
In situ and readily prepared metal catalysts and initiators for living cationic polymerization of isobutyl vinyl ether: dual-purpose salphen as a ligand framework for ZrCl4 and an initiating proton source†
Sensho
Kigoshi
 ,
Arihiro
Kanazawa
,
Shokyoku
Kanaoka
and
Sadahito
Aoshima
*
,
Arihiro
Kanazawa
,
Shokyoku
Kanaoka
and
Sadahito
Aoshima
*
Department of Macromolecular Science, Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka 560-0043, Japan. E-mail: aoshima@chem.sci.osaka-u.ac.jp; Fax: +81-6-6850-5448; Tel: +81-6-6850-5448
First published on 2nd September 2014
Abstract
A salphen complex [salphen = N,N′-o-phenylene-bis(3,5-di-tert-butyl-salicylidene-imine)], synthesized in situ by mixing a salphen ligand with a metal chloride is shown to be an effective catalyst in cationic polymerization for the first time. Furthermore, the salphen/ZrCl4 system induced the living cationic polymerization of isobutyl vinyl ether in toluene at 0 °C with quantitative initiation from HCl simultaneously generated upon the complex formation.
The expansion of the scope of substrates or monomers in organic reactions or polymerization, respectively, is often sparked by the development of new metal catalysts with well-designed ligands. For example, various elaborately designed polymerization catalysts have enhanced tolerance towards polar groups and catalytic activity, widening the choice of polymerizable monomers in the field of olefin metathesis polymerization,1,2 transition metal-catalyzed living radical polymerization or ATRP,3–5 and the coordination polymerization of polar vinyl monomers.6–8 This explosive growth in precise organic synthesis and polymerization can be attributed to the variable and/or controllable activity of metal complex catalysts with various carefully designed ligands. Their diverse activity is attained by tuning the electron density of the central metals and/or the redox potential of the complexes through the interaction of the designed ligands with the central metals.
The rather limited scope of monomers in cationic polymerization for high polymers may be improved if well-defined metal catalysts with various ligands are available. A distinct opportunity for the utility of designed metal complexes as catalysts in cationic polymerization was shown by our recent development of initiating systems for base-assisting living cationic polymerization. These systems consist of a metal halide and an externally-added base, the interaction of which is key to successful living polymerization.9 Among various combinations, alcohol or acetylacetone with an appropriate metal halide permitted in situ ligand exchange, generating a real catalytic species inducing living polymerization.10,11 These results encouraged us to pursue the goal of designing metal complex catalysts for precise polymer synthesis using cationic polymerization. Despite the potency of metal complex catalysts, catalyst design using various ligands has been limited in cationic polymerization,9,12,13 although ligand design or in situ complex formation was demonstrated to be effective for achieving the stereoselective cationic polymerization of alkyl vinyl ethers14 or styrene derivatives.15,16
The difficulty in using metal-complex catalysts for cationic polymerization lies in maintaining sufficient catalytic activity with a highly nucleophilic ligand and/or preventing the termination. In fact, the amount of nucleophilic additives is a decisive factor for controlled polymerization, particularly when an ammonium salt or an amine/amide compound is employed as the additive.17–19 The polymerization is readily inhibited by an excess of such relatively strong nucleophilic additives, even if their absolute concentration is very low. In addition, our previous study showed that an alkoxy group on certain Lewis acid catalysts dissociates from the central metal to react with the propagating carbocation.10 Hence, catalyst design to date in this field has focused on relatively simple compounds containing alkoxy or phenoxy groups.10,11,14,20
To circumvent the abovementioned problems, the use of a Schiff base ligand would be effective because the chelating effect of the ligand is expected to suppress any ligand exchange reactions that might lead to the deactivation of the polymerization. Furthermore, a family of Schiff base ligands, known as one of the most useful ligands, can coordinate with many different metals,21,22 yielding catalysts capable of achieving a variety of synthetic transformations. In particular, various transition metals are available for complex formation with Schiff base ligands, which permits the precise tuning of catalytic activity23 based upon the unique properties of each transition metal. Another feature of the ligand is that the electronic and geometric characteristics of catalysts are readily adjusted22,24 because Schiff base ligands are prepared by a condensation reaction between aldehydes and amines with various substituents. In this study, therefore, we examined catalyst design using a chelating Schiff base ligand for the cationic polymerization of vinyl ether.
As a first step toward catalyst design, an in situ complexation method was investigated for catalyst modification using a salphen ligand [salphen = N,N′-o-phenylene-bis(3,5-di-tert-butyl-salicylidene-imine)]. The salphen ligand, a tetradentate Schiff base ligand, is easily synthesized by the condensation of equivalent quantities of a specific salicylaldehyde and o-phenylenediamine. A salphen complex was synthesized simply by mixing the ligand and a metal chloride in dichloromethane, and the resulting solution was directly added to a monomer solution in toluene. In addition, the ligand performs another role: during complex formation, HCl is generated and can function as a protonogen, or an initiator, for the cationic polymerization of vinyl ether.
The polymerization was conducted in toluene using an as-prepared salphen complex solution, obtained simply by mixing equivalent amounts of a metal chloride and a salphen ligand at 0 °C in the presence of ethyl acetate in dichloromethane (Scheme 1). The advantage of this method over a common, previously reported method22 is the greatly facilitated synthetic procedure without purification and the in situ generation of toxic HCl as a proton source, as observed with the systems using alcohol or acetylacetone in conjunction with metal chlorides.10,11
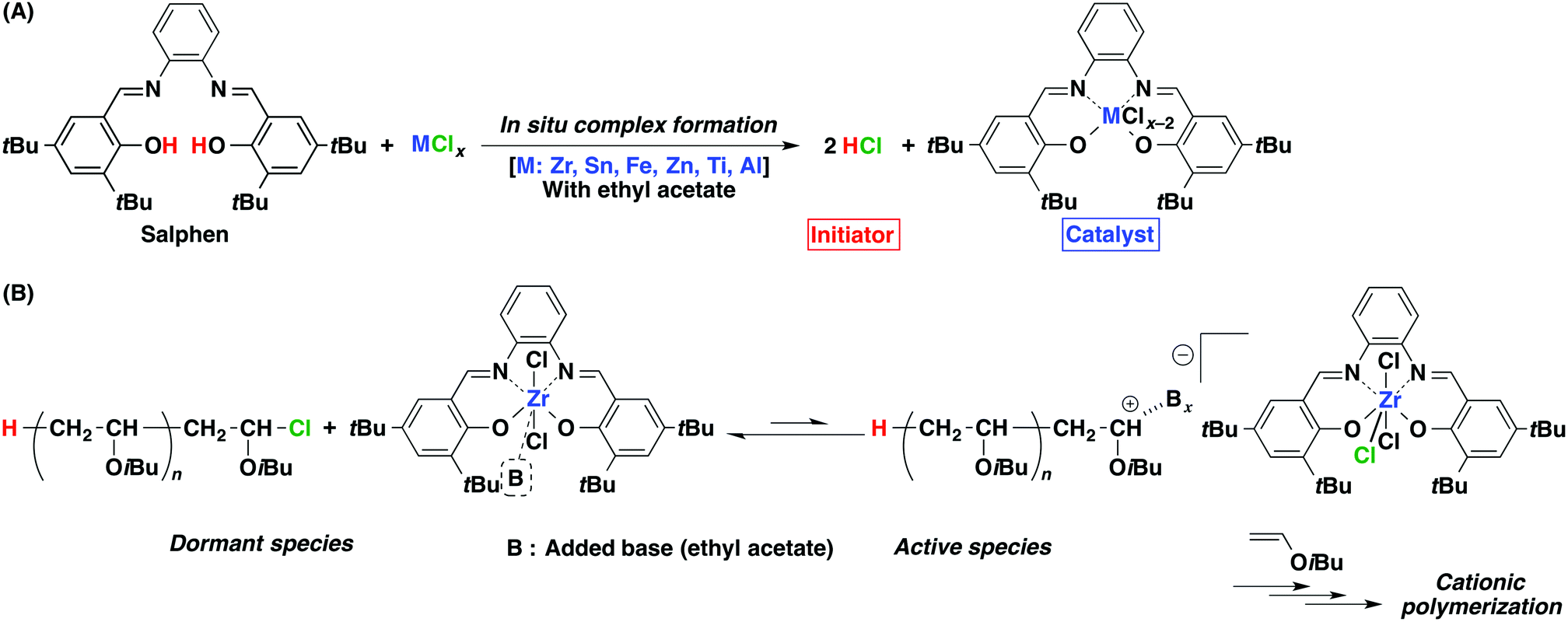 | ||
| Scheme 1 (A) In situ complex formation between the salphen ligand and a metal chloride and (B) polymerization mechanism of IBVE using the salphen/ZrCl4 initiating system. | ||
The choice of metal halide was crucial for achieving controlled polymerization. Among the metal halides examined, ZrCl4 combined with the ligand catalyzed the polymerization in the best-controlled manner (Table 1, entry 1). The reaction proceeded quantitatively at a moderate rate25 in a homogeneous system in the presence of ethyl acetate as an added base26 in toluene at 0 °C. The obtained polymers had narrow molecular weight distributions (MWDs), which shifted toward higher molecular weight regions with increasing monomer conversion. It is noteworthy that their Mn values increased linearly along the theoretical line calculated based on the molar ratio of IBVE to the phenoxy groups in the salphen ligand27 (Fig. S1†). These results also indicate the quantitative formation of the metal complex from the mixture of a salphen ligand and ZrCl4 along with a protonogen to initiate polymerization (Scheme 1A). 1H NMR analysis of the product polymer also showed the suppression of undesired side reactions28,29 during the polymerization (Fig. S2†).
| Entry | MCln | Temp (°C) | Time | Convc (%) | M n × 10−3 (calcd)d | M n × 10−3 (obs)e |
M
w/Mn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e e |
meso dyadf (%) |
|---|---|---|---|---|---|---|---|---|
| a [IBVE]0 = 0.76 M, [MCln]0 = 5.0 mM for entries 1–4 and 6–11, 15 mM for entry 5, [salphen ligand]0 = 5.0 mM for entries 1–4 and 6–11, 15 mM for entry 5, [ethyl acetate] = 1.0 M, [heptane] = 5.0 vol% in toluene at 0 °C. b In dichloromethane. c Determined by gas chromatography. d Calculated from the concentration of the salphen ligand.27 e Determined by GPC (polystyrene standards). f Determined by 13C NMR analysis (see Fig. S4 for the spectra). | ||||||||
| 1 | ZrCl4 | 0 | 18.5 h | 98 | 7.6 | 7.3 | 1.15 | 66 |
| 2 | 60 | 2.5 h | 94 | 7.2 | 5.0 | 1.18 | 59 | |
| 3 | 30 | 7 h | 99 | 7.6 | 7.4 | 1.16 | 62 | |
| 4 | −30 | 98 h | 96 | 7.4 | 8.9 | 1.37 | 70 | |
| 5 | −78 | 1410 h | 59 | 1.5 | 3.2 | 1.49 | 75 | |
| 6b | 0 | 2 h | 82 | 6.3 | 7.3 | 1.26 | 61 | |
| 7 | SnCl4 | 0 | 20 min | 94 | 7.3 | 10.5 | 1.37 | 65 |
| 8 | FeCl3 | 0 | 5 s | 90 | 7.0 | 11.5 | 1.57 | 61 |
| 9 | ZnCl2 | 0 | 6 h | 97 | 7.5 | 17.3 | 1.94 | 60 |
| 10 | TiCl4 | 0 | 168 h | 12 | 0.9 | n.d. | n.d. | n.d. |
| 11 | AlCl3 | 0 | 430 h | 2 | 0.2 | n.d. | n.d. | n.d. |
Several common metal chloride catalysts induced uncontrolled or no polymerization (Table 1, entries 7–11). The combination of SnCl4, FeCl3, or ZnCl2 with the salphen ligand induced uncontrolled polymerization. The GPC profiles of the products obtained with these metal chlorides had broad MWDs with noticeable tailing. The Mn values of the polymers obtained from the salphen/SnCl4 and ZnCl2 systems increased linearly, although they were higher than the calculated values. The higher Mn values likely result from a smaller proton source concentration (HCl) due to insufficient complex formation. In sharp contrast, no polymerization occurred for the combination of the salphen ligand with TiCl4 or AlCl3. The inactivity is attributable to the absence of vacant sites that exhibit Lewis acidity on the central metals, as opposed to the Zr-salphen complexes [Zr(salphen)Cl2], which have a labile coordination (a vacant) site because of their ability to form seven-coordinate structures30–33 (Scheme 1B). Therefore, metal chlorides can be classified into three groups according to their polymerization behavior: catalysts inducing living/controlled polymerization mediated by living/long-lived species (ZrCl4); uncontrolled polymerization (SnCl4, FeCl3, and ZnCl2); and no reaction (TiCl4 and AlCl3).
To confirm the living nature of the polymerization with the salphen/ZrCl4 initiating system, a monomer addition experiment was conducted under the optimized conditions. As shown in Fig. 1, after the addition of a fresh feed of the monomer, the MWD of the products shifted toward the higher molecular weight region with no original polymer remaining. A linear increase of the Mn values against the monomer conversion was also confirmed. Moreover, the 1H NMR spectra of these polymers showed the quantitative generation of the terminal acetal group34 and the undetectable undesired side reactions28,29 (Fig. S3†). These results demonstrate the progression of living polymerization using the complex prepared in situ from salphen and ZrCl4.
 | ||
| Fig. 1 (A) Time-conversion curves for the polymerization, (B) Mn (calculated Mn, dotted line) and Mw/Mn and (C) MWD curves for poly(IBVE)s obtained using the salphen/ZrCl4 initiating system ([IBVE]0 = [IBVE]added = 0.76 M, [ZrCl4]0 = 5.0 mM, [salphen ligand]0 = 5.0 mM, [ethyl acetate] = 1.0 M, [heptane] = 5.0 vol% in toluene at 0 °C). | ||
The stereoregularity of the product polymers was determined from the peaks of the methylene carbons of the main chains in the 13C NMR spectra recorded in CDCl3 (Fig. S4†). The meso dyad values of the polymer prepared with the examined salphen/MCln systems ranged from 60 to 66%, a range similar to the range obtained using simple metal halides as catalysts (61–67%).14,35 These results indicate that the direction of the insertion of the monomer molecules to the propagating carbocation is not affected by the steric hindrance around the catalysts produced by the salphen framework employed in the present study.
Table 1 also summarizes the polymerization results under various conditions, demonstrating the importance of temperature and the polarity of the solvent. The polymerization using this initiating system in toluene was examined at various temperatures (Table 1, entries 1–5). The polymerizations proceeded successfully even at higher temperatures (0–60 °C), producing polymers with relatively narrow MWDs (Fig. S5†). However, the polymerization reactions at low temperatures (−30 and −78 °C) yielded polymers with broad MWDs and Mn values that were higher than the theoretical ones. The Mn of the polymers obtained at −30 °C and the linear first-order plots for the polymerization (Fig. S6†) indicated that the broadening of the MWDs was not attributable to undesired side reactions, but instead, to slow initiation reaction kinetics. The polymerization in dichloromethane at 0 °C quantitatively yielded a polymer with a unimodal, although slightly broader MWD (Table 1, entry 6). In addition, the Mn was higher than the theoretical values (Fig. S7†). The results obtained under various conditions demonstrate that the polymerization in toluene at 0 °C is most suitable for the salphen/ZrCl4 system.
We examined the cationic polymerization of IBVE using an isolated salphen complex to confirm the occurrence of a quantitative, fast initiating reaction in the salphen/ZrCl4 system. Because the HCl generated during the complex formation was also removed during solvent evaporation, the adduct of IBVE with HCl (IBVE–HCl) was used as a cationogen for polymerization. The cationic polymerization of IBVE using the isolated catalyst proceeded in a controlled manner in the presence of ethyl acetate in toluene at 0 °C (Fig. S8†). The obtained polymers were similar in molecular weight and MWD to the products prepared from the in situ prepared salphen complex system. This result supports the hypothesis that the salphen/ZrCl4 initiating system, prepared in situ, led to quantitative complexation and a successive initiating reaction due to the generated HCl.
Conclusions
In conclusion, a well-defined metal complex was demonstrated to be effective for catalyzing the living cationic polymerization of alkyl vinyl ether. The as-prepared salphen/ZrCl4 complex induced the living cationic polymerization of IBVE in the presence of ethyl acetate as an added base in toluene at 0 °C. The use of tetradentate Schiff base ligands, which are easily tunable electronically and sterically, is expected to expand the available synthetic strategies for the precise control of cationic polymerization reactions, such as the adjustment of the catalytic activity and stereoselectivity of the catalyst. We are currently investigating polymerization reactions using various combinations of metal chlorides and salen ligands to reveal the relationship between the structure of the complex and its catalytic activity, especially by comparing the polymerization behavior of salphen and salen ligands, which will be reported in a forthcoming study.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research (22107006) on Innovative Areas of “Fusion Materials” (2206) from MEXT and by Grant-in-Aid for Young Scientists (A) (no. 26708014) from JSPS.Notes and references
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- R. R. Schrock, Dalton Trans., 2011, 40, 7484–7495 RSC.
- (a) M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS PubMed; (b) M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS PubMed.
- (a) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed; (b) F. di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959–1021 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100, 1479–1494 CrossRef CAS PubMed.
- E. Y.-X. Chen, Chem. Rev., 2009, 109, 5157–5214 CrossRef CAS PubMed.
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215–5244 CrossRef CAS PubMed.
- (a) S. Aoshima, T. Yoshida, A. Kanazawa and S. Kanaoka, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1801–1813 CrossRef CAS; (b) S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS PubMed.
- A. Kanazawa, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2509–2516 CrossRef CAS.
- S. Kanaoka, S. Nakayama and S. Aoshima, Kobunshi Ronbunshu, 2011, 68, 349–351 CrossRef CAS.
- (a) M. Sawamoto, Prog. Polym. Sci., 1991, 16, 111–172 CrossRef CAS; (b) J. P. Kennedy and B. Ivan, in Designed Polymers by Carbocationic Macromolecular Engineering: Theory and Practice, Hanser, New York, 1992 Search PubMed; (c) K. Matyjaszewski and M. Sawamoto, in Cationic Polymerizations, ed. K. Matyjaszewski, Marcel Dekker, New York, 1996, ch. 4 Search PubMed; (d) J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2285–2293 CrossRef CAS; (e) J. E. Puskas and G. Kaszas, Prog. Polym. Sci., 2000, 25, 403–452 CrossRef CAS; (f) P. De and R. Faust, in Macromolecular Engineering. Precise Synthesis, Materials Properties, Applications, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley-VCH GmbH & Co. KGaA, Weinheim, 2007, ch. 3 Search PubMed; (g) E. Goethals and F. D. Prez, Prog. Polym. Sci., 2007, 32, 220–246 CrossRef CAS.
- (a) M. C. Baird, Chem. Rev., 2000, 100, 1471–1478 CrossRef CAS PubMed; (b) T. D. Shaffer and J. R. Ashbaugh, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 329–344 CrossRef CAS; (c) M. Bochmann, Acc. Chem. Res., 2010, 43, 1267–1278 CrossRef CAS PubMed; (d) S. Jacob, Z. Pi and J. P. Kennedy, Polym. Bull., 1998, 41, 503–510 CrossRef CAS; (e) S. V. Kostjuk, H. Y. Yeong and B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 471–486 CrossRef CAS; (f) B. E. Diebl, Y. Li, M. Cokoja, F. E. Kühn, N. Radhakrishnan, S. Zschoche, H. Komber, H. Y. Yeong, B. Voit, O. Nuyken, P. Hanefeld and H.-M. Walter, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3775–3786 CrossRef CAS.
- (a) M. Ouchi, M. Kamigaito and M. Sawamoto, Macromolecules, 1999, 32, 6407–6411 CrossRef CAS; (b) M. Ouchi, M. Kamigaito and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1060–1066 CrossRef CAS.
- B. Li, Y. Wu, H. Cheng and W. Liu, Polymer, 2012, 53, 3726–3734 CrossRef CAS.
- S. Banerjee, T. K. Paira and T. K. Mandal, Macromol. Chem. Phys., 2013, 214, 1332–1344 CrossRef CAS.
- T. Pernecker, J. P. Kennedy and B. Ivan, Macromolecules, 1992, 25, 1642–1647 CrossRef CAS.
- M. Kamigaito, Y. Maeda, M. Sawamoto and T. Higashimura, Macromolecules, 1993, 26, 1643–1649 CrossRef CAS.
- M. Yonezumi, N. Takano, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6746–6753 CrossRef CAS.
- M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 5671–5675 CrossRef CAS.
- M. D. Hobday and T. D. Smith, Coord. Chem. Rev., 1973, 9, 311–337 CrossRef CAS.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- A polymerization using 2,6-diisopropylphenol and o-phenylenediamine with ZrCl4 was conducted as a control experiment. The equimolar amounts of the three components were mixed to give a catalytic solution. The polymerization using the solution proceeded but the monomer conversion leveled off at around 30% (150 h, 31%). On the other hand, the reaction using 2,6-diisopropylphenol and ZrCl4 without the diamine induced controlled polymerization in a manner similar to the polymerization using the alcohol/MCln initiating system.10 Thus, the superiority of the salphen complex over the catalysts with these model compounds is that both the electronic and geometric characteristics can be readily tuned because of the versatility of the ligand design and the multidentate ligation.
- (a) M. Palucki, N. S. Finney, P. J. Pospisil, M. L. Güler, T. Ishida and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 948–954 CrossRef CAS; (b) D. D. Ford, L. P. C. Nielsen, S. J. Zuend, C. B. Musgrave and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 15595–15608 CrossRef CAS PubMed; (c) T. Katsuki, Chem. Soc. Rev., 2004, 33, 437–444 RSC; (d) E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed; (e) N. Iwasa, S. Katao, J. Liu, M. Fujiki, Y. Furukawa and K. Nomura, Organometallics, 2009, 28, 2179–2187 CrossRef CAS; (f) H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363–2449 CrossRef CAS PubMed.
- In this system, it is difficult to tune the polymerization rate without changing target molecular weights because the initiator and Lewis acid catalysts should be always mixed at the same concentrations.
- An “added base” is a weak Lewis base that is used for living/controlled cationic polymerization. A weak Lewis base such as ethyl acetate, 1,4-dioxane, and THF controls the polymerization through its interaction with a Lewis acid catalyst, the propagating carbocation, and/or the counteranion.
- The generated HCl during complex formation was employed as a protonogen, or an initiator, for the cationic polymerization of vinyl ether. Hence, the theoretical Mn values were calculated based on the concentration of the generated HCl ([HCl]theoretical = 2 × [salphen ligand]0 = 10 mM except for entry 5 or 30 mM for entry 5 in Table 1).
- The product polymers had no irregular structures originating from undesired side reactions, such as β-proton elimination, dealcoholization, and the generation of aldehyde groups at chain ends,29 which was confirmed by 1H NMR (Fig. S2 and S3†).
- A. Kanazawa, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3702–3708 CrossRef CAS.
- F. Corazza, E. Solari, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Chem. Soc., Dalton Trans., 1990, 1335–1344 RSC.
- T. Repo, M. Klinga, P. Pietikäinen, M. Leskelä, A.-M. Uusitalo, T. Pakkanen, K. Hakala, P. Aaltonen and B. Löfgren, Macromolecules, 1997, 30, 171–175 CrossRef CAS.
- M. Wang, H. Zhu, D. Huang, K. Jin, C. Chen and L. Sun, J. Organomet. Chem., 2004, 689, 1212–1217 CrossRef CAS.
- H. Zhu, M. Wang, C. Ma, B. Li, C. Chen and L. Sun, J. Organomet. Chem., 2005, 690, 3929–3936 CrossRef CAS.
- The functionality of the terminal acetal group was determined from the peak integral ratio of the methyl proton of the initiating group (3H, peak a in Fig. S3†) and the methine proton of the end group (1H, peak g). The integral ratio a/g was 3.0/1.03, which indicates that quantitative functionality was achieved.
- A. Kanazawa, S. Kanaoka and S. Aoshima, Macromolecules, 2009, 42, 3965–3972 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and additional data. See DOI: 10.1039/c4py01012d |
| This journal is © The Royal Society of Chemistry 2015 |