Stereoselective intramolecular cyclopropanation of α-diazoacetates via Co(II)-based metalloradical catalysis†
Joshua V.
Ruppel
ab,
Xin
Cui
a,
Xue
Xu
a and
X. Peter
Zhang
*a
aDepartment of Chemistry, University of South Florida, Tampa, Florida 33620-5250, USA. E-mail: xpzhang@usf.edu
bDivision of Natural Sciences and Engineering, University of South Carolina Upstate, Spartanburg, South Carolina 29303, USA
First published on 15th April 2014
Abstract
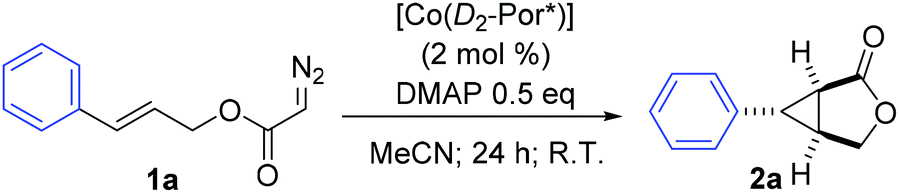
Co(II) complexes of D2-symmetric chiral porphyrins have been proven to be effective metalloradical catalysts for the asymmetric intramolecular cyclopropanation of allyl α-diazoacetates. 4-(Dimethylamino)pyridine (DMAP), through a positive trans effect, plays an important role in the enhancement of the asymmetric induction for the intramolecular cyclopropanation process. This metalloradical catalytic system is suitable for cyclopropanation of allyl α-diazoacetates with varied functional groups and substitution patterns, producing bicyclic products with complete diastereocontrol and good enantiocontrol.
Introduction
Optically pure cyclopropane and cyclopropane-containing polycyclic compounds have been attracting diverse research interest due to not only their fundamental importance, but also their potential synthetic applications.1 Among different methods, transition metal-catalyzed asymmetric olefin cyclopropanation via carbene transfer has developed into one of the most versatile methods for the stereoselective construction of these valuable three-membered ring compounds.2 The intramolecular variation of cyclopropanation allows stereoselective construction of the [n.1.0]bicyclic skeletons, which serve as important intermediates in the synthesis of many interesting biologically active compounds and natural products.3 As one class of relatively stable and readily available diazo reagents, α-diazoacetates have been widely used for the generation of carbene species in a number of carbene transfer reactions. The intramolecular cyclopropanation of allyl α-diazoacetates provides a direct way for rapid construction of the bicyclo[3.1.0]hexan-2-one structure. This important transformation, especially the asymmetric version, has been extensively studied over the past few decades, which is highlighted by the catalytic systems based on rhodium4 and ruthenium5 complexes.As stable metalloradicals with a well-defined open-shell doublet d7 electronic structure, cobalt(II) complexes of porphyrins [Co(Por)] have been demonstrated as a new class of potent metalloradical catalysts for cyclopropanation reactions.6 The unprecedented radical pathway initiated by this new system enabled the development of highly asymmetric cyclopropanation of a broad combination of olefin substrates and diazo reagents.7 Recently, Co(II) complexes of D2-symmetric chiral porphyrins [Co(D2-Por*)] were reported to be effective catalysts for the asymmetric intramolecular cyclopropanation of allyl α-diazoacetates with two electron-withdrawing α-groups, namely acceptor/acceptor-substituted diazo reagents.8 While the Co(II)-catalyzed intramolecular cyclopropanation was shown to be both general and highly stereoselective for various acceptor/acceptor-substituted diazo reagents, only one example of acceptor-substituted allyl α-diazoacetate was examined with the catalytic system for intramolecular formation of the corresponding bicyclo[3.1.0]hexan-2-one via the Co(II)-based metalloradical catalysis (MRC).9 To assess whether the Co(II)-based MRC would also be generally effective for acceptor-substituted diazo reagents, in addition to the well-demonstrated acceptor/acceptor-substituted diazo reagents, we have performed a systematic investigation on asymmetric intramolecular cyclopropanation of allyl α-diazoacetates.
Results and discussion
We herein report the study of allyl α-diazoacetate derivatives, a representative class of acceptor-substituted diazo reagents, for asymmetric intramolecular cyclopropanation utilizing a “toolbox” of first-generation Co(II)-based metalloradical catalysts (Fig. 1).10 Among them, the Co(II) complex of 3,5-DitBu-ChenPhyrin, [Co(P1)], with 4-(dimethylamino)pyridine (DMAP) as an additive, can effectively catalyze the intramolecular cyclopropanation reactions of allyl α-diazoacetates with a range of functional groups and substitution patterns. The corresponding bicyclo[3.1.0]hexan-2-one derivatives can be produced as single diastereomers in good to high yields and with significant asymmetric induction.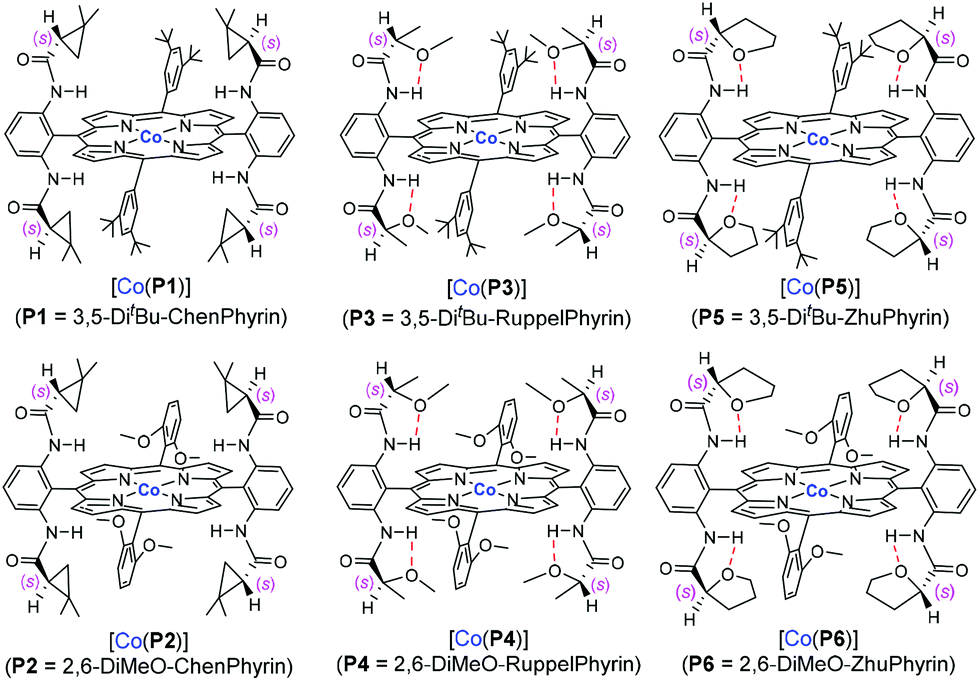 | ||
| Fig. 1 Structures of Co(II) complexes of first-generation D2-symmetric chiral amidoporphyrins. | ||
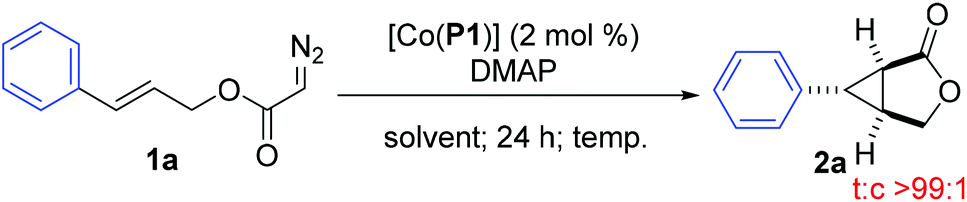
The initial experiments were focused on the intramolecular cyclopropanation of cinnamyl α-diazoacetate (1a) (Table 1) by utilizing a standard “toolbox” of first-generation metalloradical catalysts [Co(P1–P6)] (Fig. 1), which have been proven to be effective for a variety of asymmetric intermolecular cyclopropanation reactions.7 Applying similar conditions that were optimized for asymmetric intermolecular olefin cyclopropanation with α-diazoacetates, which took the advantage of the positive trans effect of DMAP as an additive,11 it was found that the use of 2 mol % of [Co(P1)] (P1 = 3,5-DitBu-ChenPhyrin) could successfully catalyze the cyclopropanation reaction of 1a in 75% yield with complete diastereocontrol and significant asymmetric induction (entry 1). In the absence of DMAP, the enantioselectivity of the intramolecular cyclopropanation process dropped dramatically while the yield was substantially increased (entry 2). This result demonstrates that the additive DMAP, a potential axial ligand for the Co center, played an important role in enhancing the asymmetric induction for this catalytic intramolecular process. Under the same conditions, the catalyst [Co(P2)] (P2 = 2,6-DiMeO-ChenPhyrin), which contains the same chiral amide units as [Co(P1)] but with more sterically hindered non-chiral substituents, appeared to be similarly stereoselective, but less active (entry 3). [Co(P3–P6)] (Fig. 1) represent a subclass of [Co(D2-Por*)] catalysts with enhanced rigidity in a chiral environment due to the presence of an intramolecular hydrogen bond in the chiral amide unit. Among them, [Co(P6)] was reported to be an excellent catalyst for the asymmetric cyclopropanation of diazosulfones.7c These catalysts, however, were found to be ineffective for the intramolecular cyclopropanation of allyl α-diazoacetate 1a (entries 4–7).

Using the most active [Co(P1)] as the catalyst, the effect of solvents was then examined for the catalytic intramolecular cyclopropanation reaction of allyl α-diazoacetate 1a (Table 2). The metalloradical cyclopropanation could proceed in a variety of solvents, including coordinating and non-coordinating, aromatic and aliphatic, polar and non-polar as well as halogenated solvents (entries 2–6). The reactivity and stereoselectivity of the cyclopropanation reaction were generally unaffected by the broad range of solvent characteristics, except the non-polar hexanes which provided insufficient solubility. Among the solvents examined, DCM was selected as the solvent of choice as it provided the highest enantioselectivity while retaining a satisfactory yield. Additional experiments were attempted to fine-tune the reaction conditions by optimizing the additive stoichiometry and reaction temperature. It was shown that either decrease or increase in the amount of DMAP led to lower yields and slight decrease in the enantioselectivity (entries 7–9), indicating that 0.5 equivalent of DMAP is optimal for the intramolecular reaction, which is consistent with the reported intermolecular reactions.6a,11 Although lower and higher temperature resulted in increase of the enantioselectivity and yield, respectively (entries 10 and 11), room temperature was chosen to be optimal since it gave the best overall performance.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Additive (equiv.) | Solvent | Temp. | Yieldb (%) | eec (%) |
| a Reactions were carried out in a one-portion protocol using 2 mol % catalyst under N2 for 24 h with [1] = 0.20 M. The trans/cis ratio was determined by 1H NMR. b Isolated yields. c Enantiomeric excess of the major diastereomer was determined by chiral HPLC. | |||||
| 1 | DMAP (0.5) | MeCN | R.T. | 75 | 68 |
| 2 | DMAP (0.5) | Toluene | R.T. | 66 | 62 |
| 3 | DMAP (0.5) | PhCl | R.T. | 75 | 61 |
| 4 | DMAP (0.5) | DCM | R.T. | 70 | 72 |
| 5 | DMAP (0.5) | Hexanes | R.T. | 31 | 65 |
| 6 | DMAP (0.5) | EtOAc | R.T. | 74 | 64 |
| 7 | DMAP (0.5) | DCM | R.T. | 70 | 72 |
| 8 | DMAP (0.25) | DCM | R.T. | 57 | 68 |
| 9 | DMAP (0.75) | DCM | R.T. | 50 | 69 |
| 10 | DMAP (0.5) | DCM | 0 °C | 37 | 76 |
| 11 | DMAP (0.5) | DCM | 40 °C | 76 | 64 |
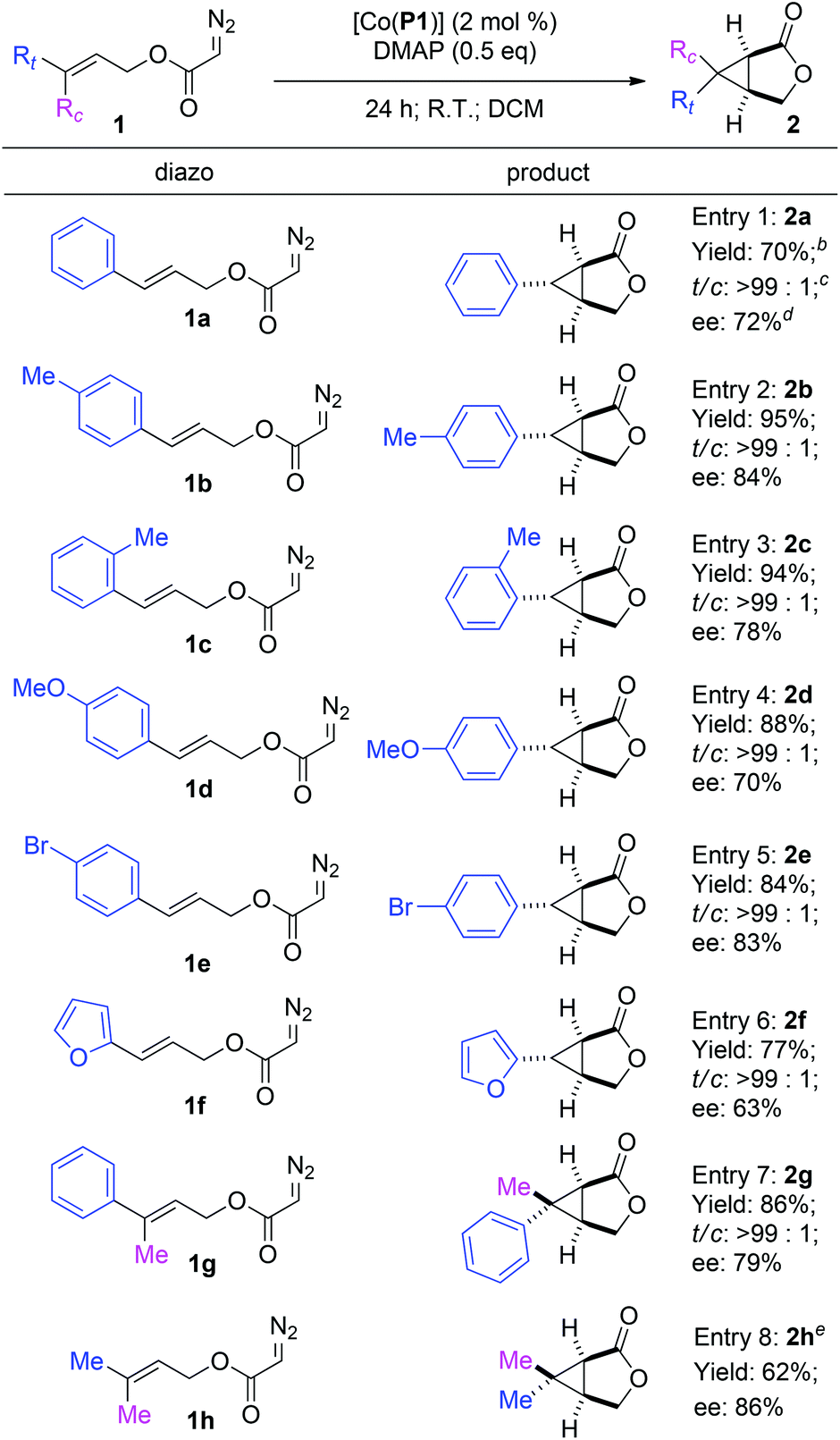
Under the optimized reaction conditions, the [Co(P1)]-based metalloradical intramolecular cyclopropanation system was found to be applicable for different allyl α-diazoacetates (Table 3). In addition to cinnamyl α-diazoacetate (1a) (entry 1), different (E)-3-aryl allyl α-diazoacetates with groups that possess different steric and electronic properties on the aromatic ring could also undergo smooth cyclopropanation reactions by [Co(P1)] to form the corresponding bicyclic products. For example, α-diazoacetates 1b and 1c with para- and ortho-methyl groups, respectively, could be converted to the bicyclic products 2b and 2c as single diastereomers in excellent yields with good enantioselectivities (entries 2 and 3). In addition, α-diazoacetate 1d containing an electron-donating MeO-group was shown to be a suitable substrate for the [Co(P1)]-catalyzed cyclopropanation process, producing the desired 2d smoothly with a good level of enantiocontrol (entry 4). The metalloradical cyclopropanation system was shown to also tolerate the presence of halogen atoms in the substrates, as exemplified for the stereoselective formation of brominated bicyclo[3.1.0]hexan-2-one 2e (entry 5). In addition to allyl α-diazoacetates with aryl substituents, the Co(II)-based catalytic system could be applied to heteroaryl-containing α-diazoacetates, as demonstrated with the intramolecular cyclopropanation of (E)-3-(furan-2-yl)allyl α-diazoacetate (1f), which was transformed into 6-(furan-2-yl)-3-oxabicyclo[3.1.0]hexan-2-one (2f) in 77% yield with complete diastereocontrol and 63% ee (entry 6). Furthermore, even the sterically demanding tri-substituted olefin-based allyl α-diazoacetates were found to be effective substrates for the intramolecular cyclopropanation via Co(II)-based MRC. For example, the reactions of allyl α-diazoacetates 1g and 1h could be effectively catalyzed by [Co(P1)] to form the corresponding 6,6-disubstituted 3-oxabicyclo[3.1.0]hexan-2-one products 2g and 2h, respectively, in good yields and stereoselectivities (entries 7 and 8).
| a Reactions were carried out in a one-portion protocol using 2 mol % catalyst under N2 for 24 h with [1] = 0.20 M. b Isolated yields. c The trans/cis ratio was determined by 1H NMR. d Enantiomeric excess of the major diastereomer was determined by chiral HPLC. e Reaction was carried out at 40 °C. Reaction under room temperature resulted in 35% yield with 90% ee. |
|---|
|
Conclusions
In summary, the Co(II)-based metalloradical system has been proven to enantioselectively catalyze intramolecular cyclopropanation of allyl α-diazoacetates, a representative class of acceptor-substituted diazo reagents. Using readily accessible first-generation metalloradical catalyst [Co(P1)], allyl α-diazoacetates have been transformed into [3.1.0]bicyclic structures in good to high yields, with complete diastereocontrol and good asymmetric induction. Together with acceptor/acceptor-substituted diazo reagents,8 the Co(II)-based intramolecular cyclopropanation has been shown to be suitable for diazo substrates with a wide range of electronic properties. This feature of broad substrate scope, which is uncommon for catalytic systems involved with electrophilic Fischer-type carbene intermediates, is in good accord with the proposed radical mechanism of the Co(II)-based MRC.7 Further investigation is currently underway to address remaining issues in asymmetric olefin cyclopropanation, both intermolecular and intramolecular reactions, via Co(II)-based metalloradical catalysis.Experimental section
General considerations
All catalytic reactions were performed under nitrogen in oven-dried glassware following standard Schlenk techniques unless otherwise specifically noted. Toluene was distilled under nitrogen from sodium benzophenone ketyl prior to use. Chlorobenzene, acetonitrile, and dichloromethane were dried over calcium hydride under nitrogen and freshly distilled before use. 4 Å molecular sieves were dried in a vacuum oven prior to use. Chemicals were purchased from commercial sources and used without further purification unless specifically noted. Allyl α-diazoacetates utilized in this study (1a–h) were synthesized according to known literature procedures.4f,5d,9b Thin layer chromatography was performed on Merck TLC plates (silica gel 60 F254). Flash column chromatography was performed with Merck silica gel (60 Å, 230–400 mesh, 32–63 μm). 1H NMR and 13C NMR were recorded on a Varian Inova400 (400 MHz) or a Varian Inova500 (500 MHz) with chemical shifts reported relative to residual solvent. Infrared spectra were measured with a Nicolet Avatar 320 spectrometer with a Smart Miracle accessory. HRMS data were obtained on an Agilent 1100 LC/MS/TOF mass spectrometer. GC/MS measurements were carried out on a Hewitt Packard GCD system. Enantiomeric excess was measured using a Chiraldex G-TA chiral column. Optical rotation was measured on a Rudolf Autopol IV polarimeter. Configuration was determined by analogy based upon the optical rotation of known compounds in the literature.4f,5d,9bGeneral procedure for intramolecular cyclopropanation
An oven dried Schlenk tube that was previously evacuated and backfilled with nitrogen gas was charged with diazoacetate (0.2 mmol, if solid), catalyst, and DMAP. The Schlenk tube was then evacuated and back filled with nitrogen. The Teflon screw cap was replaced with a rubber septum and 0.5 mL portion of the solvent was added followed by diazo (0.2 mmol, if liquid), and the remaining solvent (total 1 mL). The Schlenk tube was then purged with nitrogen for 1 minute and the rubber septum was replaced with a Teflon screw cap. The Schlenk tube was then placed in an oil bath for the desired time and temperature. Following completion of the reaction, the reaction mixture was purified by flash chromatography. The fractions containing the product were collected and concentrated by rotary evaporation to afford the compound. In most cases, the product was visualized on TLC using the cerium ammonium molybdate (CAM) stain.
(1R,5S,6S)-6-Phenyl-3-oxabicyclo[3.1.0]hexan-2-one (
2a
) was obtained using the general procedure in 70% yield (24.3 mg). [α]20D = 61 (c = 2.05, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.31–7.21 (m, 3H), 7.05–7.04 (m, 2H), 4.45 (dd, J = 4.8, 9.6 Hz, 1H), 4.39 (d, J = 9.6 Hz, 1H), 2.53–2.49 (m, 1H), 2.33–2.30 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 174.9, 137.1, 128.7, 127.1, 125.9, 69.69, 29.34, 27.36, 26.12. IR (neat, cm−1): 2923 (C–H), 2852 (C–H), 1740 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). HRMS (ESI): calcd for C11H11O2 ([M + H]+) m/z 175.0759, found 175.0765. GC/MS: Chiraldex G-TA (initial temperature: 150 °C; isothermal for 34 min; temperature increased 5.0 °C min−1 to a final temperature of 180 °C): 72% ee; 19.7 min (major) 21.8 min (minor).
O). HRMS (ESI): calcd for C11H11O2 ([M + H]+) m/z 175.0759, found 175.0765. GC/MS: Chiraldex G-TA (initial temperature: 150 °C; isothermal for 34 min; temperature increased 5.0 °C min−1 to a final temperature of 180 °C): 72% ee; 19.7 min (major) 21.8 min (minor).
6-(p-Tolyl)-3-oxabicyclo[3.1.0]hexan-2-one (
2b
) was obtained using the general procedure in 95% yield (35.9 mg). [α]20D = 66 (c = 3.5, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.11 (d, J = 8.4 Hz, 2H), 6.96 (d, J = 8.4 Hz, 2H), 4.45 (dd, J = 4.8, 9.6 Hz, 1H), 4.40 (d, J = 9.6 Hz, 1H), 2.51–2.48 (m, 1H), 2.32 (s, 3H), 2.30–2.29 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 175.1, 136.8, 134.1, 129.3, 125.8, 69.69, 29.12, 27.24, 25.94, 20.93. IR (neat, cm−1): 2979 (C–H), 2848 (C–H), 1766 (CO). HRMS (ESI): calcd for C12H13O2 ([M + H]+) m/z 189.0915, found 189.0919. GC/MS: Chiraldex G-TA (initial temperature: 150 °C; isothermal for 60 min; temperature increased 4.0 °C min−1 to a final temperature of 180 °C; isothermal for 30 min): 84% ee; 30.6 min (major) 33.2 min (minor).
6-(o-Tolyl)-3-oxabicyclo[3.1.0]hexan-2-one (
2c
) was obtained using the general procedure in 94% yield (35.2 mg). [α]20D = 24 (c = 1.16, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.19–7.13 (m, 3H), 6.95 (d, J = 6.8 Hz, 1H), 4.48 (dd, J = 4.8, 9.6 Hz, 1H), 4.43 (d, J = 9.6 Hz, 1H), 2.58–2.54 (m, 1H), 2.43 (s, 3H), 2.35–2.33 (m, 1H), 2.31–2.29 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 175.2, 137.5, 134.7, 130.2, 127.3, 126.1, 125.4, 69.68, 27.62, 25.99, 24.59, 19.55. IR (neat, cm−1): 2979 (C–H), 2904 (C–H), 1766 (CO). HRMS (ESI): calcd for C12H13O2 ([M + H]+) m/z 189.0915, found 189.0905. GC/MS: Chiraldex G-TA (initial temperature: 150 °C; isothermal for 60 min; temperature increased 4.0 °C min−1 to a final temperature of 180 °C; isothermal for 30 min): 78% ee; 25.0 min (major) and 26.9 min (minor).
6-(4-Methoxyphenyl)-3-oxabicyclo[3.1.0]hexan-2-one (
2d
) was obtained using the general procedure in 88% yield (36.1 mg). [α]20D = 62 (c = 0.72, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.00 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 8.4 Hz, 2H), 4.45 (dd, J = 4.4, 9.6 Hz, 1H), 4.39 (d, J = 9.6 Hz, 1H), 3.81 (s, 3H), 2.49–2.46 (m, 1H), 2.29–2.25 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 175.1, 158.7, 129.0, 127.1, 114.1, 69.69, 55.29, 28.91, 27.18, 25.72. IR (neat, cm−1): 2927 (C–H), 2851 (C–H), 1760 (CO). HRMS (ESI): calcd for C12H13O3 ([M + H]+) m/z 205.0864, found 205.0868. GC/MS: Chiraldex G-TA (initial temperature: 150 °C; isothermal for 60 min; temperature increased 4.0 °C min−1 to a final temperature of 180 °C; isothermal for 30 min): 70% ee; 66.1 min (major) and 67.8 min (minor).
6-(4-Bromophenyl)-3-oxabicyclo[3.1.0]hexan-2-one (
2e
) was obtained using the general procedure in 84% yield (42.3 mg). [α]20D = 54 (c = 0.70, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.42 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 4.45 (dd, J = 4.8, 9.6 Hz, 1H), 4.40 (d, J = 9.6 Hz, 1H), 2.52–2.48 (m, 1H), 2.31–2.26 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 174.5, 136.2, 131.8, 127.6, 120.9, 69.58, 28.69, 27.23, 26.09. IR (neat, cm−1): 2922 (C–H), 2852 (C–H), 1743 (CO). HRMS (ESI): calcd for C11H10BrO2 ([M + H]+) m/z 252.9864, found 252.9869. GC/MS: Chiraldex G-TA (initial temperature: 150 °C; isothermal for 60 min; temperature increased 4.0 °C min−1 to a final temperature of 180 °C; isothermal for 30 min): 83% ee; 75.9 min (major) and 79.3 min (minor).
6-(Furan-2-yl)-3-oxabicyclo[3.1.0]hexan-2-one (
2f
) was obtained using the general procedure in 77% yield (25.3 mg). [α]20D = 85 (c = 0.36, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.25 (d, J = 1.2 Hz, 1H), 6.29–6.27 (m, 1H), 6.12 (d, J = 3.2 Hz, 1H), 4.42 (dd, J = 4.8, 9.6 Hz, 1H), 4.36 (d, J = 9.6 Hz, 1H), 2.63–2.60 (m, 1H), 2.42–2.40 (m, 1H), 2.34–2.32 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 174.4, 149.9, 141.7, 110.6, 106.3, 69.28, 25.15, 24.03, 22.73. IR (neat, cm−1): 2979 (C–H), 2910 (C–H), 1762 (CO). HRMS (ESI): calcd for C9H9O3 ([M + H]+) m/z 165.0551, found 165.0549. GC/MS: Chiraldex G-TA (initial temperature: 150 °C; isothermal for 60 min; temperature increased 4.0 °C min−1 to a final temperature of 180 °C): 63% ee; 8.8 min (major) and 9.8 min (minor).
6-Methyl-6-phenyl-3-oxabicyclo[3.1.0]hexan-2-one (
2g
) was obtained using the general procedure in 86% yield (32.5 mg). [α]20D = 71 (c = 0.46, CHCl3). 1H NMR (400 MHz, CDCl3): 7.33–7.21 (m, 5H), δ 4.51 (dd, J = 5.2, 10.0 Hz, 1H), 4.34 (d, J = 10.0 Hz, 1H), 2.53–2.50 (m, 1H), 2.44–2.43 (m, 1H), 1.46 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 174.4, 143.3, 128.7, 127.2, 127.1, 66.54, 31.02, 30.63, 29.32, 15.59. IR (neat, cm−1): 2980 (C–H), 2902 (C–H), 1762 (CO). HRMS (ESI): Calcd for C12H13O2 ([M + H]+) m/z 189.0916, found 189.0922. GC/MS: Chiraldex G-TA (initial temperature: 150 °C; isothermal for 60 min; temperature increased 4.0 °C min−1 to a final temperature of 180 °C; isothermal for 30 min): 79% ee; 16.9 min (minor) and 18.0 min (major).
6,6-Dimethyl-3-oxabicyclo[3.1.0]hexan-2-one (
2h
) was obtained using the general procedure in 35% yield (9.0 mg). [α]20D = 67 (c = 0.15, CHCl3). 1H NMR (400 MHz, CDCl3): δ 4.35 (dd, J = 5.6, 10.0 Hz, 1H), 4.15 (d, J = 10.0 Hz, 1H), 2.05–2.02 (m, 1H), 1.95–1.94 (m, 1H), 1.18 (s, 3H), 1.17 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 174.9, 66.52, 30.49, 30.02, 25.20, 14.40. IR (neat, cm−1): 2980 (C–H), 2902 (C–H), 1766 (CO). HRMS (ESI): Calcd for C9H9O3 ([M + H]+) m/z 127.0759, found 127.0748. GC/MS: Chiraldex G-TA (initial temperature: 150 °C; isothermal for 3 min; temperature increased 5.0 °C min−1 to a final temperature of 180 °C; isothermal for 11 min): 90% ee; 20.9 min (minor) and 22.3 min (major).
Acknowledgements
We are grateful for the financial support by the National Science Foundation (CHE-1152767; X.P.Z.); the National Institutes of Health (R01-GM098777; X.P.Z.), and the Office of Sponsored Awards and Research Support of USC Upstate (J.V.R.).Notes and references
- (a) J. Pietruszka, Chem. Rev., 2003, 103, 1051 CrossRef CAS PubMed; (b) L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625 CrossRef CAS PubMed; (c) J. Salaun, Chem. Rev., 1989, 89, 1247 CrossRef CAS.
- (a) H. Pellissier, Tetrahedron, 2008, 64, 7041 CrossRef CAS PubMed; (b) H. Lebel, J. F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed.
- (a) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC; (b) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (c) H. U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed.
- (a) M. P. Doyle, Y. H. Wang, P. Ghorbani and E. Bappert, Org. Lett., 2005, 7, 5035 CrossRef CAS PubMed; (b) P. F. Teng, T. S. Lai, H. L. Kwong and C. M. Che, Tetrahedron: Asymmetry, 2003, 14, 837 CrossRef CAS; (c) M. P. Doyle, W. H. Hu and T. M. Weathers, Chirality, 2003, 15, 369 CrossRef CAS PubMed; (d) C. M. Che, J. S. Huang, F. W. Lee, Y. Li, T. S. Lai, H. L. Kwong, P. F. Teng, W. S. Lee, W. C. Lo, S. M. Peng and Z. Y. Zhou, J. Am. Chem. Soc., 2001, 123, 4119 CrossRef CAS PubMed; (e) M. P. Doyle, Q. L. Zhou, A. B. Dyatkin and D. A. Ruppar, Tetrahedron Lett., 1995, 36, 7579 CrossRef CAS; (f) M. P. Doyle, R. E. Austin, S. Bailey, M. P. Dwyer, A. B. Dyatkin, A. V. Kalinin, M. M. Y. Kwan, S. Liras, C. J. Oalmann, R. J. Pieters, M. N. Protopopova, C. E. Raab, G. H. P. Roos, Q.-L. Zhou and S. F. Martin, J. Am. Chem. Soc., 1995, 117, 5763 CrossRef CAS.
- (a) A. M. Abu-Elfotoh, P. T. N. Diem, S. Chanthamath, K. Phomkeona, K. Shibatomi and S. Iwasa, Adv. Synth. Catal., 2012, 354, 3435 CrossRef CAS; (b) J. Ito, S. Ujiie and H. Nishiyama, Chem. – Eur. J., 2010, 16, 4986 CrossRef CAS PubMed; (c) A. M. Abu-Elfotoh, K. Phomkeona, K. Shibatomi and S. Iwasa, Angew. Chem., Int. Ed., 2010, 49, 8439 CrossRef CAS PubMed; (d) Z. J. Xu, R. Fang, C. Zhao, J. S. Huang, G. Y. Li, N. Zhu and C. M. Che, J. Am. Chem. Soc., 2009, 131, 4405 CrossRef CAS PubMed; (e) G. Y. Li, J. Zhang, P. W. H. Chan, Z. J. Xu, N. Y. Zhu and C. M. Che, Organometallics, 2006, 25, 1676 CrossRef CAS.
- (a) Y. Chen, K. B. Fields and X. P. Zhang, J. Am. Chem. Soc., 2004, 126, 14718 CrossRef CAS PubMed; (b) Y. Chen and X. P. Zhang, J. Org. Chem., 2007, 72, 5931 CrossRef CAS PubMed.
- (a) Y. Chen, J. V. Ruppel and X. P. Zhang, J. Am. Chem. Soc., 2007, 129, 12074 CrossRef CAS PubMed; (b) S. F. Zhu, J. A. Perman and X. P. Zhang, Angew. Chem., Int. Ed., 2008, 47, 8460 CrossRef CAS PubMed; (c) S. F. Zhu, J. V. Ruppel, H. J. Lu, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2008, 130, 5042 CrossRef CAS PubMed; (d) W. I. Dzik, X. Xu, X. P. Zhang, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2010, 132, 10891 CrossRef CAS PubMed; (e) S. F. Zhu, X. Xu, J. A. Perman and X. P. Zhang, J. Am. Chem. Soc., 2010, 132, 12796 CrossRef CAS PubMed; (f) X. Xu, S. F. Zhu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2013, 52, 11857 CrossRef CAS PubMed.
- X. Xu, H. J. Lu, J. V. Ruppel, X. Cui, S. L. de Mesa, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 15292 CrossRef CAS PubMed.
- For other cobalt-based stereoselective cyclopropanation of allyl α-diazoacetates, see: (a) B. K. Langlotz, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2008, 47, 4670 CrossRef CAS PubMed; (b) B. Saha, T. Uchida and T. Katsuki, Tetrahedron: Asymmetry, 2003, 14, 823 CrossRef CAS; (c) T. Uchida, B. Saha and T. Katsuki, Tetrahedron Lett., 2001, 42, 2521 CrossRef CAS.
- In this report, we focus on the application of readily accessible first-generation D2-symmetric chiral porphyrin catalysts, which are directly synthesized from commercially available chiral building blocks. One example in our previous report (ref. 8) showed that our second-generation catalyst [Co(3,5-DitBu-QingPhyrin)], which contains more complex synthetic chiral amide units, could catalyze cyclopropanation of cinnamyl α-diazoacetate in 95% yield with 99% ee.
- Y. Chen and X. P. Zhang, Synthesis, 2006, 1697 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00041b |
| This journal is © the Partner Organisations 2014 |