Synthesis and photophysics of a broadband absorbing texaphyrin derivative bearing a Rhodamine 6G motif†
Lei
Hu
ab,
Chengkui
Pei
a,
Zhongjing
Li
a,
Chengzhe
Wang
a,
Guichun
Yang
b and
Wenfang
Sun
*a
aDepartment of Chemistry and Biochemistry, North Dakota State University, Fargo, ND 58108-6050, USA. E-mail: Wenfang.Sun@ndsu.edu
bCollege of Chemistry and Chemical Engineering, Hubei University, Wuhan 430062, P. R. China
First published on 15th April 2014
Abstract
A texaphyrin derivative with Rhodamine 6G attached (complex 11) via a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond was synthesized and characterized. The UV-vis absorption, emission and nanosecond transient absorption (TA) characteristics of this complex were systematically studied in acetone solutions. The photophysics of this complex was also compared with those of its precursor compounds texaphyrins 12 and 13 and the Rhodamine 6G derivative 3. The UV-vis absorption spectrum of 11 consists of both the characteristic Soret and Q bands of the texaphyrin derivative 13 and the identical absorption band from the Rhodamine 6G derivative 3. When excited at 550 nm (the major absorption band of Rhodamine 6G), 11 exhibits fluorescence bands from both the Rhodamine 6G component (582 nm) and texaphyrin component (802 nm), but the intensity of the 582 nm band dramatically reduced accompanied by a significant increase of the 802 nm band compared to those from 3 and 13, indicating electron/energy transfer from the singlet excited state of Rhodamine 6G. The ns TA spectrum of 11 resembles that of the texaphyrin derivative 13 but with both the bleaching band and absorption band red-shifted. The triplet lifetimes deduced from the decay of ns TA are quite similar for 11, 12 and 13, indicating the lack of interactions between the triplet excited states of the texaphyrin component and the Rhodamine 6G component. The broadband ground-state absorption of 11 from the visible to the near-IR region, and the possible electron/energy transfer from the singlet excited state of the Rhodamine 6G component to the texaphyrin component suggest that this complex could potentially be a broadband photosensitizer for dye-sensitized solar cell applications.
C bond was synthesized and characterized. The UV-vis absorption, emission and nanosecond transient absorption (TA) characteristics of this complex were systematically studied in acetone solutions. The photophysics of this complex was also compared with those of its precursor compounds texaphyrins 12 and 13 and the Rhodamine 6G derivative 3. The UV-vis absorption spectrum of 11 consists of both the characteristic Soret and Q bands of the texaphyrin derivative 13 and the identical absorption band from the Rhodamine 6G derivative 3. When excited at 550 nm (the major absorption band of Rhodamine 6G), 11 exhibits fluorescence bands from both the Rhodamine 6G component (582 nm) and texaphyrin component (802 nm), but the intensity of the 582 nm band dramatically reduced accompanied by a significant increase of the 802 nm band compared to those from 3 and 13, indicating electron/energy transfer from the singlet excited state of Rhodamine 6G. The ns TA spectrum of 11 resembles that of the texaphyrin derivative 13 but with both the bleaching band and absorption band red-shifted. The triplet lifetimes deduced from the decay of ns TA are quite similar for 11, 12 and 13, indicating the lack of interactions between the triplet excited states of the texaphyrin component and the Rhodamine 6G component. The broadband ground-state absorption of 11 from the visible to the near-IR region, and the possible electron/energy transfer from the singlet excited state of the Rhodamine 6G component to the texaphyrin component suggest that this complex could potentially be a broadband photosensitizer for dye-sensitized solar cell applications.
Introduction
Dye-sensitized solar cells (DSSCs) are considered as the most promising innovative solar energy conversion technology due to high incident solar light-to-electricity conversion efficiency (η) and low cost of production.1 To date, ruthenium polypyridyl-based complexes have been one of the most efficient sensitizers with the highest η value of 11.5%.2 However, the ruthenium sensitizers are toxic, expensive and display relatively low molar absorptivity especially in the near-IR region.3,4 To expand the absorption spectral region of DSSCs, great efforts have been put to the development of cyclic tetrapyrrole-based molecules,5,6 including porphyrins,7 chlorins,8 bacteriochlorins,9 and phthalocyanines.10 The interest in tetrapyrrole-based molecules is based on their extremely intense Soret band in the visible spectral region and their readily tunable Q bands. It is possible to red-shift the Q bands to the near-IR region by expanding the π-conjugation of the macrocyclic ligand,11 introducing different functional groups on the pyrrole moiety, axial ligation of the central metal,12 and coordination with different central metals (Mg, Zn, Fe, Ni, Cu, Pd, etc.).13 Although the Q bands could be red-shifted, the highest η value of the cell based on bacteriochlorin sensitizers was no more than 7.1%.9 It was reported that the increased probability of exciton annihilation from porphyrins in proximity could be accounted for their lower efficiency because porphyrins have an inherent tendency to aggregate.14 However, in recent years, Grätzel and co-workers reported that the efficiency of mesoscopic dye-sensitized solar cells based on donor–acceptor-substituted porphyrins could reach 11–12%,15,16 which exceeds the efficiency of Ru complex based DSSCs.Texaphyrins are pentaazadentate porphyrin-like aromatic macrocycles with extended π-conjugations and approximately 20% larger core size than porphyrins.17–19 They can coordinate with large metal ions to form almost coplanar configurations. As a result, the Q bands of texaphyrins could be bathochromically shifted to above 800 nm with an appropriate diaminoarene precursor,20,21 which makes it possible to efficiently absorb the solar energy in both the high energy and near-IR regions. However, due to the red-shifted Q bands, there appears to be a larger window (∼500–700 nm) between the Soret and the Q bands, which makes it insufficient to harvest light in the visible spectral region between 500 and 680 nm.22 For an ideal DSSC sensitizer, it is required that the absorption of the sensitizer should cover the full visible to near-IR spectral region. To improve the light-harvesting efficiency in the visible spectral region, we propose to introduce Rhodamine 6G that absorbs light intensely between 500 and 600 nm23,24 into the texaphyrin macrocycle with a CC linker to facilitate the conjugation between the texaphyrin macrocycle and Rhodamine 6G and allow for the possible electron or energy transfer from the Rhodamine 6G to the texaphyrin or vice versa to occur. It is expected that the absorption band from the attached Rhodamine 6G could fill in the gap between the Soret and Q bands in texaphyrins and thus could sufficiently increase the light-harvesting ability of texaphyrins in the visible region.
The structure of the target texaphyrin derivative (complex 11) with the attached Rhodamine 6G is shown in Chart 1 and the synthetic route to this complex is outlined in Scheme 1. The UV-vis absorption, emission and triplet transient absorption characteristics of complex 11 are investigated and reported in this paper. For comparison purposes, the photophysics of the parent texaphyrin complexes (12 and 13) and ethynyl Rhodamine 6G (compound 3) were also studied and reported herewith. A point worth mentioning is that this paper only focuses on the synthesis and photophysics of the new texaphyrin derivative 11; its application in DSSCs will be studied and reported later.
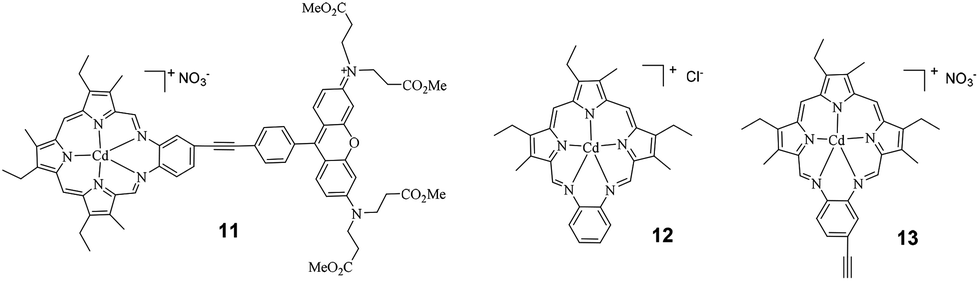 | ||
| Chart 1 Structures of the target complex 11 and the parent texaphyrins 12 and 13. | ||
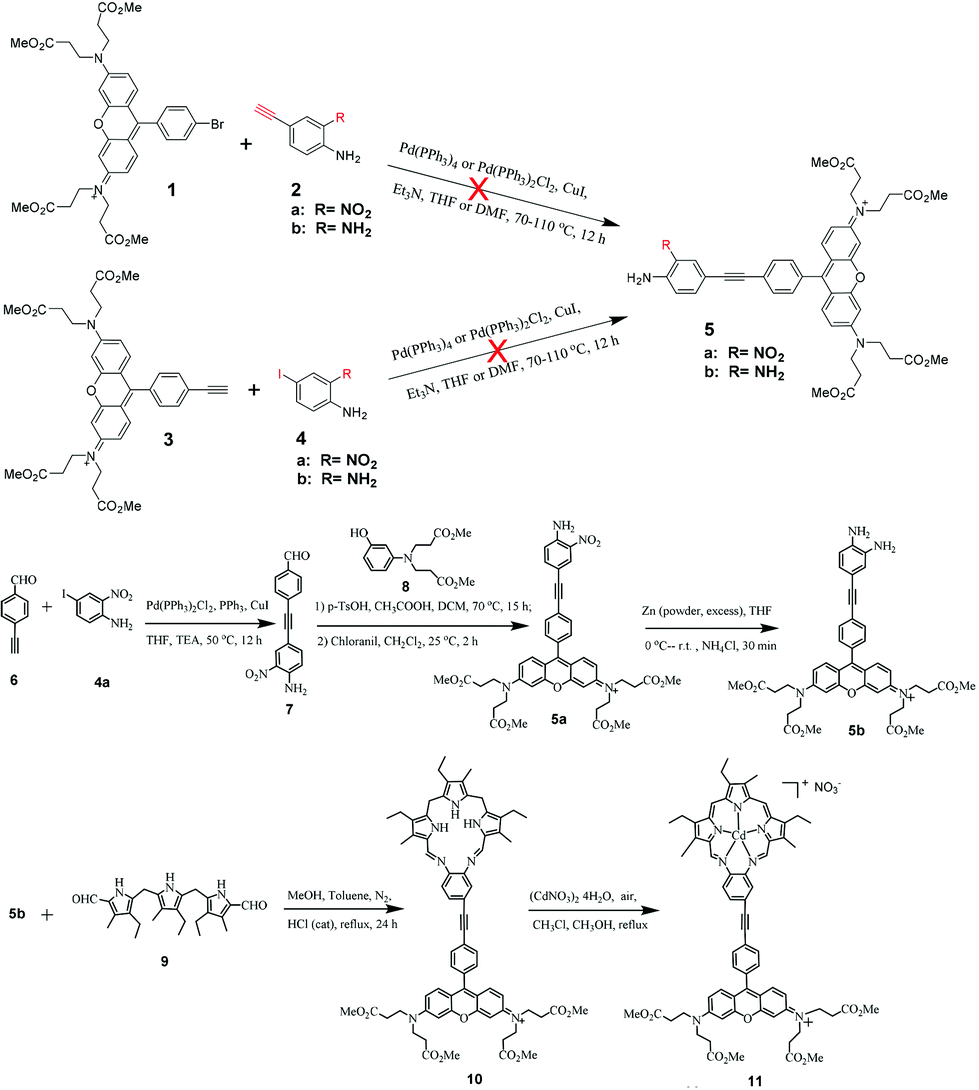 | ||
| Scheme 1 Synthetic route to the Rhodamine 6G pendant texaphyrin complex (11). | ||
Results and discussion
Synthesis
The general procedure for the synthesis of the Rhodamine 6G pendant texaphyrin derivative 11 follows the procedure originally reported by Sessler et al. for texaphyrins,17 namely using the acid-catalyzed Schiff base condensation reaction between the Rhodamine 6G ethynyl substituted benzene diamine (5b) and diformyltripyrrane (9) to form the macrocyclic ligand (10). Then 10 reacts with cadmium salt at room temperature in air to form 11. In this step, oxidation of the ligand and coordination with the Cd2+ ion occur simultaneously. As shown in Scheme 1, the key precursor for the synthesis of 11 is the diamine precursor 5b. Initially, we attempted to use the Sonogashira coupling reaction between brominated Rhodamine 6G (1) and 4-ethynyl-2-nitroaniline (2a) or 4-ethynylbenzene-1,2-diamine (2b), or use ethynyl Rhodamine 6G (3) to react with 4-iodo-2-nitroaniline (4a) or 4-iodobenzene-1,2-diamine (4b). Unfortunately, none of the aforementioned combinations yielded the desired product 5 regardless whether we used the standard Sonogashira coupling reaction conditions or modified conditions. Therefore, we adopted a new strategy to form the aldehyde precursor 7 with 4-ethynyl-2-nitroaniline attached first and then convert it to the Rhodamine 6G derivative 5a. Reduction of the nitro substituent resulted in the desired precursor 5b.Compounds 1,252a,26,274a,286,22,298,30 and 917 were synthesized according to the procedures reported in the literature with some modifications. The Sonogashira coupling reaction between 4-ethynylbenzaldehyde 6 and 4a afforded compound 7 as an orange powder. Reaction of compounds 7 and 8 using p-TsOH as the catalyst, followed by oxidation using chloranil at room temperature for 2 hours gave compound 5a in a very low yield (3.4%). 5a was then reduced with Zn powder in the presence of NH4Cl to afford the key precursor 5b. During the synthesis of the macrocyclic ligand 10, a water segregator was used to remove the water generated from the acid-catalyzed Schiff base condensation reaction between 5b and diformyltripyrrane 9 in order to move the reaction equilibrium forward towards the product.15 Because 10 was very difficult to be purified even after several times of column purification and recrystallization, the crude product confirmed by HRMS was directly used for the next step reaction without further purification. Complex 11 was synthesized from 10 following the procedure reported for texaphyrins.17,31 The reaction was monitored by UV-vis spectroscopy. During the reaction, the 375 nm peak corresponding to the ligand 10 gradually disappeared, while the Q band at ∼770 nm originating from the conjugated metal complex kept increasing. The reaction was stopped when no more change of the Q band intensity was observed. The crude product was purified by silica gel column chromatography with 3.3–20% methanol in dichloromethane (v/v) as the eluent, and then recrystallized from methanol and ethyl acetate. The structure and purity of 11 were verified by 1H and 13C NMR, HRMS and elemental analysis.
The synthesis and characterization of the reference complex 12 have been reported previously.32 The other reference complex 13 was obtained by the acid-catalyzed Schiff base condensation reaction between the 4-ethynyl-1,2-diaminobenzene (2b) and diformyltripyrrane (9) to form the macrocyclic ligand first; then complexation with cadmium salt at room temperature in air yielded complex 13.
Electronic absorption
The UV-vis absorption spectra of 3, 11, 12 and 13 in acetone are displayed in Fig. 1a. Complex 11 exhibits three characteristic absorption bands: the Soret bands appear in the 400–500 nm region (λmax = 464 nm, ε = 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 390 L mol−1 cm−1), while the Q bands are in the near-IR region (λmax = 772 nm, ε = 24920 L mol−1 cm−1). Compared to those of complex 12, the Soret and Q bands of 11 exhibit a 45 and 15 nm bathochromic shift, respectively, and the molar extinction coefficients are also significantly enhanced. This should be attributed to the increased π-conjugation in the texaphyrin through triplet bond connection with Rhodamine 6G, which is confirmed by the similar energies and molar extinction coefficients to those of 13. The most intense absorption band at 553 nm (ε = 89780 L mol−1 cm−1) appears at the identical position of the absorption band of 3, and thus is assigned to the 1π,π* transition from the Rhodamine 6G component. To understand whether the UV-vis absorption spectrum of 11 is a simple addition of the absorption bands from 3 and 12 or any interactions occur between these two components, we measured the absorption spectra of the mixture of equivalent 3 and 12 in acetone at the identical concentration of 1 × 10−5 mol L−1, and the spectrum is compiled in Fig. 1a as well. Comparing this spectrum with that of 11, we find that the Soret and Q bands in 11 are pronouncedly red-shifted and the molar extinction coefficients are higher in comparison with those of the mixed solution of 3 and 12; however, the absorption band at 553 nm remains at the same position but with a slightly decreased molar extinction coefficient. The red-shift and increased molar extinction coefficient of 11 are again due to the extended π-conjugation by the triple bond. This is confirmed by the similar energies and molar extinction coefficients of the absorption bands in 11 to those in the mixed equivalent 3 and 13 in acetone at the identical concentration of 1 × 10−5 mol L−1 (see the inset in Fig. 1a). However, the new band at 464 nm in 11 should be due to the interactions between the Rhodamine 6G component and the texaphyrin component.
390 L mol−1 cm−1), while the Q bands are in the near-IR region (λmax = 772 nm, ε = 24920 L mol−1 cm−1). Compared to those of complex 12, the Soret and Q bands of 11 exhibit a 45 and 15 nm bathochromic shift, respectively, and the molar extinction coefficients are also significantly enhanced. This should be attributed to the increased π-conjugation in the texaphyrin through triplet bond connection with Rhodamine 6G, which is confirmed by the similar energies and molar extinction coefficients to those of 13. The most intense absorption band at 553 nm (ε = 89780 L mol−1 cm−1) appears at the identical position of the absorption band of 3, and thus is assigned to the 1π,π* transition from the Rhodamine 6G component. To understand whether the UV-vis absorption spectrum of 11 is a simple addition of the absorption bands from 3 and 12 or any interactions occur between these two components, we measured the absorption spectra of the mixture of equivalent 3 and 12 in acetone at the identical concentration of 1 × 10−5 mol L−1, and the spectrum is compiled in Fig. 1a as well. Comparing this spectrum with that of 11, we find that the Soret and Q bands in 11 are pronouncedly red-shifted and the molar extinction coefficients are higher in comparison with those of the mixed solution of 3 and 12; however, the absorption band at 553 nm remains at the same position but with a slightly decreased molar extinction coefficient. The red-shift and increased molar extinction coefficient of 11 are again due to the extended π-conjugation by the triple bond. This is confirmed by the similar energies and molar extinction coefficients of the absorption bands in 11 to those in the mixed equivalent 3 and 13 in acetone at the identical concentration of 1 × 10−5 mol L−1 (see the inset in Fig. 1a). However, the new band at 464 nm in 11 should be due to the interactions between the Rhodamine 6G component and the texaphyrin component.
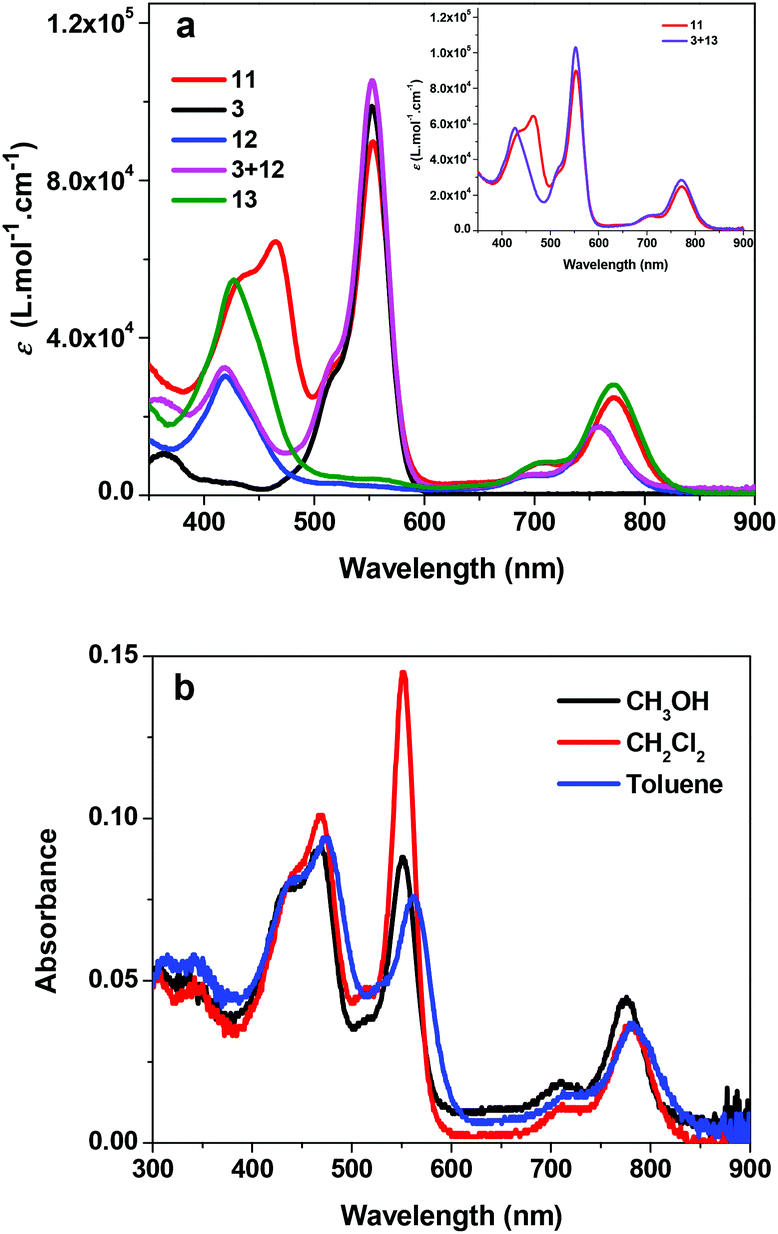 | ||
| Fig. 1 (a) UV-vis absorption spectra of 3, 11, 12, 13, and the mixture of equivalent 3 and 12, as well as 3 + 13 at the concentration of 1 × 10−5 mol L−1 in acetone. (b) UV-vis absorption spectra of 11 in different solvents. | ||
The concentration-dependence study shows that the UV-vis absorption of 11 obeys Lambert–Beer's law in the concentration range used in our study (1 × 10−6 to 1 × 10−4 mol L−1), indicating that no ground-state aggregation occurs in this concentration range. The solvent-dependent UV-vis absorption spectra of 11 were also measured and the results are shown in Fig. 1b. It appears that the absorption bands are slightly red-shifted in the less polar solvent toluene. In CH2Cl2 solution, only the Q band shows somewhat red-shift in comparison with that in methanol. Although the minor solvatochromic effect of 11 is in accordance with the π,π* transition nature of these bands, the observed negative solvatochromic effect implies that the excited state of 11 is slightly less polar than its ground state.
Photoluminescence
Fig. 2a shows the fluorescence spectra of 3, 11, 12, 13 and the mixture of equivalent 3 and 12 at the concentration of 1 × 10−5 mol L−1 in acetone when excited at the Q(0,0) band (i.e. 771 nm). For the Rhodamine 6G derivative 3, a strong emission appears at 582 nm, which is attributed to the two-photon induced upconverted fluorescence from 3.33 For the reference complexes 12 and 13, the emission occurs at ca. 787 nm and 802 nm, respectively, which originates from the respective 1π,π* states of 12 and 13. The equal–equivalent mixture of 3 and 12 at the same concentration of 1 × 10−5 mol L−1 gives an identical emission band at 787 nm to that of 12 and a slightly reduced intensity band at 582 nm. The slightly weaker emission at 582 nm for the mixed 3 and 12 solution compared to that of 3 should be attributed to the reduced efficient excitation energy towards the upconversion fluorescence because both the texaphyrin and Rhodamine 6G are excited with the 771 nm light. Taking this factor into account, we can conclude that no interactions occur between 3 and 12 when they are physically mixed with each other. In contrast, when the texaphyrin and Rhodamine 6G are covalently bonded to each other via a triple bond in complex 11, the emission bands appear at 582 nm and 802 nm with the emission intensity of the 582 nm band dramatically reduced. The red-shifted fluorescence at 802 nm should be attributed to the extended π-conjugation of the texaphyrin induced by the triplet bond, which is confirmed by the same emission energy as that of 13. The reduced intensity of the Rhodamine 6G upconverted fluorescence at 582 nm in 11 upon excitation at 771 nm implies that some degrees of interactions occur between the texaphyrin and Rhodamine 6G components. To verify the interaction between the texaphyrin and Rhodamine 6G in complex 11, the emissions of 3, 11, 12 and the mixed solution of 3 and 12 at the identical concentration are studied at 550 nm excitation, at which wavelength both the texaphyrin and Rhodamine 6G can be directly excited. As shown in Fig. 2b and in ESI Fig. S1,† upon excitation at 550 nm, 3 exhibits a very strong fluorescence at 582 nm, while 12 and 13 show a very weak fluorescence at 787 nm and 802 nm, respectively. The fluorescence from the mixed solution of 3 and 12 is predominantly from the Rhodamine 6G emission at 582 nm. However, the emission intensity at 582 nm drastically decreased while the emission at 802 nm significantly increased in the emission spectrum of 11 compared to that in respective 3, 12 and 3 + 12 solutions. This indicates that electron transfer or energy transfer from Rhodamine 6G to the texaphyrin component occurs. It has been reported that the reduction potential for [(TXP)Cd]+ in acetonitrile is −0.31 V vs. Ag/AgCl (corresponding to −0.33 V vs. SSCE) and the oxidation potential is 1.10 V vs. Ag/AgCl (corresponding to 1.08 V vs. SSCE),34 while the reduction potential for Rhodamine 6G in acetonitrile is −0.50 V vs. SSCE and the oxidation potential is 0.55 V vs. SSCE.35 According to the equation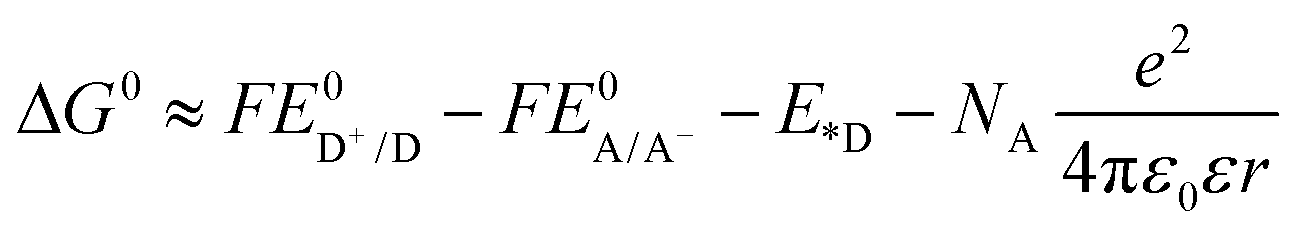 (where F is the Faraday constant),36 the free energy change for electron transfer from the singlet excited state of Rhodamine 6G to the texaphyrin is estimated to be approximately −250 kJ mol−1; while the back electron transfer from the singlet excited state of the texaphyrin to Rhodamine 6G is approximately −220 kJ mol−1. Therefore, electron transfer from Rhodamine 6G to the texaphyrin is more exothermic and thus feasible in acetonitrile. Although the solvent used in our emission study is acetone, the reduction potentials for Rhodamine 6G and the texaphyrin would be different from those in acetonitrile, and thus the ΔG0 value in acetone would be different from −250 kJ mol−1, this estimation still provides valuable information on the feasibility of electron transfer from Rhodamine 6G to the texaphyrin in 11. However, energy transfer from Rhodamine 6G to the texaphyrin is also likely to occur in view of the overlap of the emission band of Rhodamine 6G and the weak absorption from the texaphyrin in the same spectral region. To estimate the fluorescence resonance energy transfer (FRET) efficiency, the quantum yields of the fluorescence band at 582 nm of 3 and 11 in acetone solutions were measured using Rhodamine 6G in ethanol as the reference (Φf = 0.95 at λex = 480 nm).37 This gives an Φf of 0.29 for 3 and 0.033 for 11 upon excitation at 530 nm (this excitation wavelength was chosen to ensure the primary excitation being the Rhodamine 6G component in 11), corresponding to an approximately 89% of energy transfer efficiency.
(where F is the Faraday constant),36 the free energy change for electron transfer from the singlet excited state of Rhodamine 6G to the texaphyrin is estimated to be approximately −250 kJ mol−1; while the back electron transfer from the singlet excited state of the texaphyrin to Rhodamine 6G is approximately −220 kJ mol−1. Therefore, electron transfer from Rhodamine 6G to the texaphyrin is more exothermic and thus feasible in acetonitrile. Although the solvent used in our emission study is acetone, the reduction potentials for Rhodamine 6G and the texaphyrin would be different from those in acetonitrile, and thus the ΔG0 value in acetone would be different from −250 kJ mol−1, this estimation still provides valuable information on the feasibility of electron transfer from Rhodamine 6G to the texaphyrin in 11. However, energy transfer from Rhodamine 6G to the texaphyrin is also likely to occur in view of the overlap of the emission band of Rhodamine 6G and the weak absorption from the texaphyrin in the same spectral region. To estimate the fluorescence resonance energy transfer (FRET) efficiency, the quantum yields of the fluorescence band at 582 nm of 3 and 11 in acetone solutions were measured using Rhodamine 6G in ethanol as the reference (Φf = 0.95 at λex = 480 nm).37 This gives an Φf of 0.29 for 3 and 0.033 for 11 upon excitation at 530 nm (this excitation wavelength was chosen to ensure the primary excitation being the Rhodamine 6G component in 11), corresponding to an approximately 89% of energy transfer efficiency.
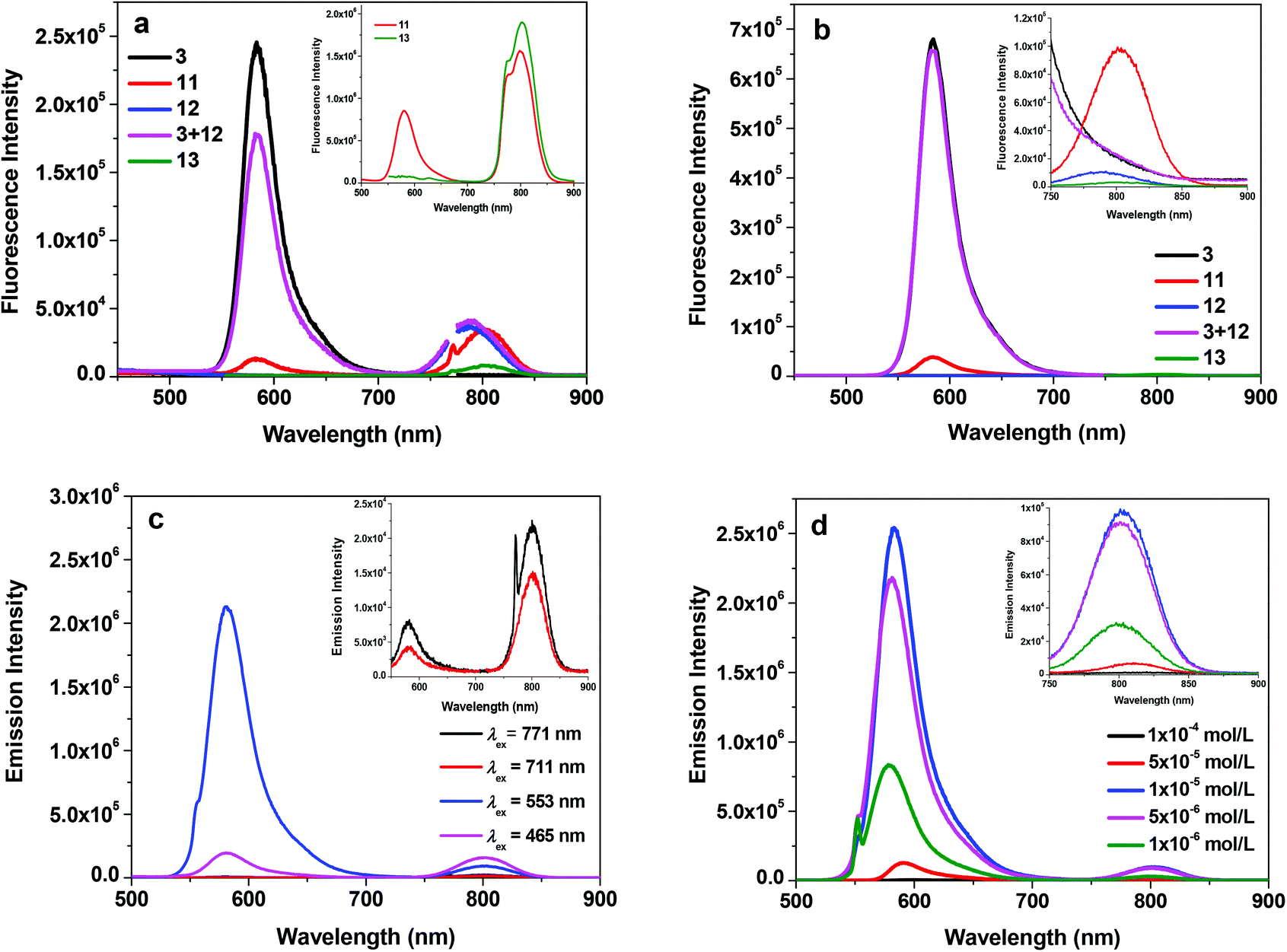 | ||
| Fig. 2 (a) Fluorescence spectra of 3, 11, 12, 13 and the mixture of equivalent 3 and 12 at the concentration of 1 × 10−5 mol L−1 in acetone, λex = 771 nm for all of the samples. The inset shows the comparison of the fluorescence intensity of 11 and 13 under the identical excitation conditions of A771 nm = 0.05 in a 1 cm cuvette. (b) Fluorescence spectra of 3, 11, 12, 13 and the mixture of equivalent 3 and 12 at the concentration of 1 × 10−5 mol L−1 in acetone, λex = 550 nm for all of the samples. The inset shows the NIR emission band at a larger slit width upon 550 nm excitation. (c) Emission spectra of 11 in acetone at different excitation wavelengths (c = 1 × 10−5 mol L−1). (d). Emission spectra of 11 at different concentrations in acetone, λex = 550 nm. | ||
It is worth noting that except for excitation at 550 nm, excitation at Soret band (465 nm) and Q bands (711 and 771 nm) all gives rise to comparable or stronger fluorescence at 802 nm than that at 582 nm (Fig. 2c) for complex 11 due to direct excitation of the texaphyrin component. The fluorescence quantum yields of 11 (including both the emission bands at 582 nm and 802 nm) at different excitation wavelengths were identified to be 0.006 at λex = 465 nm, 0.027 at λex = 550 nm, 0.003 at λex = 711 nm, and 0.0016 at λex = 771 nm.
The concentration-dependent fluorescence study was also carried out for 11 in acetone upon excitation at 550 nm and the results are shown in Fig. 2d. The fluorescence intensities of 11 at both 582 nm and 802 nm increase from the concentration of 1 × 10−6 mol L−1 to 1 × 10−5 mol L−1. However, the intensity starts to decrease at the concentration of 5 × 10−5 mol L−1. Meanwhile, the emission bands are slightly red-shifted. The decreased fluorescence intensity and slightly red-shifted fluorescence bands are a clear indication of the inner-filter effect. However, the self-quenching effect cannot be excluded, which could also contribute to the reduced fluorescence intensity at higher concentrations.
Fig. 3 shows the fluorescence spectra of complex 11 in different solvents. The energy of the near-IR band is obviously red-shifted in less polar solvents, which is in line with that observed from the UV-vis absorption spectra (Fig. 1b). The energy of the emission band at ca. 580 nm does not exhibit a significant change; however, the intensity of this band is dramatically decreased in methanol and toluene.
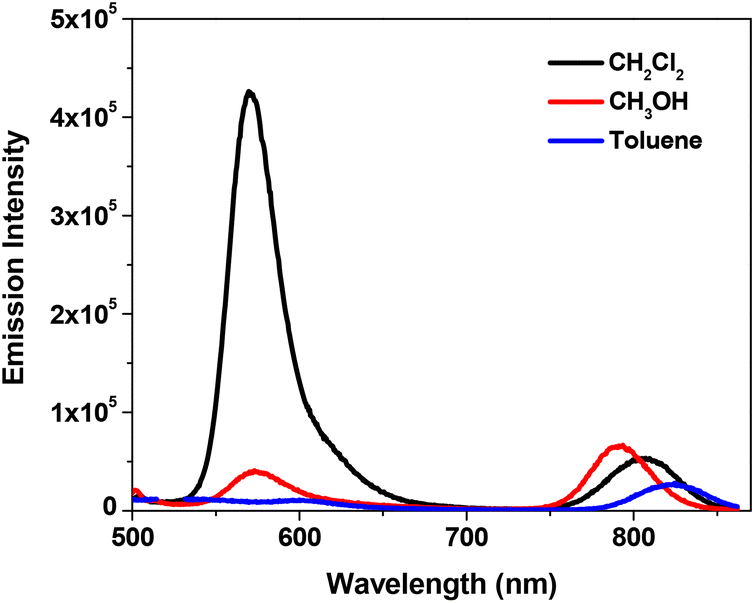 | ||
| Fig. 3 Fluorescence spectra of 11 in different solvents (λex = 436 nm). The concentrations of the sample solutions were adjusted in order to obtain the same absorbance of 0.08 at 436 nm in a 1 cm cuvette. | ||
Triplet transient absorption (TA)
In order to understand the triplet excited-state characteristics, nanosecond transient absorption spectra and the decay characteristics of 3, 11, 12, 13 and the mixture of 3 and 12 were investigated. Fig. 4 shows the TA spectra of 3, 11–13 and the mixture of 3 and 12 in acetone at zero delay after 355 nm laser excitation. The TA spectrum of 3 featured a very weak positive band between 400 and 500 nm and a strong bleaching band at ∼560 nm. The TA spectra of 11–13 and 3 + 12 all feature two bleaching bands that correspond to the Soret and Q bands in their respective UV-vis absorption spectra and a broad, positive absorption band between these two bleaching bands. These features imply that the excited state that gives rise to the observed TA spectra should be the 3π,π* state from the texaphyrin. Similar to the trend observed from the UV-vis absorption, the bleaching bands and the positive absorption band of 11 are red-shifted compared to 12, attributing to the extended π-conjugation by the triple bond. However, the triplet lifetimes obtained from the decay of the TA for all complexes are similar, all on the order of ∼12 μs. This indicates that no energy or electron transfer occurs between the texaphyrin and Rhodamine 6G components at the triplet excited state.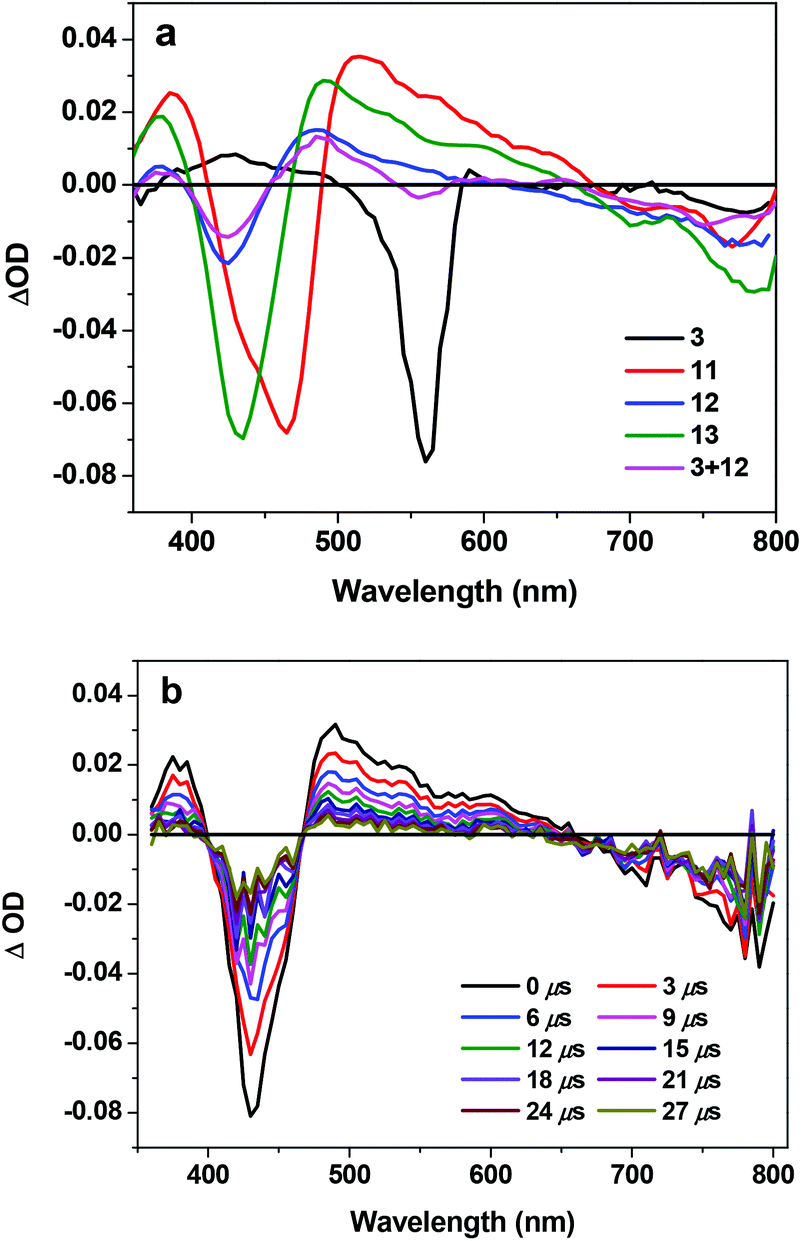 | ||
| Fig. 4 (a) Nanosecond transient difference absorption spectra of 3, 11–13 and the mixture of 3 and 12 in acetone at zero delay after 355 nm laser excitation. A355 nm = 0.4 in a 1 cm cuvette for all of the samples. (b) Time-resolved ns TA spectra of 13 in acetone. λex = 355 nm, A355 nm = 0.4 in a 1 cm cuvette. | ||
Conclusions
A texaphyrin derivative with the pendant Rhodamine 6G component (complex 11) via a CC bond was synthesized and characterized. The photophysics of this complex and its parent complexes 12 and 13 and the Rhodamine 6G derivative 3 were all systematically investigated in acetone solutions under identical experimental conditions. It is found that the UV-vis absorption spectrum of 11 is essentially a simple addition of the absorption spectra of the texaphyrin derivative 13 and the Rhodamine 6G derivative 3 except for the new band at 464 nm, suggesting that little interaction occurs between the texaphyrin component and the Rhodamine 6G component in the ground state. However, when excited at the major absorption band of Rhodamine 6G (550 nm), the intensity of the 582 nm fluorescence band from the Rhodamine 6G component is dramatically reduced accompanied by a significant increase of the 802 nm band from the texaphyrin component, indicating the occurrence of electron/energy transfer from the singlet excited state of the Rhodamine 6G component to the texaphyrin component in 11. The ns TA study suggests that the triplet transient absorption of 11 is dominated by the texaphyrin component, with a long triplet excited-state lifetime (τT ≈ 12 μs), which implies the lack of interactions between the triplet excited states of the Rhodamine 6G component and the texaphyrin component. The broadband ground-state absorption of 11 that covers most of the visible to the near-IR region and the possible electron/energy transfer from the singlet excited state of the Rhodamine 6G component to the texaphyrin component suggest that this complex could potentially be used as a broadband photosensitizer for dye-sensitized solar cell applications. Future work will be focused on covering the gap between 600 and 670 nm in the absorption spectrum of 11 and replacing the cadmium central metal ion with the environmentally benign Zn metal ion. For DSSC application, it is also necessary to hydrolyze the ester groups on the Rhodamine 6G component to carboxyl groups in order to anchor this sensitizer to the TiO2 layer.
Experimental section
Synthesis and characterization
All reagents and solvents were purchased from commercial sources and used as is unless otherwise mentioned. THF and benzene were distilled over sodium. Dichloromethane was distilled over CaH2.The synthetic scheme for complex 11 was outlined in Scheme 1. The synthesis of 1, 2a, 4a, 6, 8, and 9 followed the literature procedures.17,25–30 Diamine compounds 2b, 4b, and 5b were synthesized according to the procedures reported by Bahmanyar et al.38 The synthesis of 2a and 7 followed the procedures reported by Tour et al.27 Compound 3 was synthesized by the reaction of 4-ethynylbenzaldehyde with diester 8 under acidic condition followed by oxidation with chloranil. Compounds 10 and 11 were obtained following the procedures reported by Sessler et al.17,31 The synthesis and characterization of complex 12 were reported previously.32 The synthesis of complex 13 also followed the literature procedure.17,31 The synthetic details and the characterization data are provided below.
1H NMR spectra were recorded on a 400 or 500 MHz NMR spectrometer at room temperature. ESI-HRMS analysis was conducted on an electrospray ionization/time of flight (TOF) mass spectrometer. Elemental analyses were carried out by a commercial company.
2b. To a rapidly stirred suspension of 1.55 g powdered Zn in 10 mL of concentrated ammonium hydroxide, a 10 mL THF solution containing 4-ethynyl-2-nitroaniline (2a) (0.5 mg, 3.07 mmol) was added. The mixture was stirred at room temperature for 2 hours. After the reaction, the reaction mixture was filtered and the mother liquor was washed with diethyl ether. The solvent was then removed, and the residual oil was purified by silica gel column chromatography. The impurity was first removed by the CH2Cl2 eluent, then elution with ethyl acetate yielded 0.23 g brown oil as the product (yield: 57%). 1H NMR (400 MHz, CDCl3): δ 6.85 (d, J = 7.6 Hz, 1H), 6.76 (s, 1H), 6.50 (d, J = 7.6 Hz, 1H), 3.47 (s, 4H), 2.97 (s, 1H). 13C NMR (100 MHz, CDCl3, TMS) δ 74.5, 84.5, 112.8, 115.9, 120.3, 125.0, 133.9, 136.2. ESI-HRMS calcd for C16H17N4 [2M + H]+: 265.1148, found: 265.1147.
3. Compounds 4-ethynylbenzaldehyde (0.75 g, 5.98 mmol), diester 8 (3.36 g, 11.95 mmol), and p-TsOH (0.057 g, 2.98 mmol) were dissolved in 10 mL acetic acid. The reaction mixture was heated to 70 °C and stirred for 12 hours. After the reaction mixture was cooled to r.t., 50 mL water was added. The mixture was extracted with CH2Cl2 three times. The CH2Cl2 layers were combined and dried with MgSO4. Then chloranil (0.22 g, 0.90 mmol) was added and stirred at room temperature for 2 hours. After removal of the solvent, the residue was purified by column chromatography (silica gel; CH3OH–CH2Cl2 (1:10–1:5 v/v) was used as the eluent) to yield 0.2 g sticky purple solid as the crude product (yield: 5%). Part of the crude product was further purified by preparative TLC (silica gel, CH3OH–CH2Cl2 (1:10 v/v)) to afford the pure compound 3 as a purple powder (∼10 mg), which was used for the photophysical studies. 1H NMR (500 MHz, CDCl3): δ 7.72 (d, J = 7.5 Hz, 2H), 7.39–7.36 (m, 4H), 7.06–7.02 (m, 4H), 3.99 (s, 8H), 3.68 (s, 12H), 3.29 (s, 1H), 2.78 (s, 8H). 13C NMR (100 MHz, CDCl3, TMS) δ 32.1, 47.5, 52.1, 77.2, 98.0, 113.9, 114.9, 117.9, 129.5, 130.3, 131.7, 131.9, 132.6, 155.9, 158.1, 171.5. ESI-HRMS calcd for [C37H39N2O9]+: 655.2650; found: 655.2632. Anal. calcd for C37H39N2O9·CH2Cl2·C6H14·1.5H2O: C, 61.89; H, 6.85; N, 3.28; found: C, 61.53, H, 6.61; N, 3.95.
4b. Compounds 4-iodo-2-nitroaniline (1.00 g, 3.78 mmol) and NH4Cl (2.00 g, 37.8 mmol) were dissolved in a mixture of MeOH (20 mL) and THF (20 mL). Zn powder (2.46 g, 37.8 mmol) was then gradually added at 0 °C. The mixture was stirred at room temperature for 40 min, and then filtered and washed with diethyl ether. After that, the solvent was removed and the residue was recrystallized in MeOH–CH2Cl2 and hexane to obtain 0.65 g gray solid as the product (yield: 74%). 1H NMR (500 MHz, CDCl3): δ 6.98 (d, J = 8.0 Hz, 1H), 6.94 (s, 1H), 6.41 (d, J = 8.0 Hz, 1H), 3.42 (s, 4H). 13C NMR (100 MHz, CDCl3, TMS) δ 81.3, 118.3, 124.7, 128.7, 134.4, 136.4. Anal. calcd for C6H7N2I: C, 30.79; H, 3.01; N, 11.97; found: C, 31.21, H, 3.43; N, 11.77.
7. Compounds 4-ethynylbenzaldehyde (0.2 g, 1.5 mmol), 5-iodo-2-nitroaniline (0.4 g, 1.5 mmol), Pd(PPh3)2Cl2 (53 mg, 0.077 mmol), PPh3 (20 mg, 0.077 mmol) and CuI (14.7 mg, 0.077 mmol) were all added in 10 mL THF and 5 mL triethylamine, and the reaction mixture was degassed. The reaction mixture was stirred at 50 °C for 20 hours. After the reaction, the mixture was washed with brine and dried over MgSO4. Then the solvent was removed, and the residue was purified by column chromatography (silica gel; hexane–CH2Cl2 (1:1 v/v) was used as the eluent) to obtain 0.3 g orange powder as the product (yield: 75%). 1H NMR (400 MHz, CDCl3): δ 9.99 (s, 1H), 8.34 (s, 1H), 7.84 (d, J = 8 Hz, 2H), 7.62 (d, J = 8 Hz, 2H), 7.48 (d, J = 8.8 Hz, 1H), 6.79 (d, J = 8.8 Hz, 1H), 6.26 (s, 2H). 13C NMR (100 MHz, CDCl3, TMS) δ 86.2, 87.9, 91.7, 111.2, 119.0, 129.6, 130.0, 131.9, 135.4, 138.2, 144.6, 191.4. Anal. calcd for C15H10N2O3·0.1CH3CH2OCH2CH3·0.1C6H14·0.8H2O: C, 64.77; H, 4.76; N, 9.44; found: C, 64.32, H, 4.82; N, 9.92.
5a. A mixture of compound 7 (2.00 g, 7.50 mmol), diester 8 (4.21 g, 15.02 mmol), and p-TsOH (210 mg, 1.12 mmol) in mixed CH2Cl2 (20 mL) and acetic acid (50 mL) was heated to 80 °C and stirred for 15 hours. After the reaction mixture was cooled to r.t., 50 mL water was added. Then the mixture was extracted with CH2Cl2 three times. The CH2Cl2 layer was combined and dried over MgSO4. After that, chloranil (46 mg, 0.19 mmol) was added to the CH2Cl2 solution and the mixture was stirred at r.t. for 2 hours. After removal of the solvent, the residue was purified by column chromatography four times (silica gel; CH3OH–CH2Cl2 (1:10–1:5 v/v) was used as the eluent) to yield 0.2 g purple solid (yield: 3.4%). 1H NMR (CDCl3, 500 MHz) δ 8.01 (s, 1H), 7.66 (s, 2H), 7.65 (d, J = 7.5 Hz, 2H), 7.43 (d, J = 9.5 Hz, 2H), 7.39 (d, J = 7.5 Hz, 2H), 7.13–7.18 (m, 2H), 7.07 (J = 9.5 Hz, 2H), 7.00 (s, 2H), 4.03 (t, J = 6.0 Hz, 8H), 3.73 (s, 12H), 2.81 (t, J = 6.0 Hz, 8H). 13C NMR (125 MHz, CDCl3, TMS) δ 32.1, 47.6, 52.2, 87.1, 91.6, 97.7, 109.1, 113.7, 114.9, 120.2, 126.1, 129.1, 130.0, 130.1, 130.6, 132.0, 137.2, 146.4, 155.8, 157.9, 171.5. ESI-HRMS calcd for [C43H43N4O11]+: 791.2923, found: 791.2910.
5b. Compound NH4Cl (70 mg, 1.30 mmol) and Zn powder (85 mg, 1.30 mmol) were added to a mixture of methanol (10 mL) and THF (10 mL) and stirred vigorously first. Then the solution of 5a (0.1 g, 0.13 mmol) in 10 mL THF was gradually added at 0 °C. The mixture was stirred at room temperature for 30 min, and then was filtered and the mother liquor was washed with diethyl ether three times. After removal of the solvent, the residue was purified by silica gel column chromatography, with mixed CH3OH–CH2Cl2 (1:10–1:3 v/v) being used as the eluent. 80 mg purple solid was obtained as the product 5b (yield: 81%). 1H NMR (400 MHz, CDCl3): δ 7.63 (d, J = 8 Hz, 2H), 7.43 (d, J = 9.2 Hz, 2H), 7.32 (d, J = 8 Hz, 2H), 7.04 (d, J = 9.6 Hz, 2H), 6.97 (s, 2H), 6.82 (d, J = 8.4 Hz, 2H), 6.57 (d, J = 8 Hz, 1H), 3.97 (s, 8H), 3.67 (s, 12H), 2.75 (t, J = 6.4 Hz, 8H). 13C NMR (125 MHz, CDCl3, TMS) δ 32.1, 47.6, 52.2, 91.4, 93.0, 97.9, 109.7, 111.4, 113.7, 114.8, 119.9, 126.0, 129.4, 129.9, 130.66, 130.7, 131.8, 132.0, 137.8, 145.9, 155.9, 158.0, 171.5. ESI-HRMS calcd for [C43H45N4O9]+: 761.3181, found: 761.3195.
10. Compounds 9 (95 mg, 0.21 mmol) and 5b (163 mg, 0.21 mmol) were dissolved in a degassed mixture of 100 mL dry toluene and 25 mL absolute methanol. Concentrated HCl (0.05 mL) was then added and the resulting dark brown solution was heated to reflux for 24 hours under an argon atmosphere. A water segregator was used to remove the water generated from the reaction. After cooling, K2CO3 (50 mg) was added to neutralize the HCl and the solution was then filtered through MgSO4. The solvent was then removed and the residue was dissolved in 10 mL CH2Cl2. Addition of 30 mL hexane precipitated out some sticky dark brown solid. This solid was recrystallized with CH2Cl2–hexane multiple times, which yielded dark red powder of 103 mg (yield: 43%). This compound was directly used for the next step reaction without further purification. ESI-HRMS calcd for [C68H74N7O9]+: 1132.5543, found: 1132.5508.
11. Compound 10 (100 mg, 0.088 mmol) was dissolved in 20 mL CHCl3, and Cd(NO3)2·4H2O (81.7 mg, 0.26 mmol) was dissolved in 50 mL methanol. The two solutions were mixed together and were heated to reflux for 72 hours while bubbling with air. UV-vis spectroscopy was used to monitor the reaction. During the reaction, the 375 nm peak corresponding to the ligand 10 gradually disappeared, while the Q band at ∼770 nm originating from 11 kept increasing. The reaction was stopped when no more change of the Q band intensity was observed. After the reaction, the solvent was removed and the residue was dissolved in CH2Cl2 and washed with water, and then dried over MgSO4. CH2Cl2 was removed and the residue was purified by silica gel column chromatography with CH3OH–CH2Cl2 (1:30–1:5 v/v) being used as the eluent, which yielded 30 mg purple solid. Further purification of these solids using preparative TLC plates (silica gel, CH3OH–CH2Cl2 (1:5 v/v) was used as the eluent), and recrystallization from methanol and ethyl acetate afforded 10 mg reddish brown powder as the pure product (yield: 8.7%). 1H NMR (400 MHz, CDCl3): δ 10.58 (s, 1H), 10.50 (s, 1H), 9.02 (s, 3H), 8.72 (s, 1H), 7.89 (s, 2H), 7.77 (s, 1H), 7.51 (s, 4H), 7.14 (s, 2H), 6.99 (s, 2H), 3.94 (s, 8H), 3.68 (s, 12H), 3.35 (s, 6H), 2.92 (s, 3H), 2.77 (s, 8H), 2.61 (s, 3H), 2.42 (s, 3H), 1.53 (m, 9H). 13C NMR (125 MHz, CDCl3, TMS) δ 17.3, 19.1, 29.3, 29.7, 32.2, 47.7, 52.1, 87.9, 90.8, 97.4, 109.2, 113.61, 113.64, 113.9, 115.35, 115.42, 116.77, 116.81, 125.66, 125.69, 127.8, 130.5, 132.5, 136.9, 138.7, 139.5, 145.6, 146.56, 146.62, 149.3, 150.9, 154.6, 155.7, 157.7, 171.7. ESI-HRMS calcd for [C68H69CdN7O9]+: 620.7100; found: 620.7089. Anal. calcd for C68H69CdN8O12·2CH2Cl2·6CH3OH: C, 55.52; H, 6.19; N, 6.56; found: C, 55.63; H, 6.21; N, 6.24.
13. Compounds 9 (200 mg, 0.49 mmol) and 2b (64 mg, 0.49 mmol) were dissolved in a degassed mixture of 100 mL dry toluene and 25 mL absolute methanol. 0.05 mL concentrated HCl was then added and the resulting yellowish brown solution was heated to reflux for 24 h under a nitrogen atmosphere. A water segregator was used to remove the water generated from the reaction. After cooling, K2CO3 (20 mg) was added to neutralize the HCl and the solution was then filtered through MgSO4. After removal of the solvent, the residue was recrystallized from CH2Cl2–hexane to give 200 mg dark brown powder, which was confirmed by MS (m/z = 503.29) to be the desired macrocyclic ligand (yield: 81%). This ligand was directly used for the next step reaction without further purification.
The obtained ligand (200 mg, 0.40 mmol) was dissolved in 20 mL CHCl3, and Cd(NO3)2·4H2O (320 mg, 1.04 mmol) was dissolved in 50 mL methanol. These two solutions were mixed together and the reaction mixture was heated to reflux for 72 hours while bubbling with air. During the reaction, the 365 nm peak corresponding to the ligand gradually disappeared, while the Q band at ∼770 nm emanating from 13 kept increasing. The reaction was stopped when the intensity of the Q band no longer changed. After the reaction, the solvent was removed and the residue was purified by silica gel column chromatography with CH2Cl2–acetone (1:20 v/v) being used as the eluent. Further purification via recrystallization from CH2Cl2–hexane yielded 100 mg dark yellow solid (yield: 37%). 1H NMR (500 MHz, CDCl3): δ 11.28 (s, 2H), 9.58 (m, 1H), 9.41 (m, 1H), 9.28 (d, 1H), 8.38 (m, 2H), 3.55–3.50 (m, 12H), 3.08–3.07 (m, 4H), 1.66–1.52 (m, 9H). ESI-HRMS calcd for [C33H32CdN5 + H2O]+: 630.1800; found: 630.1766.
Photophysical measurements
UV-vis spectra were measured on a UV-VIS recording spectrophotometer in a 1 cm quartz cuvette. Steady-state fluorescence spectra at room temperature were measured on a fluorometer. The excitation wavelength was selected at the respective absorption band maxima. Spectrophotometric grade acetone was used as the solvent. A comparative method39 was used to determine the fluorescence quantum yield, with Rhodamine 6G in ethanol (Φf = 0.95 at λex = 480 nm)37 being used as the reference. The nanosecond transient difference absorption (TA) spectra and triplet excited-state lifetimes were measured in degassed acetone solutions on a laser flash photolysis spectrometer. The third harmonic output (355 nm) of a Nd:YAG laser (4.1 ns, repetition rate was set at 1 Hz) was used as the excitation source. Each sample was purged with argon for 30 min prior to measurement.Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS
.
- C.-Y. Chen, M. Wang, J.-Y. Li, N. Pootrakulchote, L. Alibabaei, C.-H. Ngoc-le, J.-D. Decoppet, J.-H. Tsai, C. Grätzel, C.-G. Wu, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2009, 3, 3103 CrossRef CAS PubMed
- F. Odobel and H. Zabri, Inorg. Chem., 2005, 44, 5600 CrossRef CAS PubMed
-
K. Kalyanasundaram, Photochemistry of Polypyridine and Porphyrin Complexes, Academic Press, London, 1992 Search PubMed
- X.-F. Wang, L. Wang, N. Tamai, O. Kitao, H. Tamiaki and S.-I. Sasaki, J. Phys. Chem. C, 2011, 115, 24394 CAS
- X.-F. Wang and H. Tamiaki, Energy Environ. Sci., 2010, 3, 94 CAS
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809 CrossRef CAS PubMed
- X.-F. Wang, H. Tamiaki, L. Wang, N. Tamai, O. Kitao, H. Zhou and S.-I. Sasaki, Langmuir, 2010, 26, 6320 CrossRef CAS PubMed
- X.-F. Wang, O. Kitao, H. Zhou, H. Tamiaki and S.-I. Sasaki, J. Phys. Chem. C, 2009, 113, 7954 CAS
- M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 760 CrossRef
- V. S. Lin, S. G. DiMagno and M. J. Therien, Science, 1994, 264, 1105 CAS
- M. Nappa and J. S. Valentine, J. Am. Chem. Soc., 1978, 100, 5075 CrossRef CAS
- P. Bhyrappa and V. Krishnan, Inorg. Chem., 1991, 30, 239 CrossRef CAS
- Y. Tachibana, S. A. Haque, I. P. Mereer, J. R. Durrant and D. R. Klug, J. Phys. Chem. B, 2000, 104, 1198 CrossRef CAS
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., 2010, 122, 6796 (
Angew. Chem., Int. Ed.
, 2010
, 49
, 6646
) CrossRef
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, Md. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed
- J. L. Sessler, M. R. Johnson and V. Lynch, J. Org. Chem., 1987, 52, 4394 CrossRef CAS
- J. L. Sessler, T. D. Mody, G. Hemmi and V. Lynch, Inorg. Chem., 1993, 32, 3175 CrossRef CAS
- J. L. Sessler, G. Hemmi, T. D. Mody, T. Murai, A. Burrell and S. W. Young, Acc. Chem. Res., 1994, 27, 43 CrossRef CAS
- T. Lu, P. Shao, I. Mathew, A. Sand and W. Sun, J. Am. Chem. Soc., 2008, 130, 15782 CrossRef CAS PubMed
- B. G. Maiya, T. E. Mallouk, G. Hemmi and J. L. Sessler, Inorg. Chem., 1990, 29, 3738 CrossRef CAS
- Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903 CrossRef CAS
- W. Lin, L. Yuan, Z. Cao, Y. Feng and J. Song, Angew. Chem., 2010, 122, 385 (
Angew. Chem., Int. Ed.
, 2010
, 49
, 375
) CrossRef
- K. Krishnamoorthy and T. P. Begley, J. Am. Chem. Soc., 2010, 132, 11608 CrossRef CAS PubMed
- L. Huang, L. Zeng, H. Guo, W. Wu, W. Wu, S. Ji and J. Zhao, Eur. J. Inorg. Chem., 2011, 4527 CrossRef CAS
- D. W. Price Jr., S. M. Dirk, F. Maya and J. M. Tour, Tetrahedron, 2003, 59, 2497 CrossRef
- F. Maya, S. H. Chanteau, L. Cheng, M. P. Stewart and J. M. Tour, Chem. Mater., 2005, 17, 1331 CrossRef CAS
- F. Maya and J. M. Tour, Tetrahedron, 2004, 60, 81 CrossRef CAS PubMed
- Y. Chen, Y. Wu, P. Henklein, K. Nakanishi, X. Li, O. P. Ernst and K. P. Hofmann, Chem. – Eur. J., 2010, 16, 7389 CrossRef CAS PubMed
- R. Bandichhor, A. D. Petrescu, A. Vespa, A. B. Kier, F. Schroeder and K. Burgess, Bioconjugate Chem., 2006, 17, 1219 CrossRef CAS PubMed
- J. L. Sessler, T. Murai, V. Lynch and M. Cyr, J. Am. Chem. Soc., 1988, 110, 5586 CrossRef CAS
- W. F. Sun and D. Y. Wang, Chin. Chem. Lett., 1993, 4, 225 CAS
- P. J. Schuck, K. A. Willets, D. P. Fromm, R. J. Twieg and W. E. Moerner, Chem. Phys., 2005, 318, 7 CrossRef CAS PubMed
- B. G. Maiya, A. Harriman, J. L. Sessler, G. Hemmi, T. Murai and T. E. Mallouk, J. Phys. Chem., 1989, 93, 8111 CrossRef CAS
- L. Bahadur and P. Srivastava, Sol. Energy Mater. Sol. Cells, 2003, 79, 235 CrossRef CAS
-
N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, CA, 2010, ch. 7, p. 421 Search PubMed
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1982, 27, 455 CrossRef
-
S. Bahmanyar, R. J. Bates, K. Blease, A. A. Calabrese, T. O. Daniel, M. Delgado, J. Elsner, P. Erdman, B. Fahr, G. Fercuson, B. Lee, L. Nadolny, G. Packard, P. Papa, K. V. Plantevin, J. Riggs, P. Rohanf, S. Sankar, J. Sapienza, Y. Satoh, R. Stevens, L. Tehrani, J. Tikhe, E. Torres, A. Wallace, B. W. Whitefield and J. Zhao, PCT Int. Appl, 2010027500, 2010 Search PubMed
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Fluorescence spectra of 3, 11, and 13 in acetone under the identical excitation conditions of A550 nm = 0.05 in a 1 cm cuvette. 1H and 13C NMR spectra of the reported compounds 2b, 3, 4b, 7, 5a, 5b, and 11, and the 1H NMR spectrum of 13. See DOI: 10.1039/c4qo00018h |
| This journal is © the Partner Organisations 2014 |