DOI:
10.1039/C4QO00056K
(Research Article)
Org. Chem. Front., 2014,
1, 657-660
Direct cleavage of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N bond of azobenzenes by MeOTf leading to N-arylbenzimidazoles†
N bond of azobenzenes by MeOTf leading to N-arylbenzimidazoles†
Received
27th February 2014
, Accepted 9th May 2014
First published on 12th May 2014
Introduction
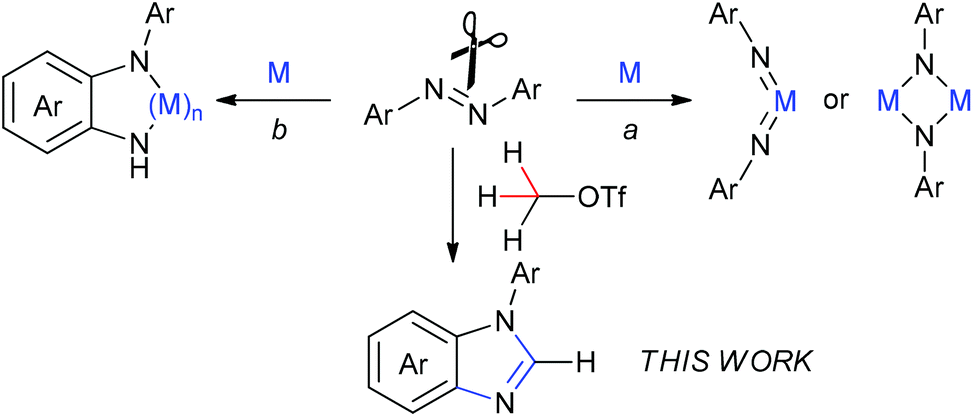
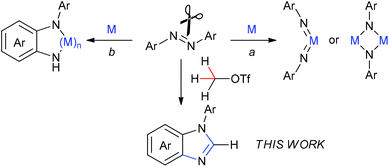
The cleavage of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond is of considerable importance in understanding the mechanisms of dinitrogen fixation as well as developing new transformations using azo compounds as the [RN] unit.1 To date, most investigations on the NN bond cleavage of the azo compounds need the assistance of transition metals. Among them, much attention has been paid to utilization of low-valent metal complexes for cleavage of the NN bond to result in imido or μ2/μ3-imido metal complexes (Scheme 1, route a).2 In addition, some transition-metal complexes mediated the NN bond cleavage to result in the corresponding metal complexes with the o-semidine (N-phenyl-o-phenylenediamine) ligand (Scheme 1, route b).3 There are also several examples of the NN bond cleavage reactions of azobenzene with heavy main-group elements Al, P, and Si compounds either in the process of route a4 or route b.5 Nevertheless, there is no report of direct cleavage of azo compounds by light main-group elements, to the best of our knowledge. Herein, we describe a direct cleavage of an NN bond of azobenzenes by methyltriflate (MeOTf) leading to benzimidazole derivatives that are valuable frameworks in organic and bioorganic molecules. In this reaction, the methyl carbon atom inserts into the NN bond to form N-arylbenzimidazoles via cleavage of the NN bond and two sp3 C–H bonds as well as one sp2 C–H bond and meanwhile formation of three C–N bonds without the assistance of any metals or metalloids.
N bond is of considerable importance in understanding the mechanisms of dinitrogen fixation as well as developing new transformations using azo compounds as the [RN] unit.1 To date, most investigations on the NN bond cleavage of the azo compounds need the assistance of transition metals. Among them, much attention has been paid to utilization of low-valent metal complexes for cleavage of the NN bond to result in imido or μ2/μ3-imido metal complexes (Scheme 1, route a).2 In addition, some transition-metal complexes mediated the NN bond cleavage to result in the corresponding metal complexes with the o-semidine (N-phenyl-o-phenylenediamine) ligand (Scheme 1, route b).3 There are also several examples of the NN bond cleavage reactions of azobenzene with heavy main-group elements Al, P, and Si compounds either in the process of route a4 or route b.5 Nevertheless, there is no report of direct cleavage of azo compounds by light main-group elements, to the best of our knowledge. Herein, we describe a direct cleavage of an NN bond of azobenzenes by methyltriflate (MeOTf) leading to benzimidazole derivatives that are valuable frameworks in organic and bioorganic molecules. In this reaction, the methyl carbon atom inserts into the NN bond to form N-arylbenzimidazoles via cleavage of the NN bond and two sp3 C–H bonds as well as one sp2 C–H bond and meanwhile formation of three C–N bonds without the assistance of any metals or metalloids.
 |
| | Scheme 1 NN bond cleavage reactions of azo compounds. | |
Results and discussion
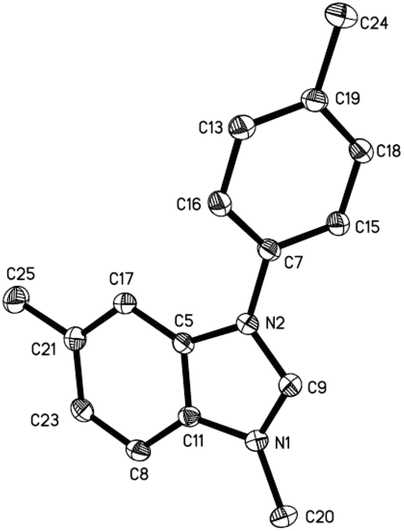
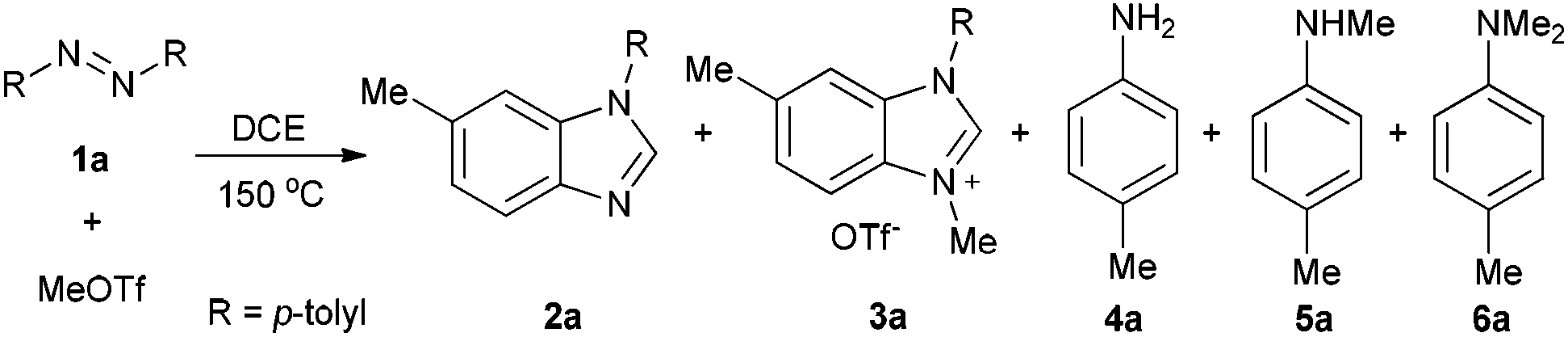
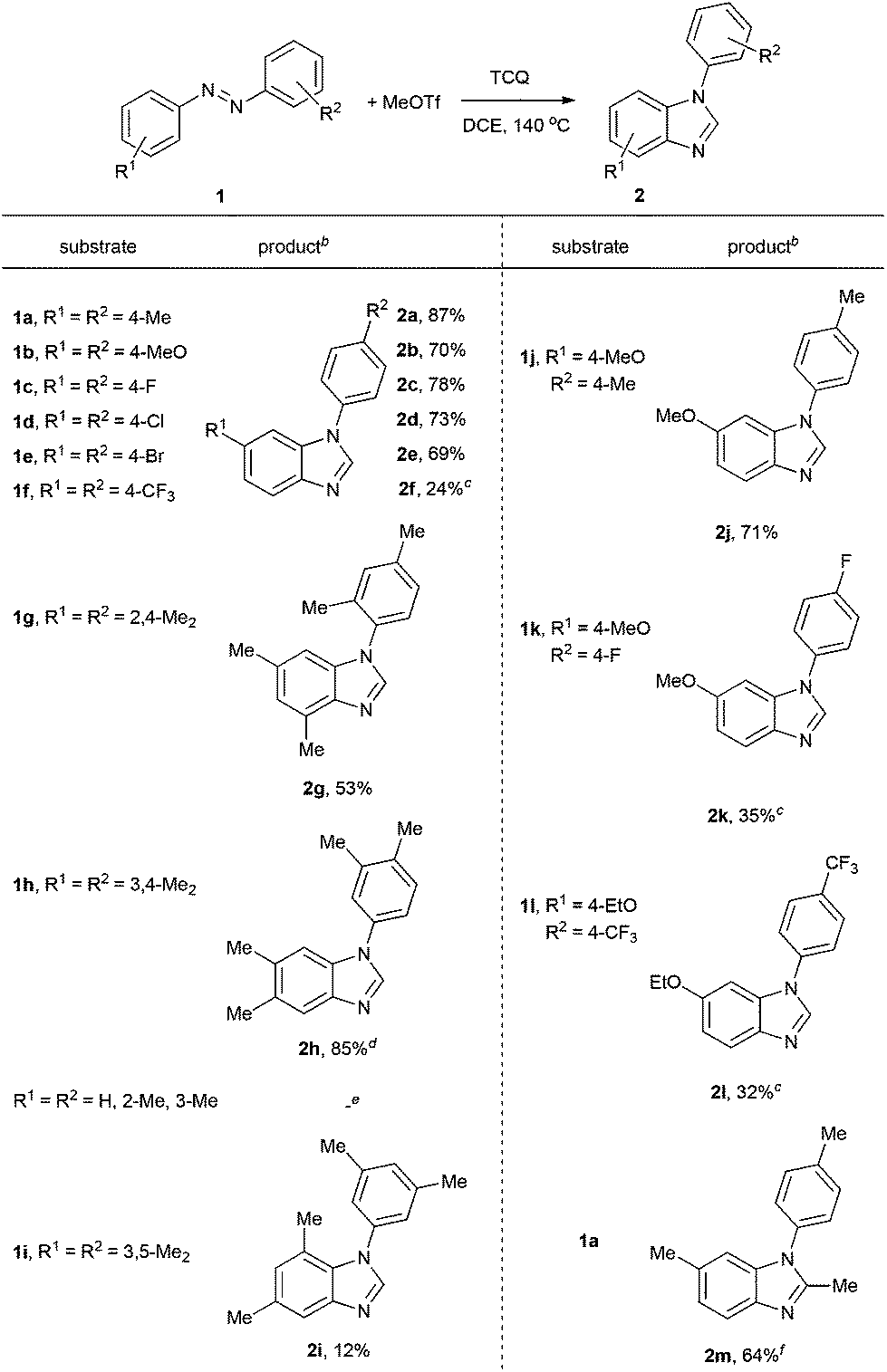
Methyltriflate was considered as an equivalent of the methyl cation (CH3+) and was frequently used in the methylation reaction of heteroatom compounds.6 During the course of our ongoing project on the construction of heterocyclic compounds,7 we initially tried the methylation of 1,2-di-p-tolyldiazene 1a by employing 1,2-dichloroethane (DCE) as the solvent in a sealed tube at 150 °C. N-Tolyl benzimidazole 2a and benzimidazolium 3a were detected in 38% and 8% NMR yields, respectively. Along with these, we also detected aniline 4a, methylaniline 5a, and dimethylaniline 6a in 13%, 10%, and 2% NMR yields, respectively (Table 1, entry 1). Then we tried different ratios of 1a and MeOTf, and we found the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 to give the best yield of 2a (entry 3). Notably, the reaction could proceed under air, and the yields were slightly decreased (entries 5 and 6). To confirm the structure of the product 2a, we isolated the product 2a and tried methylation of 2a with MeOTf. 3a formed quantitatively. The structure of 3a was confirmed by single-crystal X-ray diffraction analysis and is shown in Fig. 1.‡
:1.5 to give the best yield of 2a (entry 3). Notably, the reaction could proceed under air, and the yields were slightly decreased (entries 5 and 6). To confirm the structure of the product 2a, we isolated the product 2a and tried methylation of 2a with MeOTf. 3a formed quantitatively. The structure of 3a was confirmed by single-crystal X-ray diffraction analysis and is shown in Fig. 1.‡
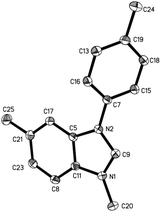 |
| | Fig. 1 The X-ray crystal structure of 3a. Thermal ellipsoids are shown at 30% probability level; hydrogen atoms and TfO− counter ions have been omitted for clarity. | |
Table 1 Reaction of 1a with MeOTf in DCE solutiona
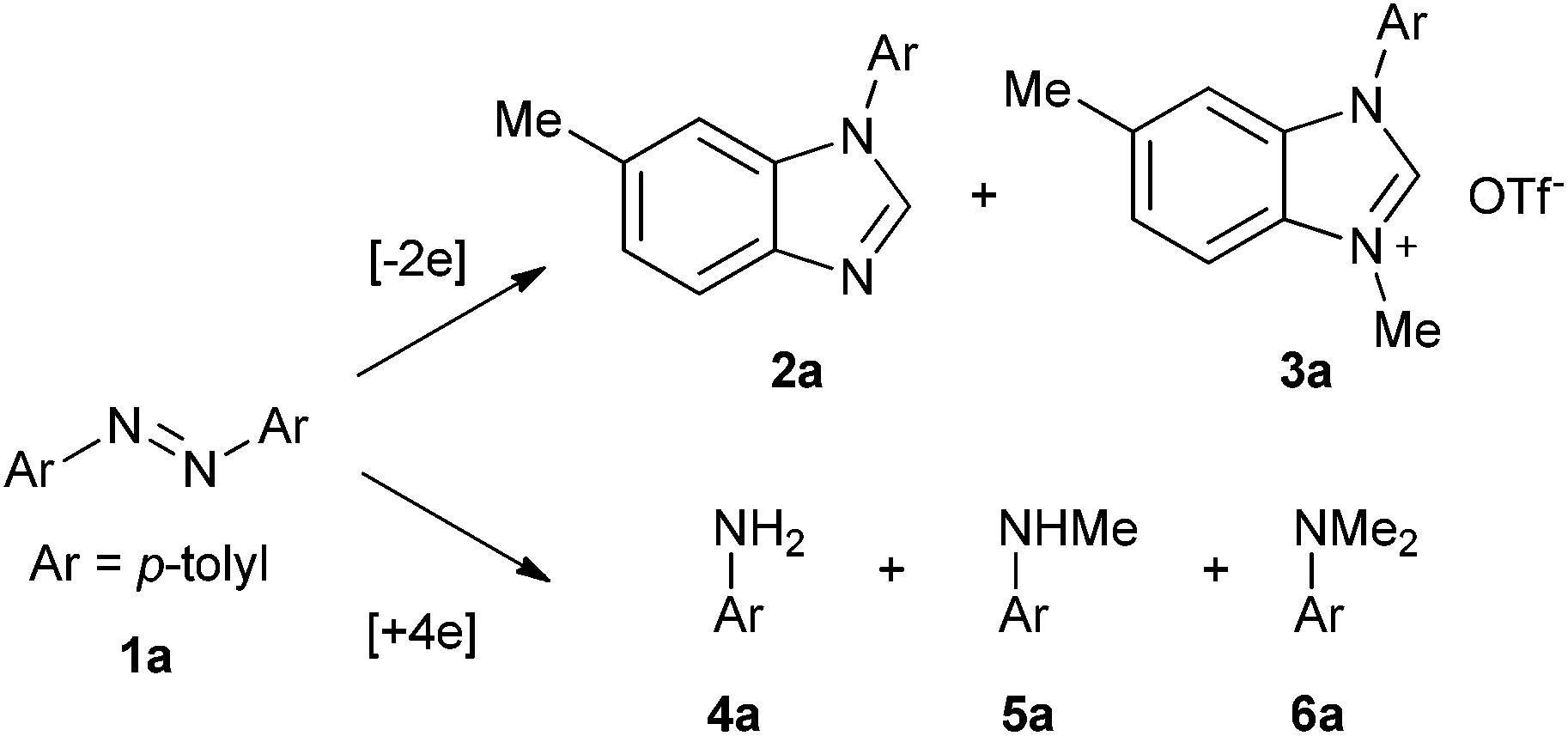
Based on the above results and redox analysis, transformation from one molecule of 1a to 2a and 3a needs loss of two electrons, and transformation from 1a to 4a, 5a, and 6a needs addition of four electrons. So in this reaction, about two thirds of 1a was converted to 2a and 3a, and one third of 1a was reduced to aniline derivatives 4a, 5a, and 6a (Scheme 2). To prevent the formation of 4a, 5a, and 6a and enhance the yield of 2a, we tried to add an oxidant in this reaction. To our delight, when 1.2 equivalents of tetrachloro-1,4-benzoquinone (TCQ) were added and the reaction mixture was heated at 140 °C for 4 h, the yield of 2a increased to 87%.
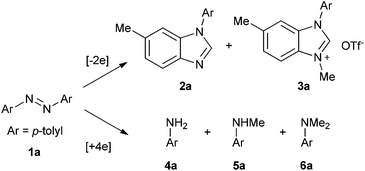 |
| | Scheme 2 Redox analysis of the reaction. | |
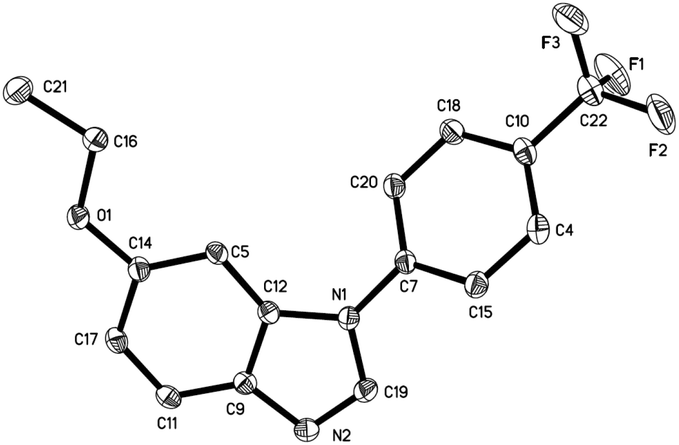
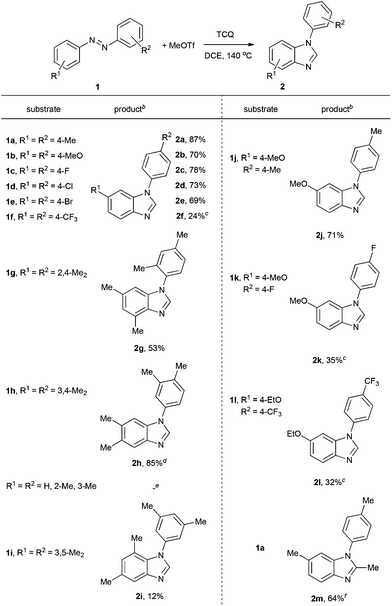
Under these conditions, a study on the substrate scope was carried out, and the representative results are summarized in Table 2. Symmetrically para-substituted azobenzenes with methyl, methoxyl, fluoro, chloro, and bromo substituents afforded the desired benzimidazoles 2a–2e in 69% to 87% isolated yields. 4,4′-Bis(trifluoromethyl) azobenzene 1f only afforded benzimidazole 2f in 24% yield even by increasing the temperature, prolonging the reaction time, and using two equivalents of MeOTf. 2,2′,4,4′-Tetramethyl azobenzene 1g afforded benzimidazole 2g in 53% yield. The lower yield may be due to the stereohindrance of the ortho-methyl group. 3,3′,4,4′-Tetramethyl azobenzene 1h afforded benzimidazole 2h in 85% yield with two isomers in a 4:1 ratio. It is noteworthy that when azobenzenes without substituents in the para-position, such as azobenzene, 2,2′-dimethyl azobenzene, and 3,3′-dimethyl azobenzene were used, complex and insoluble solids were observed without observation of benzimidazoles. Notably, when 3,3′,5,5′-tetramethyl azobenzene 1i was used, which does not have para-substituents but stereohindrance in the para-position, benzimidazole 2i was formed in 12% yield. Next we tried unsymmetrically substituted azobenzenes 1j–1l, and only one product was formed. The cyclization always occurred in the electron-rich anisolyl ring, and benzimidazoles 2j–2l were formed in 32% to 71% isolated yields. The structure of 2l was confirmed by single-crystal X-ray diffraction analysis and is shown in Fig. 2.‡ When EtOTf was used instead of MeOTf, the desired 2-methylbenzimidazole 2m was also formed in 64% yield.
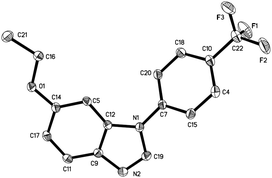 |
| | Fig. 2 The X-ray crystal structure of 2l. Thermal ellipsoids are shown at 30% probability level; hydrogen atoms have been omitted for clarity. | |
Table 2 Reaction of azo compounds with alkyl triflate using TCQ as an oxidanta
|
Reaction conditions: 0.2 mmol 1a, 0.3 mmol MeOTf, 0.24 mmol TCQ, 1 mL DCE, a sealed tube under N2, 140 °C, 4 h.
Isolated yields are shown.
2 equiv. of MeOTf, 150 °C, 12 h.
Two isomers were observed in a 4:1 ratio, the major isomer was shown.
No desired products were observed.
EtOTf was used.
|
|
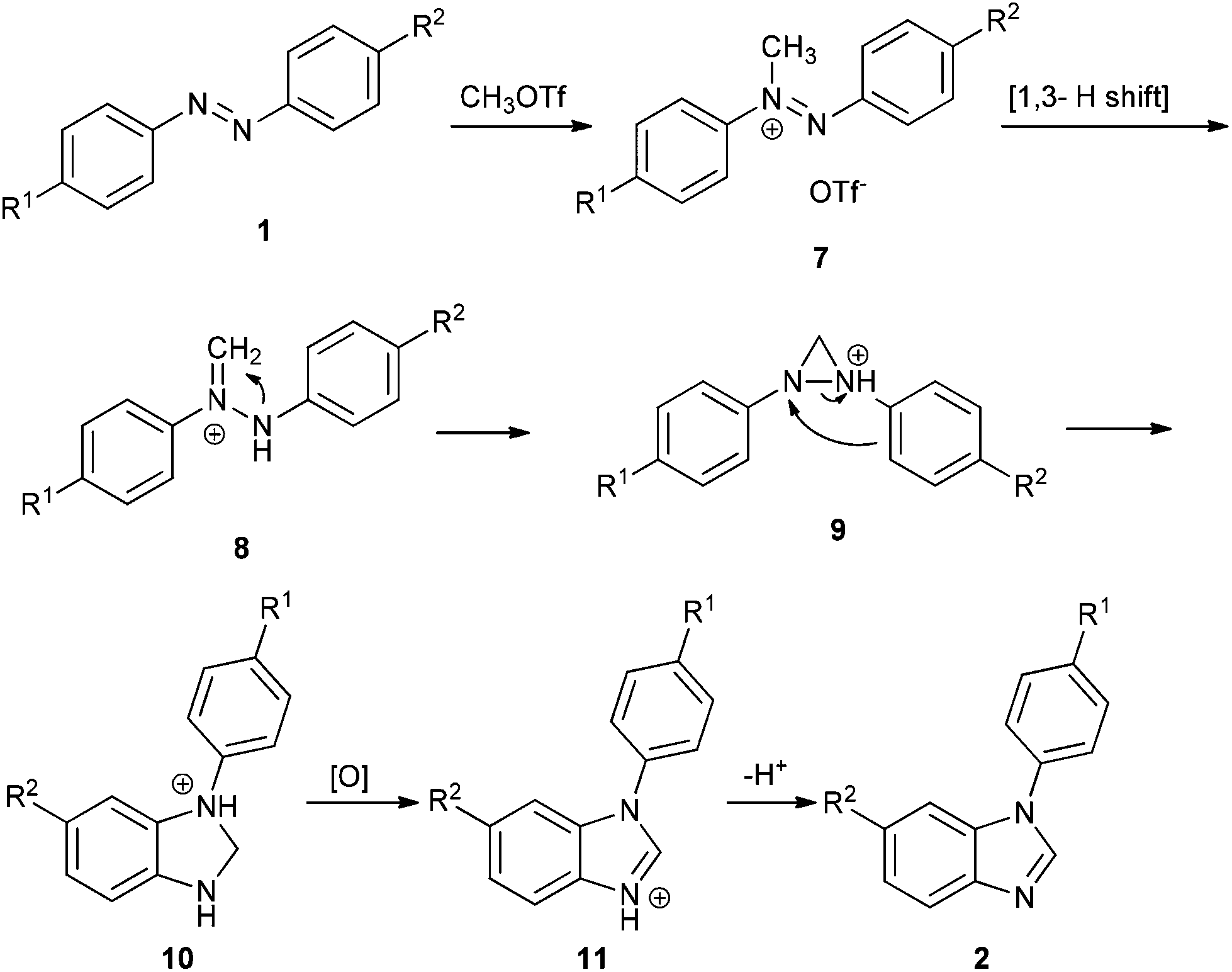
Ferguson has reported that methylation of azo compounds by MeOTf affords diazenium salts under reflux with excess MeOTf as the solvent.8 And diazenium with α-hydrogen can rearrange to the more stable hydrazonium (aminoimmonium) tautomer easily.9 On the basis of the literature and our results, a plausible mechanism for the reaction of azo compounds with MeOTf is illustrated in Scheme 3. First, methylation of 1 affords diazenium salt 7, which then tautomerizes to hydrazonium salt 8via a [1,3] hydrogen shift.9a Cyclization of 8 yields diaziridine 9, which converts to dihydrobenzoimidazole 10via o-semidine rearrangement.3d,5a,10 When unsymmetrical azobenzenes are used, the cyclization occurs in the electron-rich phenyl ring. Dihydrobenzoimidazole 10 is then oxidized to protonated benzoimidazole 11 with TCQ or azo compound.11 Workup of 11 with a base affords benzoimidazole 2.
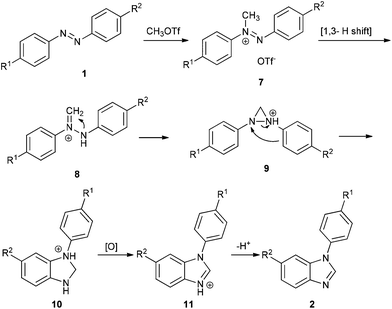 |
| | Scheme 3 Plausible reaction mechanism. | |
In general, transition-metal-mediated NN bond cleavage involves two steps: (1) formation of diazametallocycles, which are based on donation of electrons from nitrogen to transition metals as well as backdonation of d electrons from transition metals to the antibonding orbital of the NN bond; (2) reduction affords imido metal complexes, or rearrangement results in o-semidine metal complexes. In contrast, the methyl cation has one empty orbital and no lone pair, so it cannot form diaziridine directly. In our reaction, diaziridine 9 was formed by stepwise [1,3] hydrogen shift and cyclization. Apparently, the C–H bond of the methyl cation serves as a lone pair to form the diaziridine 9. In other words, in this case, methyl cations could mimic the chemical behavior of transition metals in NN bond cleavage.
Conclusion
In conclusion, we have developed a new type of NN bond cleavage reaction with methyl triflate. The methyl carbon atom inserts into the NN bond to form N-arylbenzimidazole. This is the first example of NN bond cleavage by a light main group element. Further investigations are still in progress in this area.
Experimental section
General procedures
To a 25 mL tube charged with nitrogen were added azo compounds 1 (0.2 mmol), TCQ (0.24 mmol), MeOTf (0.3 mmol), and DCE 1 mL. The tube was sealed and stirred for 4 h at 140 °C. Removing the solvent of the reaction mixture and subsequent purification by column chromatography on silica gel (petroleum ether–ethyl acetate–triethylamine: 1/1/0.05) afforded benzimidazole 2.
Acknowledgements
This work was supported by the National Key Basic Research Program of China (973 program) (2012CB933402) and the National Natural Science Foundation of China (21032004 and 21272132).
Notes and references
-
(a) M. Hidai and Y. Mizobe, Chem. Rev., 1995, 95, 1115 CrossRef CAS;
(b) B. A. MacKay and M. D. Fryzuk, Chem. Rev., 2004, 104, 385 CrossRef CAS PubMed;
(c) N. Hazari, Chem. Soc. Rev., 2010, 39, 4044 RSC.
-
(a) A. A. Trifonov, M. N. Bochkarev, H. Schumann and J. Loebel, Angew. Chem., Int. Ed. Engl., 1991, 103, 1170 CrossRef CAS;
(b) B. P. Warner, B. L. Scott and C. J. Burns, Angew. Chem., Int. Ed., 1998, 37, 959 CrossRef CAS;
(c) P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108 CrossRef CAS;
(d) G. Guillemot, E. Solari, R. Scopelliti and C. Floriani, Organometallics, 2001, 20, 2446 CrossRef CAS;
(e) M. González-Maupoey, G. M. Rodríguez and T. Cuenca, Eur. J. Inorg. Chem., 2002, 2002, 2057 CrossRef;
(f) M. H. Chisholm, D. R. Click, J. C. Gallucci and C. M. Hadad, Dalton Trans., 2003, 3205 RSC;
(g) T. Komuro, T. Matsuo, H. Kawaguchi and K. Tatsumi, Inorg. Chem., 2004, 44, 175 CrossRef PubMed;
(h) M. R. Lentz, J. S. Vilardo, M. A. Lockwood, P. E. Fanwick and I. P. Rothwell, Organometallics, 2004, 23, 329 CrossRef CAS;
(i) W. J. Evans, S. A. Kozimor and J. W. Ziller, Chem. Commun., 2005, 4681 RSC;
(j) U. J. Kilgore, X. Yang, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, Inorg. Chem., 2006, 45, 10712 CrossRef CAS PubMed;
(k) W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568 CrossRef CAS;
(l) W. H. Monillas, G. P. A. Yap, L. A. MacAdams and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 8090 CrossRef CAS PubMed;
(m) Y. Tsai, P. Wang, S. Chen and J. Chen, J. Am. Chem. Soc., 2007, 129, 8066 CrossRef CAS PubMed;
(n) Y. Tsai, P. Wang, K. Lin, S. Chen and J. Chen, Chem. Commun., 2008, 205 RSC;
(o) U. J. Kilgore, C. A. Sengelaub, H. Fan, J. Tomaszewski, J. A. Karty, M. Baik and D. J. Mindiola, Organometallics, 2008, 28, 843 CrossRef;
(p) W. J. Evans, E. Montalvo, S. A. Kozimor and K. A. Miller, J. Am. Chem. Soc., 2008, 130, 12258 CrossRef CAS PubMed;
(q) K. Kaleta, P. Arndt, T. Beweries, A. Spannenberg, O. Theilmann and U. Rosenthal, Organometallics, 2010, 29, 2604 CrossRef CAS;
(r) C. Pan, W. Chen, S. Su, Y. Pan and J. Wang, Dalton Trans., 2011, 40, 7941 RSC;
(s) C. Milsmann, Z. R. Turner, S. P. Semproni and P. J. Chirik, Angew. Chem., Int. Ed., 2012, 51, 5386 CrossRef CAS PubMed;
(t) D. P. Cladis, J. J. Kiernicki, P. E. Fanwick and S. C. Bart, Chem. Commun., 2013, 49, 4169 RSC;
(u) Y. Nakajima and H. Suzuki, Organometallics, 2005, 24, 1860 CrossRef CAS;
(v) J. M. Smith, R. J. Lachicotte and P. L. Holland, J. Am. Chem. Soc., 2003, 125, 15752 CrossRef CAS PubMed;
(w) A. R. Sadique, E. A. Gregory, W. W. Brennessel and P. L. Holland, J. Am. Chem. Soc., 2007, 129, 8112 CrossRef CAS PubMed;
(x) K. Kaleta, P. Arndt, A. Spannenberg and U. Rosenthal, Inorg. Chim. Acta, 2011, 370, 187 CrossRef CAS PubMed;
(y) I. Vidyaratne, G. B. Nikiforov, S. I. Gorelsky, S. Gambarotta, R. Duchateau and I. Korobkov, Angew. Chem., Int. Ed., 2009, 48, 6552 CrossRef CAS PubMed;
(z) P. L. Diaconescu and C. C. Cummins, Inorg. Chem., 2012, 51, 2902 CrossRef CAS PubMed.
-
(a) P. E. Baikie and O. S. Mills, Chem. Commun., 1966, 707 RSC;
(b) P. E. Baikie and O. S. Mills, Inorg. Chim. Acta, 1967, 1, 55 CrossRef CAS;
(c) T. Joh, N. Hagihara and S. Murahashi, Bull. Chem. Soc. Jpn., 1967, 40, 661 CrossRef CAS;
(d) M. I. Bruce, M. Z. Iqbal and F. G. A. Stone, J. Chem. Soc. A, 1970, 3204 RSC;
(e) M. I. Bruce, M. Z. Iqbal and F. G. A. Stone, J. Organomet. Chem., 1971, 31, 275 CrossRef CAS;
(f) Z. Dawoodi, M. J. Mays and P. R. Raithby, J. Chem. Soc., Chem. Commun., 1980, 712 RSC;
(g) A. Spencer, J. Organomet. Chem., 1985, 294, 357 CrossRef CAS.
-
(a) Y. Zhao, Y. Liu, L. Yang, J. Yu, S. Li, B. Wu and X. Yang, Chem. – Eur. J., 2012, 18, 6022 CrossRef CAS PubMed;
(b) P. Bhattacharyya, A. M. Z. Slawin and J. D. Woollins, J. Chem. Soc., Dalton Trans., 2001, 300 RSC;
(c) K. Takeuchi, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2011, 133, 12478 CrossRef CAS PubMed.
-
(a) H. Zhu, J. Chai, H. Fan, H. W. Roesky, U. N. Nehete, H. Schmidt and M. Noltemeyer, Eur. J. Inorg. Chem., 2005, 2005, 2147 CrossRef;
(b) E. Niecke, M. Link and M. Nieger, Chem. Ber., 1992, 125, 93 CrossRef CAS;
(c) M. Weidenbruch, S. Olthoff, W. Saak and H. Marsmann, Eur. J. Inorg. Chem., 1998, 1998, 1755 CrossRef.
-
(a)
R. W. Alder, J. G. E. Phillips, L. Huang and X. Huang, “Methyltrifluoromethanesulfonate”, Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, 2005 Search PubMed;
(b) T. Murai, Y. Mutoh and S. Kato, Org. Lett., 2001, 2, 1993 CrossRef PubMed;
(c) A. I. Meyers and M. E. Flanagan, Org. Synth., 1998, 9, 258 Search PubMed;
(d) X. Yan, S. Zou, P. Zhao and C. Xi, Chem. Commun., 2014, 50, 2775 RSC;
(e) P. Zhao, X. Yan, H. Yin and C. Xi, Org. Lett., 2014, 16, 1120 CrossRef CAS PubMed.
-
(a) P. Zhao, H. Yin, H. Gao and C. Xi, J. Org. Chem., 2013, 78, 5001 CrossRef CAS PubMed;
(b) P. Zhao, Q. Liao, H. Gao and C. Xi, Tetrahedron Lett., 2013, 54, 2357 CrossRef CAS PubMed;
(c) Q. Liao, W. You, Z. Lou, L. Wen and C. Xi, Tetrahedron Lett., 2013, 54, 1475 CrossRef CAS PubMed;
(d) P. Zhao, F. Wang and C. Xi, Synthesis, 2012, 1477 CAS;
(e) F. Wang, C. Chen, G. Deng and C. Xi, J. Org. Chem., 2012, 77, 4148 CrossRef CAS PubMed;
(f) F. Wang, Q. Liao and C. Xi, Synth. Commun., 2012, 42, 905 CrossRef CAS;
(g) Q. Liao, L. Zhang, S. Li and C. Xi, Org. Lett., 2011, 13, 228 CrossRef CAS PubMed;
(h) F. Wang, S. Cai, Z. Wang and C. Xi, Org. Lett., 2011, 13, 3202 CrossRef CAS PubMed;
(i) F. Wang, S. Cai, Q. Liao and C. Xi, J. Org. Chem., 2011, 76, 3174 CrossRef CAS PubMed;
(j) Y. Wang, Q. Liao, P. Zhao and C. Xi, Adv. Synth. Catal., 2011, 353, 2659 CrossRef CAS;
(k) W. You, X. Yan, Q. Liao and C. Xi, Org. Lett., 2010, 12, 3930 CrossRef CAS PubMed;
(l) Q. Liao, L. Zhang, F. Wang, S. Li and C. Xi, Eur. J. Org. Chem., 2010, 5426 CrossRef CAS.
- A. N. Ferguson, Tetrahedron Lett., 1973, 14, 2889 CrossRef.
-
(a) E. L. Allred, T. J. Chow and J. E. Oberlander, J. Am. Chem. Soc., 1982, 104, 5422 CrossRef CAS;
(b) M. I. Javed, J. M. Wyman and M. Brewer, Org. Lett., 2009, 11, 2189 CrossRef CAS PubMed.
- G. Ghigo, S. Osella, A. Maranzana and G. Tonachini, Eur. J. Org. Chem., 2011, 2326 CrossRef CAS.
- J. J. Vanden Eynde, F. Delfosse, P. Lor and Y. Van Haverbeke, Tetrahedron, 1995, 51, 5813 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, full characterization including 1H NMR and 13C NMR data for all new compounds, copies of spectra for all compounds, and the X-ray data for 2l and 3a (CIF). CCDC 978439 and 978438. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00056k |
| ‡ 3a: CCDC 978439, 2l: 978438. |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.