A turn-on fluorescent probe for detection of hydrogen sulfide in aqueous solution and living cells†
Dongjian
Zhu
ab,
Lin
Xue
a,
Guoping
Li
a,
Yanke
Che
a and
Hua
Jiang
*ac
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Photochemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: hjiang@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100190, P. R. China
cCollege of Chemistry, Beijing Normal University, Beijing 100875, P. R. China
First published on 15th April 2014
Abstract
We report the design and synthesis of a turn-on fluorescence probe FD-NO2 for H2S, utilizing H2S selective reduction of the nitro group. FD-NO2 displays good water solubility, negligible background fluorescence, high sensitivity, and high selectivity for H2S over other anions, reactive sulfur, oxygen, and nitrogen species. Further confocal imaging experiments demonstrate that FD-NO2 is capable of detecting H2S in living cells.
Introduction
Hydrogen sulfide (H2S) has been considered as a poisonous gas for many years.1 However, recent studies have demonstrated that H2S is the third endogenous gasotransmitter, in addition to nitric oxide (NO) and carbon monoxide (CO).2 At physiological concentrations, it is believed that H2S plays important roles in a number of physiological and pathological processes, including neurotransmission,3 vasorelaxation,4 cardioprotection,5 angiogenesis,6 anti-inflammation,7 and insulin signaling.8 On the other hand, H2S levels beyond the physiological range can lead to serious diseases such as Alzheimer's disease,9 Down's syndrome,10 diabetes,11 and liver cirrhosis.12 Although the important roles of H2S have been recognized, the complex contributions of H2S to both healthy and diseased states still remain unknown. Therefore, development of sensitive and selective methods for detection of H2S in aqueous solution and biological systems is strongly desired.Although traditional methods including colorimetry,13 electrochemical analysis,14 gas chromatography,15 and sulfide precipitation16 have been used for detection of H2S, these methods often require sample preprocessing and destruction of cells or tissues,17 which limit their application in biological studies. In contrast, fluorescence imaging provides a powerful method to study biomolecules in living systems because of its simplicity, high sensitivity, real-time detection, and non-destructive detection of biological samples.18 To date, various fluorescent probes for H2S detection have been reported. These probes were designed by taking advantage of several characteristic properties of H2S, including the binding affinity towards copper ions,19 thiolysis of dinitrophenyl ether,20 nucleophilicity,21 efficient reducing property towards selenoxide,22 and reduction of azide, hydroxy-amine and nitro to amine.23 It is superior to design probes with the nitro group based on the reduction feature of H2S, because the nitro group is a strong quencher for fluorophores24 and can significantly reduce the background signal in biological assays. On the other hand, rhodol has excellent photophysical properties such as high fluorescence quantum yield, photostability, good water solubility and relatively long emission wavelength.25 Hence, we present a novel rhodol-based fluorescent probe FD-NO2 for detection of H2S in aqueous solution and living cells (Scheme 1).
 | ||
| Scheme 1 Synthetic strategy for FD-NO2 and proposed reaction mechanism for H2S. | ||
Results and discussion
The synthetic strategy for preparing FD-NO2 is outlined in Scheme 1. Briefly, the synthesis of FD-NO2 was started from fluorescein, which is first conjugated with triethylene glycol monomethyl ether to yield FD-Tg. The incorporating ethylene glycolic moiety to fluorescein is in favor of the solubility of the probe in water and penetration into the cellular membrane. FD-Tg was triflated to provide FD-Tf, which was coupled with bis(pinacolato)diboron in the presence of the Pd catalyst to obtain the pinacolatoboron derivative FD-B. FD-B was then hydrolyzed to boronic acid compound, and further reacted with tert-butyl nitrite to afford FD-NO2. FD-NO2 was characterized by 1H and 13C NMR spectroscopy, and high resolution mass spectrometry.Since the nitro group is a strong fluorescence quencher, we anticipate that the closed lactone form FD-NO2 should be almost no fluorescence. In the presence of H2S, the nitro group of FD-NO2 was reduced to generate amine group, consequently leading to concomitant ring opening of the lactone so as to form the fluorescent product FA. We believe that this mechanism can be used to design a fluorescence turn-on probe for detecting H2S. To corroborate this design strategy, FD-NO2 was incubated with sodium sulphide (a H2S precursor)26 in methanol. The reaction solution was analyzed by fluorimetry and MALDI-TOF, respectively. The fluorescence experiment showed that a strong emission at 518 nm was detected (Fig. S1†), indicating the occurrence of reduction reaction and the generation of FA. On the other hand, a major mass peak m/z 478.1 detected by MALDI-TOF is assigned to the mass (M + H+) of the fluorescent conjugated product FA (Fig. S2†). The observations demonstrate that FD-NO2 can detect H2S by fluorescence enhancement. On the basis of the above results, the proposed reaction mechanism is shown in Scheme 1.
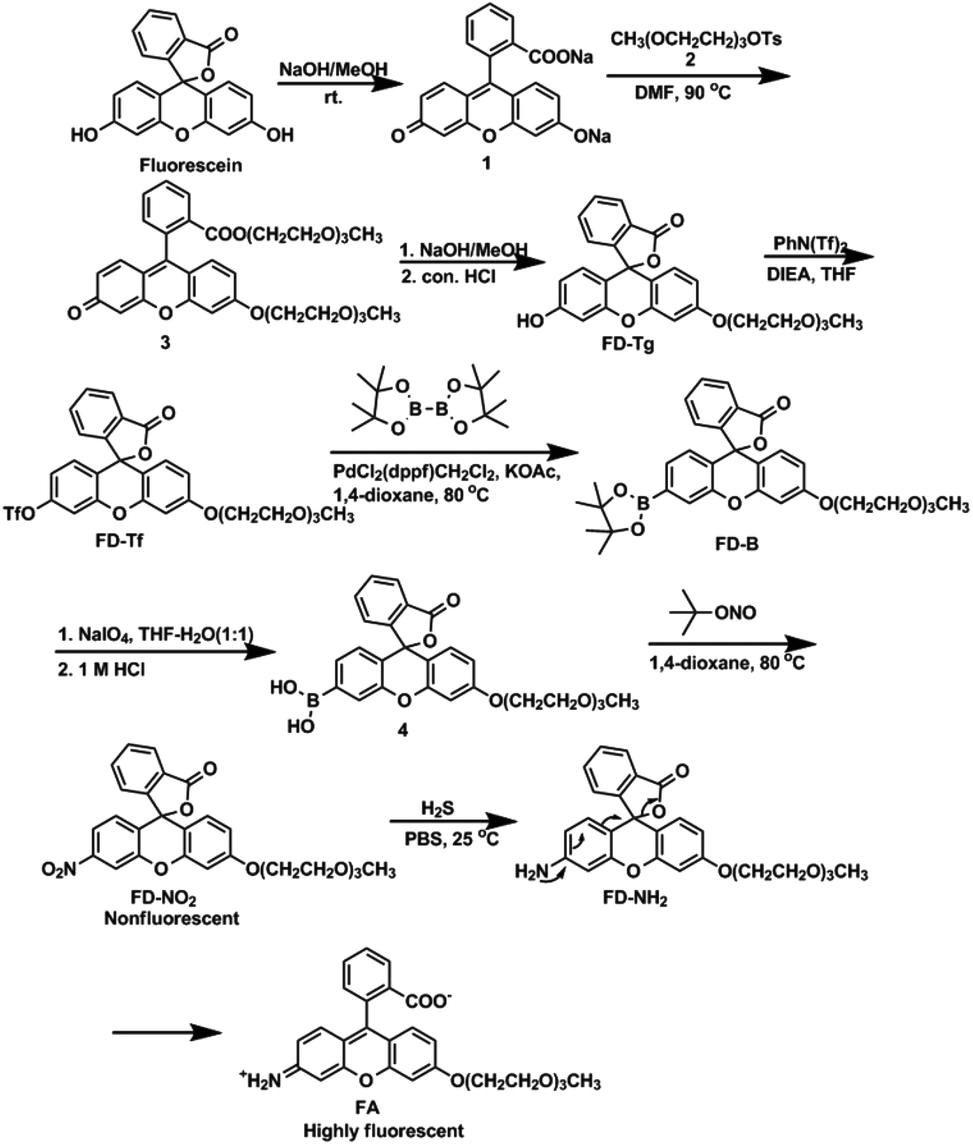
Next, we assessed the photophysical properties of FD-NO2 under simulated physiological conditions (10 mM PBS, pH 7.4, 25 °C). As expected, FD-NO2 (2 μM) has almost no fluorescence upon excitation at 480 nm. However, upon addition of H2S (100 μM) for 50 min, the FD-NO2 solution showed significant fluorescent emission, and the fluorescence intensity at 518 nm (Φ = 0.55) reached a plateau in 50 min with a 25-fold enhancement (Fig. 1).
 | ||
| Fig. 1 (a) Fluorescence response of 2 μM FD-NO2 to 100 μM H2S. Spectra were acquired every 5 min after H2S was added. (b) Fluorescence changes as a function of time for the reaction of 2 μM FD-NO2 to 100 μM H2S. Data were acquired at 25 °C in PBS buffer (pH 7.4), λex = 480 nm. | ||
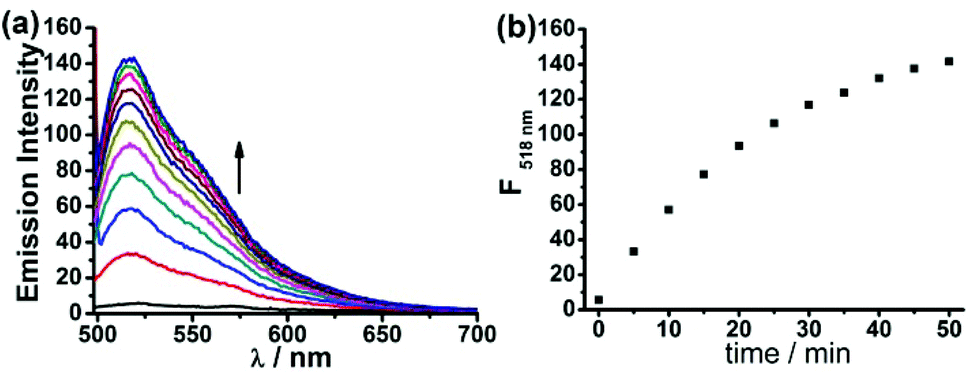
To evaluate the capability of the probe FD-NO2 in the measurement of H2S concentration, FD-NO2 was treated with H2S under various concentrations to obtain a standard curve of fluorescence intensity at 518 nm versus H2S concentration. The concentration of the compound FD-NO2 was maintained at 2 μM, while the concentrations of H2S varied from 0 to 100 μM. As shown in Fig. 2, the fluorescent signal was indeed linearly related to the concentration of H2S at this concentration range. The limit of detection (LOD) was determined to be as low as 1 μM (S/N = 3), indicating that our probe can potentially detect H2S with high sensitivity. Moreover, the effect of FD-NO2 concentration on the reaction was also studied, and the results showed very similar fluorescence response when the probe concentration is in the range of 2–10 μM (Fig. S6†).
 | ||
| Fig. 2 (a) Fluorescence spectra of FD-NO2 (2 μM) upon addition of H2S (0–100 μM). (b) Fluorescence intensities of FD-NO2 at 518 nm as a function of the concentrations of H2S in the range of 0–100 μM. Data were acquired at 25 °C in PBS buffer (pH 7.4), λex = 480 nm. | ||
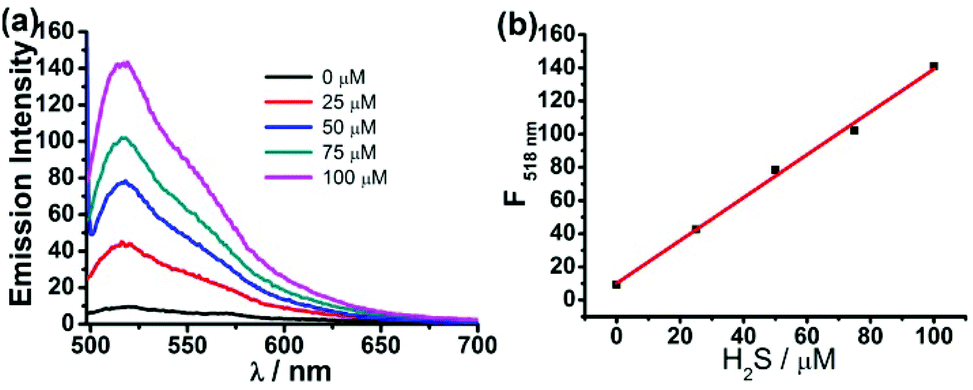
To study the selectivity profile of FD-NO2 for H2S, various potential interferences including anions, reactive sulfur species (RSS), reactive oxygen species (ROS), and reactive nitrogen species (RNS), were tested in parallel under identical conditions. As shown in Fig. 3, the fluorescence intensity was increased by over 25-fold in the presence of H2S (100 μM), while other anions (N3−, AcO−, 1 mM), RSS (SCN−, SO32−, S2O32−, 1 mM), ROS (H2O2, HClO, tBuOOH, O2−, 1 mM), RNS (NO, NO2−, NO3−, 1 mM), and in particular different thiol compounds (lipoic acid, Cys, Hcy, GSH, 1 mM) only trigger minor changes. Thus, FD-NO2 has an ideal selectivity towards H2S over other anions, RSS, ROS, and RNS.
 | ||
| Fig. 3 Fluorescence responses of FD-NO2 (2 μM) to H2S (100 μM) and other anions, RSS, ROS, and RNS (1 mM). Bars represent fluorescence intensity ratios after (F) and before (F0) addition of each anion, RSS, ROS, and RNS. Data were acquired at 25 °C in PBS buffer (pH 7.4), λex = 480 nm. | ||
To further verify whether the probe is suitable for biological applications, we examined the influence of pH in the range of 3–11. The results showed that the fluorescent intensity of FD-NO2 in the absence of H2S is hardly affected by the pH varying from 3 to 11. However, in the presence of H2S, FA develops a turn-on fluorescence in the pH range between 6.5 and 10.5, with a strongest and constant response between pH 7.5 and 9.5 (Fig. S4†). These data confirm that FD-NO2 is a very good candidate for detecting H2S in living cells.
We next tested the ability of the probe FD-NO2 to be used to visualize H2S in living cells (Fig. 4). After the incubation of NIH 3T3 cells with 10 μM probe FD-NO2 in serum-free DMEM for 30 min at 37 °C, the emission channel of 500–600 nm only exhibited faint fluorescence upon excitation at 488 nm. When we treated the FD-NO2-loaded NIH 3T3 cells with H2S (250 μM) in serum-free DMEM for another 30 min at 37 °C, the cells displayed obvious fluorescence enhancement. To further prove whether the fluorescence enhancement in the cells arises from H2S, ZnCl2 (an efficient H2S scavenger)26 was used to pretreat the NIH 3T3 cells. As expected, on addition of H2S to the Zn2+-pretreated NIH 3T3 cells, no obvious fluorescence enhancement was observed. Therefore, we can conclude that fluorescence enhancement originated from the H2S reduction of the nitro group and FD-NO2 can be used to detect H2S variations in living cells. Considering its good membrane permeability, high sensitivity and selectivity, we also anticipate that FD-NO2 can contribute as a useful molecular probe for the investigations on H2S-relevant biological processes.
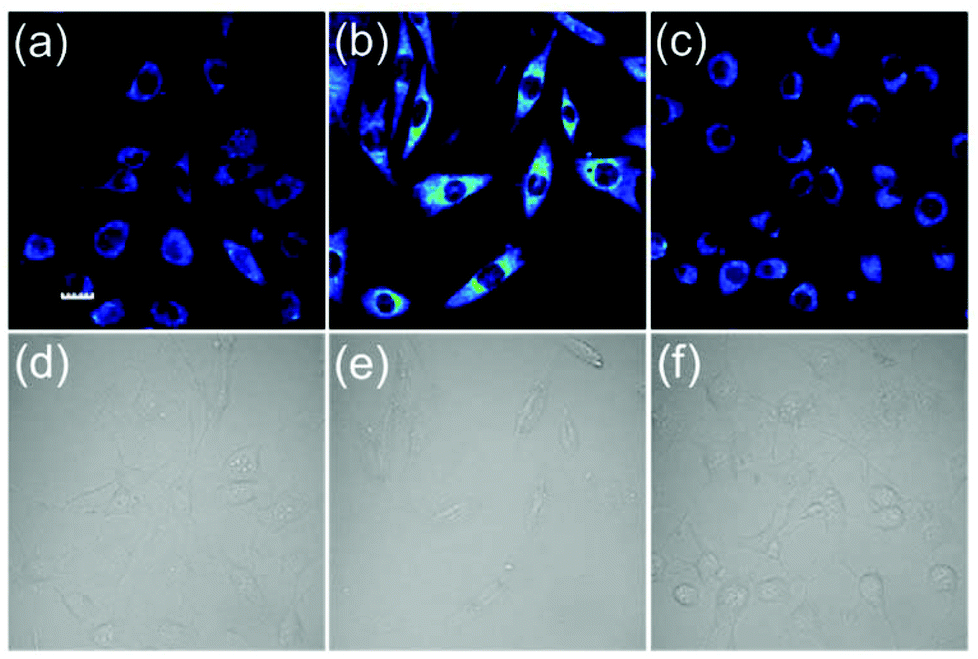 | ||
| Fig. 4 Fluorescent imaging of H2S in FD-NO2-labeled NIH 3T3 cells. (a) Cells were stained with 10 μM FD-NO2 at 37 °C for 30 min in serum-free DMEM. (b) Cells were pretreated with FD-NO2 (10 μM) at 37 °C for 30 min and then incubated with 250 μM H2S at 37 °C for 30 min. (c) Cells were pretreated with 500 μM ZnCl2 at 37 °C for 30 min, incubated with FD-NO2 (10 μM) at 37 °C for 30 min and then incubated with 250 μM H2S at 37 °C for 30 min. (d), (e) and (f) are the bright-field images of (a), (b) and (c), respectively. Emission intensities were collected in an optical window 500–600 nm, λex = 488 nm, Scale bar: 20 μm. | ||
Conclusions
In summary, we have developed a novel turn-on fluorescent probe, FD-NO2, for the detection of H2S based on H2S-selective reduction of the nitro group. The probe exhibits significant turn-on fluorescence for H2S and high selectivity for H2S over other anions, reactive sulfur, oxygen, and nitrogen species in aqueous solutions. Moreover, we have also demonstrated that FD-NO2 is suitable for imaging H2S in living cells, providing a potentially efficient tool for detection of H2S.Experimental
General methods
All the chemicals and solvents were purchased from Alfa Aesar, J&K Chemical Ltd, or Beijing Chemical Reagents, and used as received with the following exceptions. Tetrahydrofuran (THF) and 1,4-dioxane were distilled from sodium. 1H and 13C NMR spectra were measured on a Bruker Avance-400 400 MHz NMR spectrometer and referenced to solvent signals. MALDI-TOF and HRMS-ESI were measured on Autotlex III and Bruker Apex IV Fourier transform mass spectrometers respectively. 2 was prepared according to the literature.27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). NaIO4 (534.7 mg, 2.5 mmol) was added, and the resulting solution was stirred at room temperature for 2 h. Then 1 M HCl (20 mL) was added, and stirred at room temperature for 5 h. The mixture was extracted with ethyl acetate (3 × 20 mL), and the combined extracts were washed with brine (20 mL), dried over Na2SO4, and evaporated to give the crude product 4 which was used without further purification. The crude product 4 was dried in vacuo, then 5 mL anhydrous 1,4-dioxane and tert-butyl nitrite (0.6 mL, 5.0 mmol) were added, and the resulting solution was heated at 80 °C overnight. The reaction mixture was dried by rotary evaporation, and purified by silica gel column chromatography using ether–ethyl acetate as the eluent to afford the desired product (50.7 mg, 20%). 1H NMR (400 MHz, d6-acetone, ppm) δ 8.18–8.17 (d, J = 2.0 Hz, 1H), 8.07–8.05 (d, J = 7.6 Hz, 1H), 7.98–7.95 (dd, J = 8.8 Hz, J = 2.0 Hz, 1H), 7.87–7.83 (m, 1H), 7.81–7.77 (m, 1H), 7.39–7.37 (d, J = 7.6 Hz, 1H), 7.24–7.22 (d, J = 8.8 Hz, 1H), 7.01–7.00 (d, J = 2.0 Hz, 1H), 6.86–6.83 (m, 2H), 4.27–4.25 (t, J = 4.4 Hz, 2H), 3.87–3.85 (m, 2H), 3.67–3.65 (m, 2H), 3.61–3.56 (m, 4H), 3.47–3.45 (m, 2H), 3.27 (s, 3H). 13C NMR (400 MHz, d6-acetone, ppm) δ 169.12, 162.20, 153.57, 152.72, 152.18, 150.00, 136.62, 131.42, 130.72, 129.95, 126.93, 126.89, 125.92, 124.92, 119.10, 114.14, 113.42, 111.63, 102.50, 81.64, 72.68, 71.49, 71.29, 71.13, 70.15, 69.08, 58.80. HRMS (ESI) for C27H26NO9+ ([M + H]+): calcd: 508.16021, found: 508.15946.
:1). NaIO4 (534.7 mg, 2.5 mmol) was added, and the resulting solution was stirred at room temperature for 2 h. Then 1 M HCl (20 mL) was added, and stirred at room temperature for 5 h. The mixture was extracted with ethyl acetate (3 × 20 mL), and the combined extracts were washed with brine (20 mL), dried over Na2SO4, and evaporated to give the crude product 4 which was used without further purification. The crude product 4 was dried in vacuo, then 5 mL anhydrous 1,4-dioxane and tert-butyl nitrite (0.6 mL, 5.0 mmol) were added, and the resulting solution was heated at 80 °C overnight. The reaction mixture was dried by rotary evaporation, and purified by silica gel column chromatography using ether–ethyl acetate as the eluent to afford the desired product (50.7 mg, 20%). 1H NMR (400 MHz, d6-acetone, ppm) δ 8.18–8.17 (d, J = 2.0 Hz, 1H), 8.07–8.05 (d, J = 7.6 Hz, 1H), 7.98–7.95 (dd, J = 8.8 Hz, J = 2.0 Hz, 1H), 7.87–7.83 (m, 1H), 7.81–7.77 (m, 1H), 7.39–7.37 (d, J = 7.6 Hz, 1H), 7.24–7.22 (d, J = 8.8 Hz, 1H), 7.01–7.00 (d, J = 2.0 Hz, 1H), 6.86–6.83 (m, 2H), 4.27–4.25 (t, J = 4.4 Hz, 2H), 3.87–3.85 (m, 2H), 3.67–3.65 (m, 2H), 3.61–3.56 (m, 4H), 3.47–3.45 (m, 2H), 3.27 (s, 3H). 13C NMR (400 MHz, d6-acetone, ppm) δ 169.12, 162.20, 153.57, 152.72, 152.18, 150.00, 136.62, 131.42, 130.72, 129.95, 126.93, 126.89, 125.92, 124.92, 119.10, 114.14, 113.42, 111.63, 102.50, 81.64, 72.68, 71.49, 71.29, 71.13, 70.15, 69.08, 58.80. HRMS (ESI) for C27H26NO9+ ([M + H]+): calcd: 508.16021, found: 508.15946.
Acknowledgements
We thank the National Natural Science Foundation of China (21102148 and 21125205), National Basic Research Program of China (2011CB935800), and the State Key Laboratory of Fine Chemicals, Department of Chemical Engineering, Dalian University of Technology for financial supports.Notes and references
- (a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed; (b) R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef PubMed.
- R. Wang, Physiol. Rev., 2012, 92, 791 CrossRef CAS PubMed.
- K. Abe and H. J. Kimura, Neurosci., 1996, 16, 1066 CAS.
- D. J. Elsey, R. C. Fowkes and G. F. Baxter, Cell Biochem. Funct., 2010, 28, 95 CrossRef CAS PubMed.
- G. Szabó, G. Veres, T. Radovits, D. Gerö, K. Módis, C. Miesel-Gröschel, F. Horkay, M. Karck and C. Szabó, Nitric Oxide, 2011, 25, 201 CrossRef PubMed.
- C. Szabó and A. Papapetropoulos, Br. J. Pharmacol., 2011, 164, 853 CrossRef PubMed.
- L. Li, M. Bhatia, Y. Z. Zhu, Y. C. Zhu, R. D. Ramnath, Z. J. Wang, F. B. Anuar, M. Whiteman, M. Salto-Tellez and P. K. Moore, FASEB J., 2005, 19, 1196 CAS.
- S. Taniguchi and I. Niki, J. Pharm. Sci., 2011, 116, 1 CrossRef CAS.
- K. Eto, T. Asada, K. Arima, T. Makifuchi and H. Kimura, Biochem. Biophys. Res. Commun., 2002, 293, 1485 CrossRef CAS.
- P. Kamoun, M.-C. Belardinelli, A. Chabli, K. Lallouchi and B. Chadefaux-Vekemans, Am. J. Med. Genet. A, 2003, 116, 310 CrossRef PubMed.
- W. Yang, G. Yang, X. Jia, L. Wu and R. Wang, J. Physiol., 2005, 569, 519 CrossRef CAS PubMed.
- S. Fiorucci, E. Antonelli, A. Mencarelli, S. Orlandi, B. Renga, G. Rizzo, E. Distrutti, V. Shah and A. Morelli, Hepatology, 2005, 42, 539 CrossRef CAS PubMed.
- (a) D. Jiménez, R. Martínez-Máñez, F. Sancenón, J. V. Ros-Lis, A. Benito and J. Soto, J. Am. Chem. Soc., 2003, 125, 9000 CrossRef PubMed; (b) M. G. Choi, S. Cha, H. Lee, H. L. Jeon and S. K. Chang, Chem. Commun., 2009, 7390 RSC.
- (a) N. S. Lawrence, J. Davis, L. Jiang, T. G. J. Jones, S. N. Davies and R. G. Compton, Electroanalysis, 2000, 18, 1453 CrossRef; (b) J. E. Doeller, T. S. Isbell, G. Benavides, J. Koenitzer, H. Patel, R. P. Patel and J. R. Lancaster Jr., Anal. Biochem., 2005, 341, 40 CrossRef CAS PubMed.
- (a) J. Radford-Knaery and G. A. Cutter, Anal. Chem., 1993, 65, 976 CrossRef; (b) R. V. Kartha, J. Zhou, L. B. Hovde, B. W. Y. Cheung and H. Schröder, Anal. Biochem., 2012, 423, 102 CrossRef CAS PubMed.
- (a) M. Ishigami, K. Hiraki, K. Umemura, Y. Ogasawara, K. Ishii and H. Kimura, Antioxid. Redox Signaling, 2009, 11, 205 CrossRef CAS PubMed; (b) E. G. Lynn and R. C. Austin, Expert Rev. Clin. Pharmacol., 2011, 120, 97 CrossRef PubMed.
- (a) Y. Han, J. Qin, X. Chang, Z. Yang and J. Du, Cell. Mol. Neurobiol., 2006, 26, 101 CrossRef PubMed; (b) J. Furne, A. Saeed and M. D. Levitt, Am. J. Physiol., 2008, 295, R1479 CAS.
- (a) M. E. Jun, B. Roy and K. H. Ahn, Chem. Commun., 2011, 47, 7583 RSC; (b) C. Chung, D. Srikun, C. S. Lim, C. J. Chang and B. R. Cho, Chem. Commun., 2011, 47, 9618 RSC.
- (a) K. Sasakura, K. Hanaoka, N. Shibuya, Y. Mikami, Y. Kimura, T. Komatsu, T. Ueno, T. Terai, H. Kimura and T. Nagano, J. Am. Chem. Soc., 2011, 133, 18003 CrossRef CAS PubMed; (b) F. Hou, L. Huang, P. Xi, J. Cheng, X. Zhao, G. Xie, Y. Shi, F. Cheng, X. Yao, D. Bai and Z. Zeng, Inorg. Chem., 2012, 51, 2454 CrossRef CAS PubMed; (c) F. Hou, J. Cheng, P. Xi, F. Chen, L. Huang, G. Xie, Y. Shi, H. Liu, D. Bai and Z. Zeng, Dalton Trans., 2012, 41, 5799 RSC; (d) X. Qu, C. Li, H. Chen, J. Mack, Z. Guo and Z. Shen, Chem. Commun., 2013, 49, 7510 RSC.
- (a) X. Cao, W. Lin, K. Zheng and L. He, Chem. Commun., 2012, 48, 10529 RSC; (b) T. Liu, Z. Xu, D. R. Spring and J. Cui, Org. Lett., 2013, 15, 2310 CrossRef CAS PubMed.
- (a) C. R. Liu, J. Pan, S. Li, Y. Zhao, L. Y. Wu, C. E. Berkman, A. R. Whorton and M. Xian, Angew. Chem., Int. Ed., 2011, 50, 10327 CrossRef CAS PubMed; (b) Y. Qian, L. Zhang, S. Ding, X. Deng, C. He, X. E. Zheng, H.-L. Zhu and J. Zhao, Chem. Sci., 2012, 3, 2920 RSC; (c) C. Liu, B. Peng, S. Li, C.-M. Park, A. R. Whorton and M. Xian, Org. Lett., 2012, 14, 2184 CrossRef CAS PubMed; (d) Z. Xu, L. Xu, J. Zhou, Y. Xu, W. Zhu and X. Qian, Chem. Commun., 2012, 48, 10871 RSC; (e) X. Li, S. Zhang, J. Cao, N. Xie, T. Liu, B. Yang, Q. He and Y. Hu, Chem. Commun., 2013, 49, 8656 RSC; (f) Y. Chen, C. Zhu, Z. Yang, J. Chen, Y. He, Y. Jiao, W. He, L. Qiu, J. Cen and Z. Guo, Angew. Chem., Int. Ed., 2013, 52, 1688 CrossRef CAS PubMed; (g) J. Liu, Y.-Q. Sun, J. Zhang, T. Yang, J. Cao, L. Zhang and W. Guo, Chem. – Eur. J., 2013, 19, 4717 CrossRef CAS PubMed; (h) Q. Huang, X.-F. Yang and H. Li, Dyes Pigm., 2013, 99, 871 CrossRef CAS PubMed; (i) J. Zhang, Y.-Q. Sun, J. Liu, Y. Shi and W. Guo, Chem. Commun., 2013, 49, 11305 RSC.
- (a) B. Wang, P. Li, F. Yu, P. Song, X. Sun, S. Yang, Z. Lou and K. Han, Chem. Commun., 2013, 49, 1014 RSC; (b) B. Wang, P. Li, F. Yu, J. Chen, Z. Qu and K. Han, Chem. Commun., 2013, 49, 5790 RSC.
- (a) A. R. Lippert, E. J. New and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 10078 CrossRef CAS PubMed; (b) M.-Y. Wu, K. Li, J.-T. Hou, Z. Huang and X.-Q. Yu, Org. Biomol. Chem., 2012, 10, 8342 RSC; (c) S. Chen, Z. Chen, W. Ren and H. Ai, J. Am. Chem. Soc., 2012, 134, 9589 CrossRef CAS PubMed; (d) F. Yu, P. Li, P. Song, B. Wang, J. Zhao and K. Han, Chem. Commun., 2012, 48, 2852 RSC; (e) L. A. Montoya and M. D. Pluth, Chem. Commun., 2012, 48, 4767 RSC; (f) S. K. Das, C. S. Lim, S. Y. Yang, J. H. Han and B. R. Cho, Chem. Commun., 2012, 48, 8395 RSC; (g) Z. Wu, Z. Li, L. Yang, J. Han and S. Han, Chem. Commun., 2012, 48, 10120 RSC; (h) W. Xuan, R. Pan, Y. Cao, K. Liu and W. Wang, Chem. Commun., 2012, 48, 10669 RSC; (i) R. Wang, F. Yu, L. Chen, H. Chen, L. Wang and W. Zhang, Chem. Commun., 2012, 48, 11757 RSC; (j) C. Yu, X. Li, F. Zeng, F. Zheng and S. Wu, Chem. Commun., 2013, 49, 403 RSC; (k) Q. Wan, Y. Song, Z. Li, X. Gao and H. Ma, Chem. Commun., 2013, 49, 502 RSC; (l) W. Sun, J. Fan, C. Hu, J. Cao, H. Zhang, X. Xiong, J. Wang, S. Cui, S. Sun and X. Peng, Chem. Commun., 2013, 49, 3890 RSC; (m) S. K. Bae, C. H. Heo, D. J. Choi, D. Sen, E.-H. Joe, B. R. Cho and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 9915 CrossRef CAS PubMed; (n) M. K. Thorson, T. Majtan, J. P. Kraus and A. M. Barrios, Angew. Chem., Int. Ed., 2013, 52, 4641 CrossRef CAS PubMed; (o) D. Zhu, G. Li, L. Xue and H. Jiang, Org. Biomol. Chem., 2013, 11, 4577 RSC.
- (a) R. J. Hodgkiss, R. W. Middleton, J. Parrick, H. K. Rami, P. Wardman and G. D. Wilson, J. Med. Chem., 1992, 35, 1920 CrossRef CAS; (b) Y. Liu, Y. F. Xu, X. H. Qian, Y. Xiao, J. W. Liu, L. Y. Shen, J. H. Li and Y. X. Zhang, Bioorg. Med. Chem. Lett., 2006, 16, 1562 CrossRef CAS PubMed.
- (a) J. E. Whitaker, R. P. Haugland, D. Ryan, P. C. Hewitt, R. P. Haugland and F. G. Prendergast, Anal. Biochem., 1992, 207, 267 CrossRef CAS; (b) T. Peng and D. Yang, Org. Lett., 2010, 12, 496 CrossRef CAS PubMed.
- H. Peng, Y. Cheng, C. Dai, A. L. King, B. L. Predmore, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2011, 50, 9672 CrossRef CAS PubMed.
- S. Kohmoto, E. Mori and K. Kishikawa, J. Am. Chem. Soc., 2007, 129, 13364 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of compounds and additional spectroscopic data. See DOI: 10.1039/c4qo00048j |
| This journal is © the Partner Organisations 2014 |