dihydro-1H-indene-2,2(3H)-dicarboxylate (15a).
Synthesised from 10a (23.9 mg, 0.0590 mmol) and (E)-tributyl(prop-1-enyl)stannane 3a (29.3 mg, 0.0885 mmol) using general procedure 3. The product was purified by flash chromatography (5:1 pet. ether–ether) to afford 15a as a colourless oil (20 mg, 0.0546 mmol, 92%); Rf 0.33 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2358, 1737, 1252, 1024, 890, 822, 781; 1H NMR (500 MHz, CDCl3) δH 5.95 (1H, d, J = 3.3 Hz, H6), 3.95 (1H, septet, J = 6.3 Hz, CHMe2), 3.74 (3H, s, CO2Me), 3.74 (3H, s, CO2Me), 3.67–3.65 (1H, m, H5), 3.20 (2H, br s, H3), 3.03–3.01 (1H, m, H4), 2.96–2.94 (1H, m, H′4), 2.46–2.44 (1H, m, H3), 2.20–2.18 (1H, m, H′3), 1.13 (3H, d, J = 6.3 Hz, CHMe2), 1.12 (3H, d, J = 6.3 Hz, CHMe2), 1.04 (3H, d, J = 7.1 Hz, Me), 0.23 (6H, s, SiMe2); 13C NMR (125 MHz, CDCl3) δC 172.8, 172.8, 143.9, 132.1, 131.3, 131.1, 65.1, 58.5, 52.7, 52.7, 43.0, 41.7, 31.1, 30.0, 25.6, 20.1, −0.90; HRMS (ES+) calcd for C19H30NaO5Si [M + Na]+ 389.1743, found 389.1755.
Dimethyl 7-(isopropoxydimethylsilyl)-5-phenyl-4,5-dihydro-1H-indene-2,2(3H)-dicarboxylate (15b).
Synthesised from 10a (26.6 mg, 0.0656 mmol) and (E)-tributyl(styryl)stannane 3b (38.8 mg, 0.0987 mmol) using general procedure 3. The product was purified by flash chromatography (5:1 pet. ether–ether) to afford 15b as a colourless oil (27.4 mg, 0.0639 mmol, 97%); Rf 0.30 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2391, 1737, 1434, 1367, 1252, 1023, 782; 1H NMR (500 MHz, CDCl3) δH 7.30 (2H, m, ArH), 7.24 (3H, m, ArH), 6.10 (1H, d, J = 3.1 Hz, H6), 3.99 (1H, septet, J = 6.2 Hz, CHMe2), 3.76 (3H, s, CO2Me), 3.74 (3H, s, CO2Me), 3.65–3.63 (1H, m, H5), 3.28–3.26 (2H, m, H1), 3.10 (1H, d, J = 17.4 Hz, H4), 2.94 (1H, d, J = 17.4 Hz, H′4), 2.45–2.43 (1H, m, H3), 2.29–2.27 (1H, m, H′3), 1.16 (3H, d, J = 6.2 Hz, CHMe2), 1.14 (3H, d, J = 6.2 Hz, CHMe2), 0.25 (6H, s, SiMe2); 13C NMR (125 MHz, CDCl3) δC 172.7, 145.4, 140.7, 133.4, 131.7, 131.1, 128.5, 127.6, 126.3, 65.2, 58.5, 52.7, 42.9, 41.2, 41.7, 32.0, 25.6, −0.9; HRMS (ES+) calcd for C24H32NaO5Si [M + Na]+ 451.1892, found 451.1911.
Dimethyl 7-(diethyl(isopropoxy)silyl)-5-phenyl-4,5-dihydro-1H-indene-2,2(3H)-dicarboxylate (15c).
Synthesised from 11a (300 mg, 0.670 mmol) and (E)-tributyl(styryl)stannane 3b (435 mg, 1.11 mmol) using general procedure 3. The crude product was purified by flash chromatography (5:1 pet. ether–ether) to afford 15c as a colourless oil (237 mg, 0.519 mmol, 77%); Rf 0.42 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2955, 1736, 1434, 1381, 1243, 1005, 874, 730, 699; 1H NMR (400 MHz, CDCl3) δH 7.32–7.30 (2H, m, ArH), 7.27–7.23 (3H, m, ArH), 6.12–6.10 (1H, m, H6), 4.01 (1H, septet, J = 6.2 Hz, CHMe2), 3.76 (3H, s, CO2Me), 3.73 (3H, s, CO2Me), 3.64–3.62 (1H, m, H5), 3.25 (2H, br s, H3), 3.10 (1H, d, J = 17.5 Hz, H1), 2.94 (1H, d, J = 17.5 Hz, H′1), 2.45–2.43 (1H, m, H4), 2.27–2.25 (1H, m, H′4), 1.16 (3H, d, J = 6.2 Hz, CHMe2), 1.15 (3H, d, J = 6.2 Hz, CHMe2), 1.00–0.95 (6H, m, SiCH2Me), 0.77–0.74 (4H, m, SiCH2Me); 13C NMR (125 MHz, CDCl3) δC 172.8, 172.7, 145.5, 141.3, 131.8, 131.5, 130.9, 128.5, 127.6, 126.3, 65.2, 58.5, 52.7, 52.7, 42.9, 42.2, 41.8, 32.1, 25.7, 25.6, 6.8, 5.0, 4.9; HRMS (ES+) calcd for C27H40NaO6Si [M + MeOH + Na]+ 511.2486, found 511.2432.
7-(Diethyl(isopropoxy)silyl)-5-methyl-2-tosyl-2,3,4,5-tetrahydro-1H-isoindole (15d).
Synthesised from 12 (50.0 mg, 0.106 mmol) and (E)-tributyl(prop-1-enyl)stannane 3a (60.0 mg, 0.181 mmol) using general procedure 3. The product was purified by flash chromatography (5:1 pet. ether–ether) to afford 15d as a colourless oil (29.0 mg, 0.0670 mmol, 63%); Rf 0.29 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2958, 1436, 1378, 1230, 1015, 874, 730, 699; 1H NMR (500 MHz, CDCl3) δH 7.72 (2H, d, J = 8.0 Hz, ArH), 7.31 (2H, d, J = 8.0 Hz, ArH), 5.93 (1H, d, J = 3.5 Hz, H6), 4.22–4.20 (2H, m, H1), 4.11–4.00 (2H, m, H3), 3.95 (1H, septet, J = 5.9 Hz, CHMe2), 2.42 (4H, s, H5, ArMe), 2.15 (1H, dd, J = 17.0, 8.5 Hz, H5), 1.81–1.73 (1H, m, H′5), 1.13 (3H, d, J = 6.0 Hz, CHMe2), 1.11 (3H, d, J = 6.0 Hz, CHMe2), 1.02 (3H, d, J = 7.1 Hz, CHMe), 0.89 (6H, app. td, J = 8.0, 1.7 Hz, SiCH2Me), 0.68–0.65 (4H, m, SiCH2Me); 13C NMR (125 MHz, CDCl3) δC 145.1, 143.2, 134.4, 129.9, 129.6, 128.1, 127.8, 127.5, 65.3, 56.3, 55.4, 29.8, 28.7, 25.7, 25.7, 21.5, 19.9, 6.8, 5.0, 5.0; HRMS (ES+) calcd for C23H35NNaO3SSi [M + Na]+ 456.1999, found 456.2000.
Diethyl(isopropoxy)(6-methyl-1,3,6,7-tetrahydroisobenzofuran-4-yl)silane (15e).
Synthesised from 13 (50.0 mg, 0.157 mmol) and (E)-tributyl(prop-1-enyl)stannane 3a (88.1 mg, 0.266 mmol) using general procedure 3. The crude product was purified by flash chromatography (5:1 pet. ether–ether) to afford 15e as a colourless oil (10.0 mg, 0.0356 mmol, 23%); Rf 0.29 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2955, 1732, 1463, 1210, 866; 1H NMR (500 MHz, CDCl3) δH 5.99 (1H, d, J = 3.3 Hz, H5), 4.75–4.73 (2H, m, H1), 4.60–4.58 (2H, m, H3), 3.99 (1H, septet, J = 6.0 Hz, CHMe2), 2.59–2.48 (1H, m, H8), 2.26 (1H, dd, J = 17.0, 9.0 Hz, H7), 1.92–1.84 (1H, m, H7), 1.16 (3H, d, J = 6.0 Hz, CHMe2), 1.15 (3H, d, J = 6.0 Hz, CHMe2), 1.09 (3H, d, J = 7.0 Hz, CHMe), 0.94 (6H, t, J = 7.9 Hz, SiCH2Me), 0.75–0.65 (4H, m, SiCH2Me); 13C NMR (125 MHz, CDCl3) δC 144.6, 131.3, 129.7, 127.4, 76.3, 75.7, 65.2, 29.9, 27.7, 25.7, 20.1, 6.7, 4.9; HRMS (ES+) calcd for C16H28NaO2Si [M + Na]+ 303.1751, found 303.1749.
Dimethyl 5-(diethyl(isopropoxy)silyl)-7-methyl-3,4,7,8-tetrahydronaphthalene-2,2(1H)-dicarboxylate (15f).
Synthesised from 11b (100 mg, 0.223 mmol) and (E)-tributyl(prop-1-enyl)stannane 3a (126 mg, 0.381 mmol) using general procedure 3. The product was purified by flash chromatography (5:1 pet. ether–ether) to afford 15f as a colourless oil (68.0 mg, 0.166 mmol, 75%); Rf 0.52 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2956, 1735, 1435, 1250, 1170, 1121, 1083, 1018, 872, 731; 1H NMR (500 MHz, CDCl3) δH 5.99 (1H, d, J = 3.4 Hz, H6), 3.94 (1H, sept, J = 6.1 Hz, OCHMe2), 3.72 (3H, s, CO2Me), 3.71 (3H, s, CO2Me), 2.60 (2H, m, H1), 2.31–2.29 (3H, m, H4 and H7), 2.24–2.20 (1H, m, H3), 2.12 (1H, dt, J = 13.4, 6.3 Hz, H3), 1.98 (1H, dd, J = 16.0, 7.5 Hz, H8), 1.80–1.69 (1H, m, H8), 1.12 (6H, t, J = 6.1 Hz, OCHMe2), 1.01 (3H, d, J = 6.8 Hz, C11), 0.95–0.90 (6H, m, SiCH2Me), 0.75–0.72 (4H, m, SiCH2Me); 13C NMR (125 MHz, CDCl3) δC 172.4, 172.0, 144.9, 134.1, 127.8, 125.7, 65.2, 53.5, 52.7, 52.7, 36.4, 36.1, 29.5, 28.5, 25.8, 25.8, 25.2, 19.6, 7.0, 7.0, 5.7, 5.6; HRMS (ES+) calcd for C22H36NaO5Si [M + Na]+ 431.2224, found 431.2236. NMR data for 16a and 17a was obtained from a purified mixture of 16a and 17a (2.8:1 ratio).
Dimethyl (Z)-4-((E)-1-(diethyl(isopropoxy)silyl)but-2-en-1-ylidene)-3-methylenecyclohexane-1,1-dicarboxylate (16a).
1H NMR (500 MHz, CDCl3) δH 6.00 (1H, dd, J = 15.5, 1.5 Hz, H8), 5.32 (1H, dq, J = 15.5, 6.5 Hz, H9), 5.01 (1H, d, J = 2.3 Hz, H11), 4.87–4.83 (1H, m, H11), 4.00 (1H, sept, J = 6.0 Hz, CHMe2), 3.72 (6H, s, CO2Me), 2.81 (2H, s, H2), 2.58–2.52 (2H, m, H5), 2.11–2.06 (2H, m, H6), 1.74 (3H, dd, J = 6.5, 1.5 Hz, H10), 1.13 (6H, d, J = 6.0 Hz, CHMe2), 0.91 (6H, t, J = 7.5 Hz, SiCH2Me), 0.69 (4H, J = 7.5 Hz, SiCH2Me); 13C NMR (126 MHz, CDCl3) δC 171.2, 151.58, 146.1, 132.1, 132.1, 127.6, 115.1, 65.2, 57.1, 52.8, 41.2, 32.3, 30.3, 25.9, 18.7, 7.3, 6.7.
Dimethyl (E)-3-(1-(diethyl(isopropoxy)silyl)buta-1,3-dien-1-yl)-4-methylcyclopent-3-ene-1,1-dicarboxylate (17a).
1H NMR (500 MHz, CDCl3) δH 6.36 (1H, d, J = 10.6 Hz, H8), 6.31 (1 H, ddd, J = 16.5, 10.6, 9.7 Hz, H9), 5.26 (1H, dd, J = 16.5, 2.0 Hz, H10), 5.13 (1H, dd, J = 9.7, 2.0, H10), 4.00 (1H, sept, J = 6.0 Hz, CHMe2), 3.73 (6H, s, CO2Me), 2.61 (1H, d, J = 17.2 Hz, H2), 2.45 (1H, d, J = 17.2 Hz, H2), 2.24–2.18 (2H, m, H6 and H5), 2.14–2.11 (1H, m, H6), 1.87–1.82 (1H, m, H5), 1.50 (3H, s, H11), 1.14 (6H, d, J = 6.0 Hz, CHMe2), 0.91 (6H, t, J = 7.5 Hz, SiCH2Me), 0.69 (4H, J = 7.5 Hz, SiCH2Me); 13C NMR (126 MHz, CDCl3) δC 172.4, 172.2, 144.5, 140.2, 134.4, 130.5, 123.2, 118.9, 65.2, 53.9, 52.7, 52.7, 36.3, 28.3, 27.7, 26.0, 25.9, 20.3, 7.0, 6.8, 5.5, 5.3.
Dimethyl 4-(diethyl(isopropoxy)silyl)-2-methyl-5,6,7,9-tetrahydro-1H-benzo[7]annulene-8,8(2H)-dicarboxylate (15g).
Synthesised from 11c (51.4 mg, 0.111 mmol) and (E)-tributyl(prop-1-enyl)stannane 3a (55.0 mg, 0.166 mmol) using general procedure 3. The crude product was purified by flash chromatography (5:1 pet. ether–ether) to afford 15g as a colourless oil (27.5 mg, 0.0651 mmol, 58%, mixture with 16b and 17b); Rf 0.52 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2954, 1736, 1455, 1228, 1172, 1007, 873, 729; 1H NMR (500 MHz, CDCl3) δH 5.98 (1H, d, J = 3.2 Hz, H3), 3.95 (1H, septet, J = 5.9 Hz, CHMe2), 3.71 (3H, s, CO2Me), 3.70 (3H, s, CO2Me), 2.49–2.44 (2H, m, H9), 2.29–2.20 (2H, m, H5), 2.17–2.04 (3H, m, H5, H2, H1), 1.78–1.72 (1H, m, H1), 1.68–1.60 (2H, m, H7), 1.38–1.33 (2H, m, H6), 1.13 (3H, d, J = 6.0, CHMe2), 1.13 (3H, d, J = 6.0, CHMe2), 0.99 (3H, d, J = 7.0 Hz, CHMe), 0.96–0.87 (6H, m, SiCH2Me), 0.76–0.60 (4H, m, SiCH2Me); 13C NMR (125 MHz, CDCl3) δC 172.3, 172.0, 145.3, 137.8, 135.6, 129.2, 65.1, 55.0, 52.3, 52.1, 39.7, 39.1, 37.2, 31.4, 29.8, 28.6, 26.7, 25.6, 25.4, 22.7, 19.4, 17.3, 13.6, 6.9, 6.8, 5.2, 5.1; HRMS (ES+) calcd for C23H38NaO5Si [M + Na]+ 445.2381, found 445.2386.
Dimethyl 8-(trimethylsilyl)bicyclo[5.2.0]nona-1,7-diene-3,3-dicarboxylate (30).
To Pd(PPh3)4 (5.0 mg, 0.0045 mmol, 5 mol%) was added a degassed solution of bromoenyne 278a (35.0 mg, 0.0899 mmol) and Et3N (25.0 μl, 0.180 mmol) in toluene (1.2 mL) under Ar, and the mixture was refluxed in a preheated oil bath (110 °C) until completion of the reaction (16 h). The mixture was concentrated, and the residue purified by flash chromatography (10:1 pet. ether–Et2O) to obtain 30 as a colourless oil (25.0 mg, 0.0811 mmol, 90%); Rf 0.46 (2:1 pet. ether–Et2O); IR (thin film) νmax/cm−1 2954, 1736, 1248, 840; 1H NMR (500 MHz, MeOD) δH 4.99 (1H, s, H2), 3.71 (6H, s, CO2Me), 2.73 (2H, t, J = 2.9 Hz, H9), 2.38–2.32 (2H, m, H6), 2.29–2.25 (2H, m, H4), 1.67 (2H, app. qd, J = 6.0, 4.5 Hz, H5), 0.12 (9H, s, SiMe); 13C NMR (126 MHz, MeOD) δC 173.1, 159.9, 150.9, 145.3, 110.9, 63.2, 53.3, 36.4, 35.7, 31.9, 23.6, −1.7; HRMS (ES+) calcd for C16H24NaO4Si [M + Na]+ 331.1336, found 331.1340.
Dimethyl 8-(diethyl(isopropoxy)-λ4-sulfanyl)bicyclo[5.2.0]nona-1,7-diene-3,3-dicarboxylate (36).
To Pd(PPh3)4 (5.0 mg, 0.0045 mmol, 5 mol%) was added a degassed solution of bromoenyne 11c (32.0 mg, 0.0693 mmol) and Et3N (20.0 μl, 0.144 mmol) in toluene (1.2 mL) under Ar, and the mixture was heated in a preheated oil bath (110 °C) until completion of the reaction (16 h). The mixture was concentrated, and the residue was purified by flash chromatography (10:1 pet. ether–Et2O) to obtain 36 as a colourless oil (23.0 mg, 0.0604 mmol, 87%); Rf 0.24 (9:1 pet. ether–Et2O); IR (thin film) νmax/cm−1 2955, 1736, 1565, 1243, 1031; 1H NMR (500 MHz, CDCl3) δH 5.09 (1H, s, H2), 3.99 (1H, sept, J = 6.0 Hz, SiOCHMe), 3.74 (6H, s, CO2Me), 2.84 (2H, t, J = 2.9 Hz, H9), 2.39–2.35 (2H, m, H6), 2.33–2.30 (2H, m, H4), 1.71 (2H, dt, J = 10.8, 5.6 Hz, H5), 1.15 (6H, d, J = 6.0 Hz, SiOCHMe), 0.96 (6H, t, J = 7.9 Hz, SiCH2Me), 0.68 (4H, q, J = 7.9 Hz, SiCH2Me); 13C NMR (101 MHz, CDCl3) δC 171.8, 160.6, 146.9, 144.5, 110.5, 65.6, 62.0, 52.9, 36.9, 34.7, 31.5, 26.0, 22.6, 6.8, 5.7; HRMS (ESI+) calcd for C20H32NaO5Si [M + Na]+ 403.1911, found 403.1916.
Dimethyl 6-methyl-4-oxo-4,5,6,7-tetrahydro-1H-indene-2,2(3H)-dicarboxylate (37).
15a (16.8 mg, 0.0458 mmol) was dissolved in THF–MeOH (1:1, 400 μL). KF (5.3 mg, 0.090 mmol), KHCO3 (9.2 mg, 0.090 mmol), H2O2 (30% aq., 5.2 μL, 0.050 mmol) were added and the reaction mixture was heated to 40 °C for 3 h. The reaction was allowed to cool to rt, then it was diluted with CH2Cl2 and water. The organic extract was washed with water, dried (MgSO4), and concentrated. The product was purified by flash chromatography (2:1 pet. ether–ether) to afford 37 as a colourless oil (5.3 mg, 0.020 mmol, 43%); Rf 0.21 (2:1 pet. ether–ether); IR (thin film) νmax/cm−1 2349, 1734, 1667, 1434, 1262, 913, 745; 1H NMR (400 MHz, CDCl3) δH 3.75 (6H, s, CO2Me), 3.23–3.21 (4H, m, H1, H3), 2.42 (1H, dd, J = 17.0, 3.9 Hz, H5), 2.42–2.36 (1H, m, H7), 2.32–2.20 (1H, m, H6), 2.09 (1H, dd, J = 17.0, 12.0 Hz, H5), 2.07–2.00 (1H, m, H7), 1.07 (3H, d, J = 6.5 Hz, Me); 13C NMR (125 MHz, CDCl3) δC 194.9, 172.0 (2C), 158.7, 134.8, 57.8, 52.5 (2C), 45.8, 45.2, 38.6, 33.8, 31.0, 21.0; HRMS (ES+) calcd for C14H19O5 [M + H]+ 267.1227, found 267.1225.
Dimethyl 4-(diethyl(isopropoxy)silyl)-6-phenyl-1H-indene-2,2(3H)-dicarboxylate (38).
To a solution of 15c (20.0 mg, 0.0438 mmol) in CH2Cl2 (1 mL) was added MnO2 (19.0 mg, 0.220 mmol) and the reaction mixture was stirred at 30 °C for 12 h. The mixture was filtered through a pad of Celite and concentrated, and the residue was purified by flash chromatography (5:1 pet. ether–ether) to afford 38 as a colourless oil (17.0 mg, 0.0374 mmol, 85%); Rf 0.31 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2923, 2853, 1740, 1460, 699; 1H NMR (500 MHz, CDCl3) δH 7.60–7.40 (6H, m, H5, H7, ArH), 7.33 (1H, t, J = 7.3 Hz, ArH), 4.08 (1H, septet, J = 6.1 Hz, CHMe2), 3.76 (6H, s, CO2Me), 3.74 (2H, s, H1 or H3), 3.63 (2H, s, H1 or H3), 1.19 (6H, d, J = 6.1 Hz, CHMe2), 1.03–0.93 (4H, m, SiCH2Me); 13C NMR (125 MHz, CDCl3) δC 172.2, 144.6, 141.5, 139.9, 139.4, 132.7, 132.5, 128.7, 127.1, 127.0, 124.3, 65.5, 60.4, 52.9, 41.4, 40.1, 25.8, 6.8, 5.5; HRMS (ES+) calcd for C26H34NaO5Si [M + Na]+ 477.2068, found 477.2053.
4-(Diethyl(isopropoxy)silyl)-6-methyl-2-tosylisoindoline (39).
To a solution of 15d (29.0 mg, 0.0669 mmol) in CH2Cl2 (1 mL) was added MnO2 (50.0 mg, 0.460 mmol) and the reaction mixture was stirred at 30 °C for 12 h. The mixture was filtered through a pad of Celite and concentrated, and the residue was purified by flash chromatography (5:1 pet. ether–ether) to afford 39 as a colourless oil (27.0 mg, 0.0625 mmol, 93%); Rf 0.30 (5:1 pet. ether–ether); IR (thin film) νmax/cm−1 2958, 1733, 1596, 1433, 1259, 699; 1H NMR (500 MHz, CDCl3) δH 7.76 (2H, d, J = 8.3 Hz, ArH), 7.29 (2H, d, J = 8.3 Hz, ArH), 7.14 (1H, s, H5), 6.99 (1H, s, H7), 4.67 (2H, s, H3), 4.56 (2H, s, H1), 4.05 (1H, sept, J = 6.3 Hz, CHMe2), 2.40 (3H, s, Ts-Me), 2.31 (3H, s, Me), 1.18 (6H, d, J = 6.3 Hz, CHMe2), 0.92 (6H, t, J = 6.8 Hz, SiCH2Me), 0.88–0.82 (4H, m, SiCH2Me); 13C NMR (125 MHz, CDCl3) δC 143.4, 138.8, 136.3, 135.6, 134.4, 133.7, 131.1, 129.7, 127.6, 124.2, 65.4, 54.6, 53.3, 25.7, 21.4, 21.2, 6.8, 5.5; HRMS (ES+) calcd for C23H33NNaO3SSi [M + Na]+ 454.1843, found 454.1845.
Dimethyl 4-hydroxy-6-phenyl-1H-indene-2,2(3H)-dicarboxylate (40).
To a solution of 38 (17.0 mg, 0.0374 mmol) in THF–MeOH (1:1, 1 mL) was added and TBAF (1 M solution in THF, 13.0 μL, 0.013 mmol), KHCO3 (7.5 mg, 0.080 mmol) and H2O2 (30% aq., 13 μL, 0.12 mmol). The reaction mixture was heated to 60 °C for 3 h, then it was cooled to rt, diluted with CH2Cl2 and water. The organic phase was washed with water, dried (MgSO4), and concentrated. The residue was purified by flash chromatography (2:1 pet. ether–ether) to afford 40 (9.4 mg, 0.0288 mmol, 78%); Rf 0.15 (2:1 pet. ether–ether); IR (thin film) νmax/cm−1 2954, 1733, 1596, 1434, 1260, 1165, 864, 699; 1H NMR (500 MHz, CDCl3) δH 7.52 (2H, d, J = 7.5 Hz, ArH), 7.41 (2H, t, J = 7.5 Hz, ArH), 7.32 (1H, t, J = 7.5 Hz, ArH), 7.01 (1H, s, H7), 6.85 (1H, s, H5), 3.78 (6H, s, CO2Me), 3.66 (2H, s, H1), 3.60 (2H, s, H3); 13C NMR (125 MHz, CDCl3) δC 172.0, 151.8, 142.8, 142.5, 140.9, 128.7, 127.2, 127.1, 124.7, 115.6, 112.6, 60.3, 53.0, 40.9, 36.8; HRMS (ES−) calcd for C19H17O5 [M]− 325.1081, found 325.1081.
6-Methyl-2-tosylisoindolin-4-ol (41).
To a solution of 39 (20.0 mg, 0.0463 mmol) in THF–MeOH (1:1, 1 mL) was added and TBAF (1 M solution in THF, 16.0 μL, 0.016 mmol), KHCO3 (9.0 mg, 0.10 mmol) and H2O2 (30% aq., 17 μL, 0.15 mmol). The reaction mixture was heated to 60 °C for 3 h, then it was cooled to rt, diluted with CH2Cl2 and water. The organic phase was washed with water, dried (MgSO4), and concentrated. The residue was purified by flash chromatography (2:1 pet. ether–ether) to afford 41 as a colourless oil (14.0 mg, 0.0461 mmol, 99%); Rf 0.26 (2:1 pet. ether–ether); IR (thin film) νmax/cm−1 2954, 1733, 1596, 1434, 1260, 1165, 864, 699; 1H NMR (500 MHz, CDCl3) δH 7.76 (2H, d, J = 7.7 Hz, ArH), 7.31 (2H, d, J = 7.7 Hz, ArH), 6.54 (1H, s, H7), 6.47 (1H, s, H5), 4.57 (4H, s, H1, H3), 2.40 (3H, s, TsMe), 2.24 (3H, s, Me); 13C NMR (125 MHz, CDCl3) δC 150.6, 143.6, 139.8, 138.2, 133.6, 129.8, 127.6, 119.7, 115.2, 114.8, 54.0, 51.4, 21.5, 21.2; HRMS (ES+) calcd for C16H17NNaO3S [M + Na]+ 326.0821, found 326.0824.
Acknowledgements
We thank the EPSRC (Advanced Research Fellowship to E.A.A.; EP/E055273/1, studentship to M.-C.A.C.), the German Academic Exchange Service (DAAD) for a postdoctoral fellowship (B.G.), and A*STAR for a National Science Scholarship (S.S.G.).
Notes and references
-
(a)
J. H. P. Tyman, Synthetic and Natural Phenols, Elsevier, New York, 1996 Search PubMed;
(b)
Z. Rappoport, The Chemistry of Phenols, Wiley-VCH, Weinheim, 2003 CrossRef;
(c) I. P. Singh, S. B. Bharate and K. K. Bhutani, Curr. Sci., 2005, 89, 269–290 CAS.
-
(a) For examples of tetracycline and pyranonaphthoquinone natural products, see: A. G. Myers, D. A. Kummer, D. R. Li and A. Dion, Chem. Sci., 2011, 2, 1710–1718 RSC;
(b) A. G. Myers, C. X. Sun, Q. Wang, J. D. Brubaker, P. M. Wright, C. D. Lerner, K. Noson, M. Charest, D. R. Siegel and Y. M. Wang, J. Am. Chem. Soc., 2008, 130, 17913–17927 CrossRef PubMed;
(c) G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed;
(d) J. Sperry, P. Bachu and M. A. Brimble, Nat. Prod. Rep., 2008, 25, 376–400 RSC.
- For examples, see ref. 1a and: P. Hanson, J. R. Jones, A. B. Taylor, P. H. Walton and A. W. Timms, J. Chem. Soc., Perkin Trans. 2, 2002, 1135–1150 RSC.
-
(a) For reviews of C–X phenolation, see: M. C. Willis, Angew. Chem., Int. Ed., 2007, 46, 3402–3404 CrossRef CAS PubMed;
(b) S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912–4924 RSC;
(c) For reviews of C–H phenolation, see: D. A. Alonso, C. Najera, I. M. Pastor and M. Yus, Chem. – Eur. J., 2010, 16, 5274–5284 CrossRef CAS PubMed;
(d) M. S. Sanford and T. W. Lyons, Chem. Rev., 2010, 110, 1147–1169 CrossRef PubMed.
-
(a) The equivalent oxidation of arylboranes can also provide phenols, but in most cases these derivative are less amenable to processing through multistep synthesis. See: R. E. Maleczka, F. Shi, D. Holmes and M. R. Smith, J. Am. Chem. Soc., 2003, 125, 7792–7793 CrossRef CAS PubMed;
(b) G. A. Molander and L. N. Cavalcanti, J. Org. Chem., 2011, 76, 623–630 CrossRef CAS PubMed;
(c) X. Y. Wang, J. M. Xu, C. W. Shao, D. Y. Su, G. L. Cheng and Y. F. Hu, Org. Lett., 2010, 12, 1964–1967 CrossRef PubMed.
-
(a) S. Bracegirdle and E. A. Anderson, Chem. Commun., 2010, 46, 3454–3456 RSC;
(b) E. J. Rayment, N. Summerhill and E. A. Anderson, J. Org. Chem., 2012, 77, 7052–7060 CrossRef CAS PubMed.
-
(a) For other examples of arylsilane oxidation, see: J. F. Hartwig and E. M. Simmons, J. Am. Chem. Soc., 2010, 132, 17092–17095 CrossRef PubMed;
(b) M. Suginome and H. Ihara, J. Am. Chem. Soc., 2009, 131, 7502–7503 CrossRef PubMed;
(c) J. D. Sunderhaus, H. Lam and G. B. Dudley, Org. Lett., 2003, 5, 4571–4573 CrossRef CAS PubMed;
(d) K. Tamao, Proc. Jpn. Acad. B, Phys. Biol. Sci., 2008, 84, 123–133 CrossRef CAS;
(e) K. Tamao, T. Kakui, M. Akita, T. Iwahara, R. Kanatani, J. Yoshida and M. Kumada, Tetrahedron, 1983, 39, 983–990 CrossRef CAS;
(f) For a borodesilylation/oxidation sequence, see: V. Gevorgyan, C. H. Huang, N. Chernyak and A. S. Dudnik, Adv. Synth. Catal., 2011, 353, 1285–1305 CrossRef PubMed.
-
(a) S. B. J. Kan and E. A. Anderson, Org. Lett., 2008, 10, 2323–2326 CrossRef CAS PubMed;
(b) M.-C. A. Cordonnier, S. B. J. Kan and E. A. Anderson, Chem. Commun., 2008, 5818–5820 RSC.
-
(a) J. Suffert, B. Salem and P. Klotz, J. Am. Chem. Soc., 2001, 123, 12107–12108 CrossRef CAS;
(b) B. Salem, P. Klotz and J. Suffert, Org. Lett., 2003, 5, 845–848 CrossRef CAS PubMed;
(c) B. Salem, E. Delort, P. Klotz and J. Suffert, Org. Lett., 2003, 5, 2307–2310 CrossRef CAS PubMed;
(d) B. Salem and J. Suffert, Angew. Chem., Int. Ed., 2004, 43, 2826–2830 CrossRef CAS PubMed;
(e) C. Bour, G. Blond, B. Salem and J. Suffert, Tetrahedron, 2006, 62, 10567–10581 CrossRef CAS PubMed;
(f) For the most recent publication on this topic, and leading references, see: M. Charpenay, A. Boudhat, C. Hulot, G. Blond and J. Suffert, Tetrahedron, 2013, 69, 7568–7591 CrossRef CAS PubMed;
(g) For seminal work on alkyne carbopalladation followed by Stille cross-coupling, see: B. Burns, R. Grigg, P. Ratananukul, V. Sridharan, P. Stevenson, S. Sukirthalingam and T. Worakun, Tetrahedron Lett., 1988, 29, 5565–5568 CrossRef CAS;
(h) E. Negishi, Y. Noda, F. Lamaty and E. J. Vawter, Tetrahedron Lett., 1990, 31, 4393–4396 CrossRef CAS;
(i) J. M. Nuss, M. M. Murphy, R. A. Rennels, M. M. Heravi and B. J. Mohr, Tetrahedron Lett., 1993, 34, 3079–3082 CrossRef CAS;
(j) P. Fretwell, R. Grigg, J. M. Sansano, V. Sridharan, S. Sukirthalingam, D. Wilson and J. Redpath, Tetrahedron, 2000, 56, 7525–7539 CrossRef CAS;
(k) For a review of palladium-catalyzed cascade cyclizations, see: T. Vlaar, E. Ruijter and R. V. A. Orru, Adv. Synth. Catal., 2011, 353, 809–841 CrossRef CAS;
(l) For a general review of carbopalladation chemistry, see: E. Negishi, C. Coperet, S. Ma, S. Y. Liou and F. Liu, Chem. Rev., 1996, 96, 365–394 CrossRef CAS PubMed.
- Suffert has also noted the utility of silylalkynes in similar cascades. See: G. Blond, C. Bour, B. Salem and J. Suffert, Org. Lett., 2008, 10, 1075–1078 CrossRef CAS PubMed.
-
(a) For recent reviews and leading references, see: Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893–4901 RSC;
(b) S. E. Denmark and J. H. C. Liu, Angew. Chem., Int. Ed., 2010, 49, 2978–2986 CrossRef CAS PubMed.
- S. S. Goh, H. Baars, B. Gockel and E. A. Anderson, Org. Lett., 2012, 14, 6278–6281 CrossRef CAS PubMed.
-
(a) For other de novo routes to fused-ring phenols, see: A. S. K. Hashmi, T. Haffner, M. Rudolph and F. Rominger, Chem. – Eur. J., 2011, 17, 8195–8201 CrossRef CAS PubMed;
(b) A. S. K. Hashmi, M. Rudolph, J. W. Bats, W. Frey, F. Rominger and T. Oeser, Chem. – Eur. J., 2008, 14, 6672–6678 CrossRef CAS PubMed;
(c) A. S. K. Hashmi, R. Salathe and W. Frey, Chem. – Eur. J., 2006, 12, 6991–6996 CrossRef CAS PubMed;
(d) K. Hashmi, A. Stephen, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef;
(e) K. Tanaka, H. Hara and M. Hirano, Org. Lett., 2009, 11, 1337–1340 CrossRef PubMed;
(f) S. Akai, T. Ikawa, S. I. Takayanagi, Y. Morikawa, S. Mohri, M. Tsubakiyama, M. Egi, Y. Wada and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 7673–7676 CrossRef CAS PubMed;
(g) K. Yoshida, R. Narui and T. Imamoto, Chem. – Eur. J., 2008, 14, 9706–9713 CrossRef CAS PubMed.
- L. H. Li, D. Wang and T. H. Chan, Organometallics, 1994, 13, 1757–1761 CrossRef CAS.
-
(a) Diethylaminochlorodiethylsilane was prepared according to the procedure of Tamao: K. Tamao, A. Kawachi and Y. Ito, Organometallics, 1993, 12, 580–582 CrossRef CAS;
(b) For an alternative preparation, see: J. Beignet, P. J. Jervis and L. R. Cox, J. Org. Chem., 2008, 73, 5462–5475 CrossRef CAS PubMed.
-
(a) R. L. Miller, S. V. Maifeld and D. Lee, Org. Lett., 2004, 6, 2773–2776 CrossRef CAS PubMed;
(b) S. Chang, M. Lee and S. Ko, J. Am. Chem. Soc., 2000, 122, 12011–12012 CrossRef.
- See the ESI† for NMR spectra of a purified mixture containing predominantly 16a and 17a.
-
(a) C. Amatore, S. Bensalem, S. Ghalem and A. Jutand, J. Organomet. Chem., 2004, 689, 4642–4646 CrossRef CAS PubMed;
(b) B. H. Lipshutz and A. Krasovskiy, Org. Lett., 2011, 13, 3818–3821 CrossRef PubMed.
- M. Murakami, K. Itami and Y. Ito, J. Am. Chem. Soc., 1997, 119, 7163–7164 CrossRef CAS.
- A reductive elimination to the syn-triene followed by rapid electrocyclization cannot be ruled out; however, we have never observed such intermediates.
- J. Q. Yu, M. J. Gaunt and J. B. Spencer, J. Org. Chem., 2002, 67, 4627–4629 CrossRef CAS PubMed.
- X. Wang, H. Chakrapani, J. W. Madine, M. A. Keyerleber and R. A. Widenhoefer, J. Org. Chem., 2002, 67, 2778–2788 CrossRef CAS PubMed.
- An intermediate of this type has also been proposed in the mechanistically distinct palladium(II)-catalyzed cyclization of an allenynoate to give a fused cyclobutene. See: C. H. Oh, D. I. Park, S. H. Jung, V. R. Reddy, A. K. Gupta and Y. M. Kim, Synlett, 2005, 2092–2094 CrossRef CAS.
-
(a) A. Furstner, A. Schlecker and C. W. Lehmann, Chem. Commun., 2007, 4277–4279 RSC;
(b) N. Saito, Y. Tanaka and Y. Sato, Org. Lett., 2009, 11, 4124–4126 CrossRef CAS PubMed.
-
(a) For a Mo-catalyzed route, see: Q. Shen and G. B. Hammond, J. Am. Chem. Soc., 2002, 124, 6534–6535 CrossRef CAS PubMed;
(b) For examples of syntheses via thermal [2 + 2] cycloaddition, see: K. M. Brummond and D. T. Chen, Org. Lett., 2005, 7, 3473–3475 CrossRef CAS PubMed;
(c) H. Ohno, T. Mizutani, Y. Kadoh, A. Aso, K. Miyamura, N. Fujii and T. Tanaka, J. Org. Chem., 2007, 72, 4378–4389 CrossRef CAS PubMed;
(d) C. Mukai, Y. Hara, Y. Miyashita and F. Inagaki, J. Org. Chem., 2007, 72, 4454–4461 CrossRef CAS PubMed.
- K. Tamao, M. Kumada and K. Maeda, Tetrahedron Lett., 1984, 25, 321–324 CrossRef CAS.
- R. L. Greenaway, C. D. Campbell, O. T. Holton, C. A. Russell and E. A. Anderson, Chem. – Eur. J., 2011, 17, 14366–14370 CrossRef CAS PubMed.
- We found it possible to use a sub-stoichiometric quantity of TBAF to effect these oxidations. See ref. 6b.
- M. G. Organ, J. T. Cooper, L. R. Rogers, F. Soleymanzadeh and T. Paul, J. Org. Chem., 2000, 65, 7959–7970 CrossRef CAS PubMed.
- F. Hoffmann, J. Wagler and G. Roewer, Eur. J. Inorg. Chem., 2010, 1133–1142 CrossRef CAS.
- P. J. Parsons, P. Willis and S. C. Eyley, Tetrahedron, 1992, 48, 9461–9472 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra, and experimental details for NMR experiments. See DOI: 10.1039/c4qo00123k |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a