DOI:
10.1039/C3PY01008B
(Paper)
Polym. Chem., 2014,
5, 169-174
Synthesis of high molar mass poly(n-butyl acrylate) and poly(2-ethylhexyl acrylate) by SET-LRP in mixtures of fluorinated alcohols with DMSO
Received
27th July 2013
, Accepted 8th August 2013
First published on 9th August 2013
Abstract
SET-LRP of n-butyl acrylate (nBA) and 2-ethylhexyl acrylate (EHA) initiated with bis(2-bromopropionyl)ethane (BPE) to synthesize high molar mass poly(nBA) and poly(EHA) was carried out in binary mixtures of 2,2,2-trifluoroethanol (TFE) or 2,2,3,3-tetrafluoropropanol (TFP) with DMSO at 50 °C. Using a solvent mixture of TFP containing 30% DMSO led for the first time to the synthesis of poly(nBA) with Mn = 527![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700, Mw/Mn = 1.21 in 12 h, and poly(EHA) with Mn = 913100, Mw/Mn = 1.20 in 15 h via SET-LRP. Although these two fluorinated alcohols provide complete solubilization of the hydrophobic monomers, nBA and EHA, and of the resulting polymers in addition to an efficient disproportionation of Cu(I)Br and subsequent stabilization of the Cu(II)Br2/L complex, SET-LRP targeting high molar mass poly(nBA) and poly(EHA) in these solvents alone resulted in a relatively slow polymerization with limited conversion. By contrast, DMSO, in spite of being the preferred solvent for SET-LRP with ability to disproportionate Cu(I)Br, stabilizes the “nascent” Cu(0) nanoparticle and provides an efficient SET process, generating a non-living polymerization for these hydrophobic monomers due to the insolubility of the resulting polymers. Remarkably, by a cooperative and synergistic effect, the binary mixtures of TFE or TFP with DMSO provide excellent reaction media for the synthesis of high molar mass poly(nBA) and poly(EHA) by SET-LRP.
700, Mw/Mn = 1.21 in 12 h, and poly(EHA) with Mn = 913100, Mw/Mn = 1.20 in 15 h via SET-LRP. Although these two fluorinated alcohols provide complete solubilization of the hydrophobic monomers, nBA and EHA, and of the resulting polymers in addition to an efficient disproportionation of Cu(I)Br and subsequent stabilization of the Cu(II)Br2/L complex, SET-LRP targeting high molar mass poly(nBA) and poly(EHA) in these solvents alone resulted in a relatively slow polymerization with limited conversion. By contrast, DMSO, in spite of being the preferred solvent for SET-LRP with ability to disproportionate Cu(I)Br, stabilizes the “nascent” Cu(0) nanoparticle and provides an efficient SET process, generating a non-living polymerization for these hydrophobic monomers due to the insolubility of the resulting polymers. Remarkably, by a cooperative and synergistic effect, the binary mixtures of TFE or TFP with DMSO provide excellent reaction media for the synthesis of high molar mass poly(nBA) and poly(EHA) by SET-LRP.
Introduction
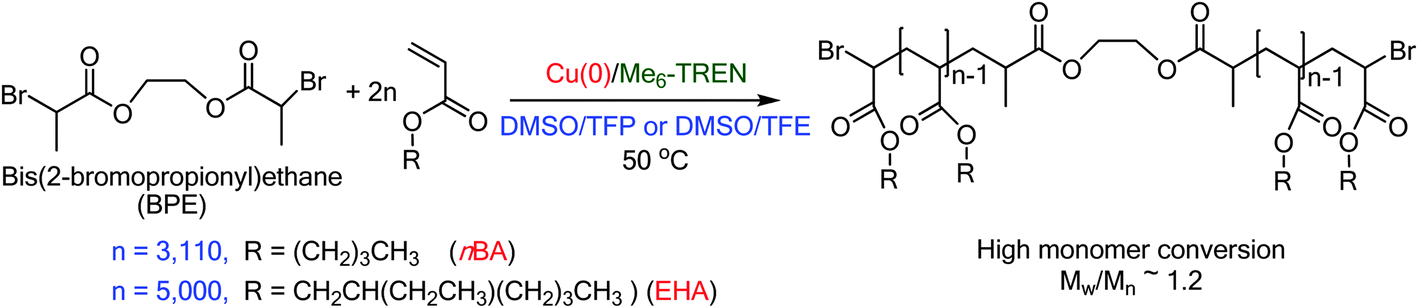
The development of Cu(0) mediated Single-Electron Transfer Living Radical Polymerization (SET-LRP)1–4 emerged as a robust technique for the synthesis of polymers3–25 with well-defined chain-end functionality even at complete monomer conversion.6,7,26–29 However, polymerization of hydrophobic monomers by SET-LRP is rather challenging since the most crucial step of SET-LRP, the disproportionation1–4,10,20,29–35 of the in situ generated Cu(I)X into a Cu(0) activator and a Cu(II)X2 deactivator in the presence of N-containing ligands (L)1–3,36 and subsequent stabilization of the Cu(II)X2/L complex is achieved in polar solvents, such as H2O,8,20,37–40 dimethyl sulfoxide (DMSO),6,7,10,14,17,20,29,41–43 dimethyl formamide (DMF),38,44 dimethyl acetamide (DMAC),38,44 ethylene and propylene carbonate,38,44 alcohols,15,41,45 and their binary mixtures,38,46 some of them exhibiting cooperative and synergistic effects.38,46 By contrast, non-polar solvents, such as toluene which mediates the disproportionation of CuBr in the presence of Me6-TREN44 but exhibits a limited solubility of the CuBr2/Me6-TREN complex gave rise to loss of chain-end functionality during SET-LRP of methyl acrylate (MA).44,47–49 Hence solvent choices for SET-LRP of monomers containing long hydrophobic chains have gained significant attention in recent years. When DMSO was employed as a solvent for a series of hydrophobic acrylates, such as n-butyl acrylate (nBA), tert-butyl acrylate (tBA), lauryl acrylate (LA), 2-ethylhexyl acrylate (EHA), phase separation of the polymer from the reaction mixture resulted in polymers with uncontrollable high molecular weight and broad molecular weight distribution as the hydrophobicity of the polymer increases.50 However, in spite of phase separation during SET-LRP of lauryl acrylate in isopropyl alcohol (IPA) living polymerization leading to polymers with narrow molecular weight distribution and high chain-end functionality resulted.22 In addition to this monomer–solvent system, binary mixtures of toluene with MeOH, and toluene with isopropyl alcohol (IPA) gave rise to excellent SET-LRP conditions for acrylates containing long hydrocarbon chains (C12–C18).22 In recent publications45,51,52 we have demonstrated that two fluorinated alcohols, TFE and TFP, which promote disproportionation of Cu(I)X and subsequent stabilization of the resultant Cu(0) species generated by disproportionation, provide suitable reaction media for SET-LRP of various hydrophobic and hydrophilic acrylates,45,51 including MA, nBA, tBA, EHA, and 2-hydroxylethyl acrylate and hydrophobic methacrylates,52 methyl methacrylate, ethyl methacrylate and n-butyl methacrylate. In both TFE and TFP, SET-LRP proceeds in a single phase without any visual sign of polymer insolubility until complete monomer conversion.45,51,52 A detailed study on solvent choices for SET-LRP has revealed the cooperative and synergistic solvent effect, which demonstrates that the addition of a solvent that promotes a higher extent of disproportionation in the reaction mixture results in a faster rate of polymerization.38,46 Therefore, we considered that it would be important to find a second solvent which would accelerate the rate of SET-LRP performed in the fluorinated alcohols, TFE and TFP that would enable us to synthesize high molar mass polymers from hydrophobic monomers. In this regard, DMSO is a unique solvent, which promotes fast disproportionation of Cu(I)Br and provides stable “nascent” Cu(0) nanoparticles and provides ultrafast polymerization of a range of vinyl monomers, such as acrylates, methacrylates, vinyl chloride in addition to the synthesis of ultra-high molar mass poly(MA),3 and even of hydrophilic poly(2-hydroxyethyl methacrylate)17 without the need for protection of the hydroxyl moiety. Hence we selected the binary mixtures of TFE and TFP with DMSO at various ratios for the synthesis of a high molar mass polymer of two hydrophobic monomers, nBA up to DP = 6220 and EHA up to DP = 10000 using bis(2-bromopropionyl)ethane (BPE) as a bifunctional initiator, Me6-TREN as a ligand, and Cu(0) wire as a catalyst. In this context it is noteworthy that the Atom Transfer Radical Polymerization (ATRP) conducted at an extremely high pressure (5 kbar) resulted in the synthesis of high molar mass poly(nBA) with Mn = 808000, Mw/Mn = 1.51 at Cu(I)/initiator = 1:1 in 4 h and Mn = 612000, Mw/Mn = 1.25 at Cu(I)/initiator = 1:4 in 6 h in MeCN at 80 °C.53 Nevertheless, the results reported in this publication demonstrate that the binary mixtures of TFE and TFP with DMSO provide suitable reaction media for SET-LRP of hydrophobic monomers, such as nBA and EHA mediating the synthesis of high molar mass polymers under mild reaction conditions such as 50 °C at atmospheric pressure (Scheme 1).
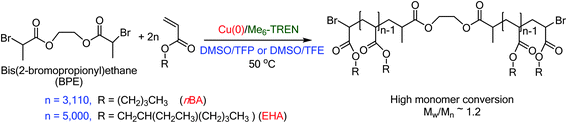 |
| | Scheme 1 Synthesis of high molar mass poly(nBA) and poly(EHA) in binary mixtures of TFE or TFP with DMSO at 50 °C. | |
Results and discussion
Synthesis of high molar mass poly(nBA)
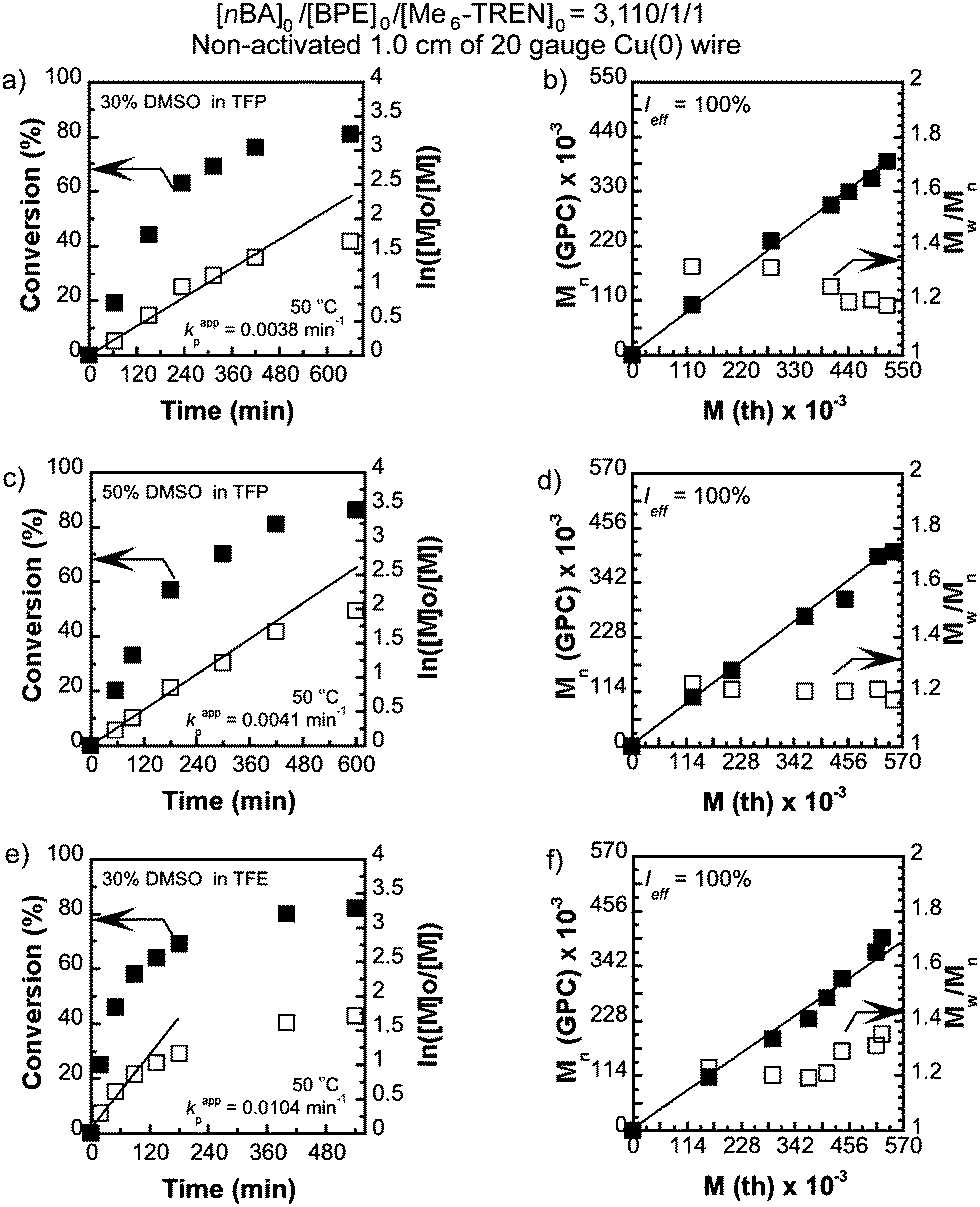
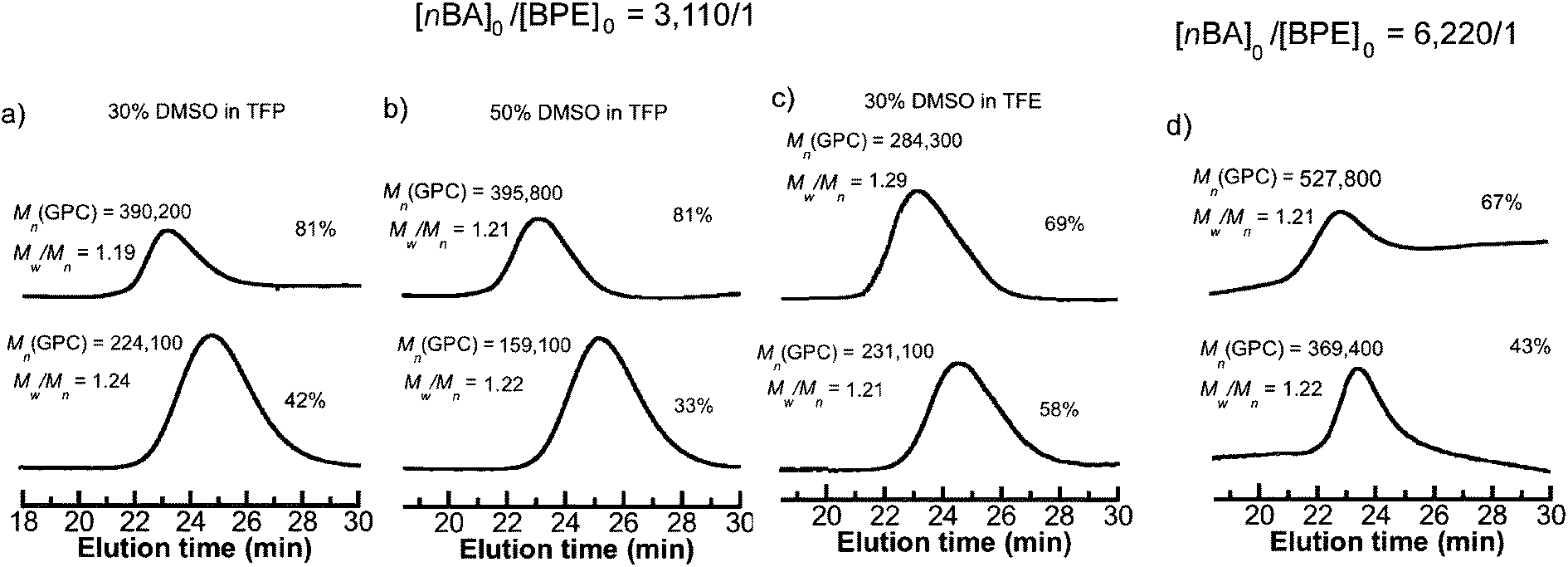
We first attempted to conduct SET-LRP of nBA in DMSO at [nBA]0/[BPE]0/[Me6-TREN]0/[CuBr2] = 3110/1/1.5/0.5 catalyzed with 1.0 cm of non-activated Cu(0) wire (0.812 mm diameter, 20 gauge) at 50 °C. This resulted in 98% monomer conversion giving rise to a polymer with Mn = 460100, Mw/Mn = 2.15 and bimodal molecular weight distribution (Table 1, entry 1). The reaction mixture showed phase separation in a similar fashion to the results reported by Boyer et al.50 during SET-LRP of nBA in DMSO at a much lower targeted molecular weight ([M]0/[I]0 = 39:1). By contrast, SET-LRP of nBA at [nBA]0/[BPE]0/[Me6-TREN]0 = 3110/1/1 under similar reaction conditions in either TFP or TFE produced polymers with low monomer conversion but narrow molecular weight distribution (entries 2 and 3, Table 1). However, when a binary mixture of TFP or TFE with DMSO was used as a solvent for SET-LRP of nBA at [nBA]0/[BPE]0/[Me6-TREN]0 = 3110/1/1 no phase separation was observed even at 50% DMSO in TFP and at least up to 30% DMSO in TFE. The kinetic evaluation of the SET-LRP experiments conducted for nBA in TFP containing 30% DMSO and 50% DMSO at [nBA]0/[BPE]0/[Me6-TREN]0 = 3110/1/1 are shown in Fig. 1a–d. While using TFE and TFP as solvents in pure form or as co-solvents with DMSO, non-activated Cu(0) wire was used instead of hydrazine-activated catalyst since these fluorinated alcohols can act as self-activating solvents resulting in comparable kappp to that of a hydrazine-activated catalyst.54 Remarkably, these mixtures of solvents provided polymerization reactions with significantly higher monomer conversion, a much faster reaction rate compared to only TFE or TFP, and resulted in poly(nBA) with narrow molecular weight distribution (Fig. 2a and b, entries 4–11, Table 1). In both cases the reaction followed first order kinetics up to monomer conversion of almost 80% with an apparent rate constant of propagation (kappp) of 0.0038 min−1 for TFP containing 30% DMSO and 0.0041 min−1 for TFP containing 50% DMSO. The number average molecular weight (Mn) of poly(nBA) follows a linear dependence on monomer conversion and narrow molecular weight distribution (Fig. 1b and d) demonstrating that the living nature of the polymerization of nBA in these two solvents results in poly(nBA) with Mn = 315200, Mw/Mn = 1.19 in TFP containing 30% DMSO and Mn = 335400, Mw/Mn = 1.17 in TFP containing 50% DMSO in 10 h. In both cases the stirring ceased at around 80% monomer conversion due to the high viscosity of the reaction mixture resulting in almost no additional polymerization even after much longer reaction times. It is noteworthy that when only TFP was used as a solvent under similar reaction conditions, polymerization reached only 58% conversion after 12 h with a kappp of 0.0014 min−1 resulting in poly(nBA) with Mn = 225100, Mw/Mn = 1.21. Hence addition of 50% DMSO to TFP resulted in an almost three fold enhancement of kappp without any broadening of the molecular weight distribution. However, addition of 70% DMSO resulted in slight phase separation and exhibited 88% monomer conversion after 9 h resulting in poly(nBA) with Mn = 381200 and broader molecular weight distribution (Mw/Mn = 1.56). This broadening could be attributed to the insolubility of the resulting polymer at higher monomer conversion in the disproportionating solvent containing the CuBr2 deactivator. Hence addition of DMSO as a co-solvent to TFP is necessary for high monomer conversion along with narrow molecular weight distribution. However, when the reaction was conducted at room temperature in TFP containing 30% DMSO a low monomer conversion (50%) was obtained after a long reaction time with Mn = 201400 and Mw/Mn = 1.29 (entry 7, Table 1).
Table 1 SET-LRP of nBA during catalysis with Cu(0)/Me6-TREN in a binary mixture of TFP or TFE and DMSO
| Entry |
[M]/[I]/[L] |
Solvent |
Temp. (°C) |
k
appp (min−1) |
Conv. (%) |
M
th
|
M
n (GPC) |
M
w/Mn |
| 1 |
3110/1/1 |
DMSO |
50 |
— |
98 |
390970 |
460100 |
2.15 |
| 2 |
3110/1/1 |
TFP |
50 |
0.0014 |
58 |
231530 |
225100 |
1.21 |
| 3 |
3110/1/1 |
TFE |
50 |
— |
68 |
271390 |
281300 |
1.32 |
| 4 |
3110/1/1 |
30% DMSO in TFP |
50 |
0.0038 |
81 |
323210 |
315200 |
1.18 |
| 5 |
3110/1/1 |
50% DMSO in TFP |
50 |
0.0041 |
86 |
343140 |
335400 |
1.17 |
| 6 |
3110/1/1 |
70% DMSO in TFP |
50 |
0.0061 |
88 |
351110 |
381200 |
1.56 |
| 7 |
3110/1/1 |
30% DMSO in TFP |
25 |
— |
50 |
199640 |
201400 |
1.29 |
| 8 |
6220/1/1 |
30% DMSO in TFP |
50 |
— |
33 |
263410 |
249300 |
1.28 |
| 9 |
6220/1/2 |
30% DMSO in TFP |
50 |
0.0027 |
67 |
534470 |
527700 |
1.21 |
| 10 |
3110/1/1 |
30% DMSO in TFE |
50 |
0.0104 |
82 |
327190 |
340900 |
1.35 |
| 11 |
6220/1/2 |
30% DMSO in TFE |
50 |
— |
52 |
414890 |
453700 |
1.52 |
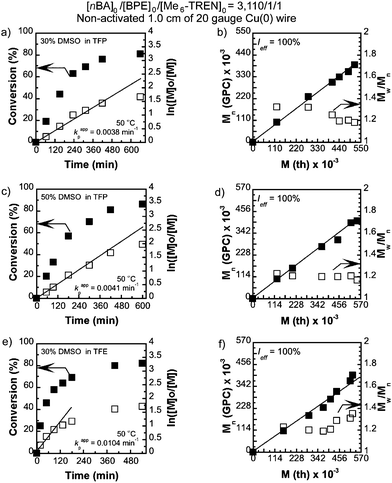 |
| | Fig. 1 Kinetic plots (left) and Mn, and Mw/Mnvs. theoretical Mth (right) for SET-LRP reactions of n-butyl acrylate (nBA) performed in a binary mixture of DMSO and trifluoroethanol (TFE) or tetrafluoropropanol (TFP). Conditions: (a and b) nBA = 1 mL, TFP containing 30% DMSO = 0.5 mL, and [nBA]0/[BPE]0/[Me6-TREN]0 = 3110/1/1, non-activated 1.0 cm Cu(0) wire, (c and d) nBA = 1 mL, TFP containing 50% DMSO = 0.5 mL, and [nBA]0/[BPE]0/[Me6-TREN]0 = 3110/1/1, non-activated 1.0 cm Cu(0) wire, (e and f) nBA = 1 mL, TFE containing 30% DMSO = 0.5 mL, and [nBA]0/[BPE]0/[Me6-TREN]0 = 3110/1/1, non-activated 1.0 cm Cu(0) wire. | |
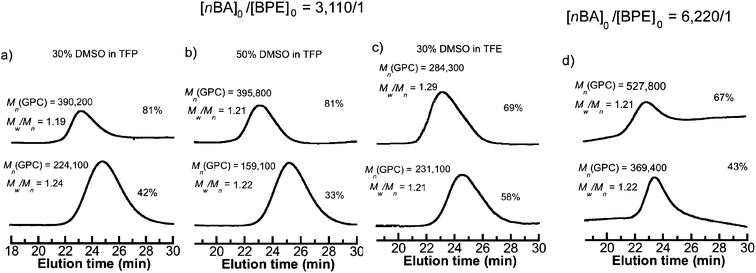 |
| | Fig. 2 GPC traces for SET-LRP reactions of nBA performed in a binary mixture of DMSO and TFE or TFP with nonactivated 1.0 cm Cu(0) wire as a catalyst at 50 °C. Conditions: (a) nBA = 1 mL, 30% DMSO in TFP = 0.5 mL, and [nBA]0/[BPE]0/[Me6-TREN]0 = 3110/1/1, (b) nBA = 1 mL, 50% DMSO in TFP = 0.5 mL, and [nBA]0/[BPE]0/[Me6-TREN]0 = 3110/1/1, (c) nBA = 1 mL, 30% DMSO in TFE = 0.5 mL, and [nBA]0/[BPE]0/[Me6-TREN]0 = 3110/1/1, (d) nBA = 1 mL, 30% DMSO in TFP = 0.5 mL, and [nBA]0/[BPE]0/[Me6-TREN]0 = 6220/1/2. The change in the slope of the baseline in the GPC chromatograms provides artificial shoulders in the GPC traces. | |
When TFE was used as the solvent under similar reaction conditions 68% conversion was obtained after 12 h resulting in poly(nBA) with Mn = 281300 and Mw/Mn = 1.32 (entry 3, Table 1). Addition of 30% DMSO to TFE gave rise to 82% conversion after 9 h resulting in poly(nBA) with Mn = 340900 and Mw/Mn = 1.35 (entry 10, Table 1). In this solvent the polymerization takes place at a higher rate (kappp = 0.0104 min−1) compared to that in TFP containing 30% DMSO (kappp = 0.0038 min−1) and a linear dependence of ln([M]0/[M]) on time was obtained up to 60% monomer conversion which then drastically deviates from linearity. Due to the faster rate of propagation than initiation, during the early stages of the polymerization29,49 when using TFE with 30% DMSO as a co-solvent and BPE as the initiator, a very small extent of bimolecular molecular weight distribution can be observed (Fig. 1e, f and 2c). However, the change in the baseline of the GPC traces as indicated in Fig. 2 can provide an artificial bimolecular weight distribution observed as a shoulder on these chromatograms.
Given the success of SET-LRP for nBA at [nBA]0/[BPE]0 = 3110/1, SET-LRP of nBA targeting a higher degree of polymerization by using [nBA]0/[BPE]0 = 6220/1 was carried out in binary mixtures of TFP and TFE containing 30% DMSO. It was observed that when 30% DMSO in TFP was employed as a solvent at 50 °C, addition of 2 equivalents of the ligand gave rise to 67% monomer conversion resulting in poly(nBA) with Mn = 527700, Mw/Mn = 1.21 (entry 9, Table 1 and Fig. 2d) which is almost two times the monomer conversion in the presence of 1 equivalent of the ligand (33%, entry 8, Table 1). However, when TFE was employed as a solvent for [nBA]0/[BPE]0 = 6220/1 a broader molecular weight distribution was obtained (entry 11, Table 1) with Mn = 453700, Mw/Mn = 1.52, similar to the case of [nBA]0/[BPE]0 = 3110/1.
Synthesis of high molar mass poly(EHA)
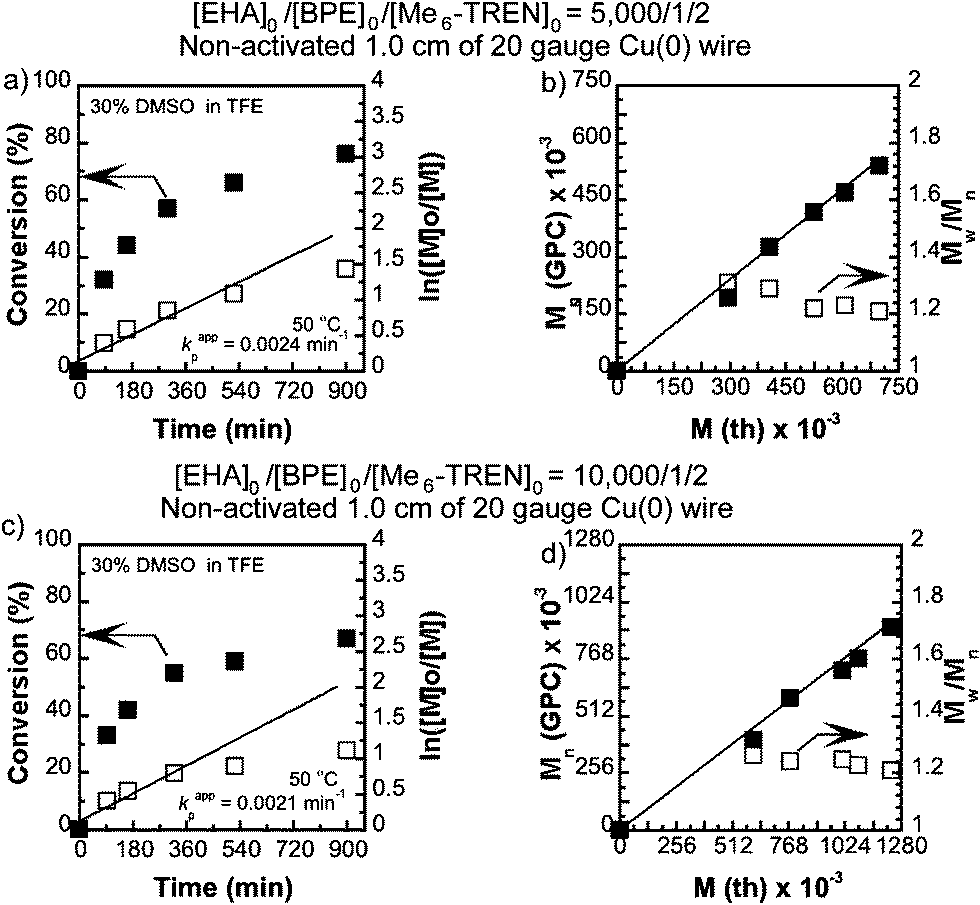
The synthesis of high molar mass poly(EHA) was carried out by SET-LRP in the same binary solvent mixtures targeting [EHA]0/[BPE]0 = 5000 and 10000. It is noteworthy that, although SET-LRP of EHA in both TFE and TFP continues in a single phase, unlike nBA the addition of 30% DMSO results in phase separation after ∼60% monomer conversion (Table 2 and Fig. 3). It was observed that for TFE containing 30% of DMSO at [EHA]0/[BPE]0 = 5000/1 addition of 2 equivalents of the ligand exhibited higher conversion compared to when 1 equivalent of the ligand was used (entry 1, 2, Table 2). Kinetic plots for [EHA]0/[BPE]0/[Me6-TREN]0 = 5000/1/2 and [EHA]0/[BPE]0/[Me6-TREN]0 = 10000/1/2 in 30% DMSO in TFE are shown in Fig. 3. In both cases ln([M]0/[M]) showed near-linear increment with time until ∼60% monomer conversion. At higher conversion a deviation from linearity was observed. However, the number average molecular weight (Mn) of poly(EHA) follows a linear dependence on monomer conversion with narrow molecular weight distribution (Mw/Mn ∼1.2). For [EHA]0/[BPE]0/[Me6-TREN]0 = 5000/1/2 in TFE containing 30% DMSO poly(EHA) was obtained with Mn = 539500, Mw/Mn = 1.20, entry 4, Table 2 and for [EHA]0/[BPE]0/[Me6-TREN]0 = 10000/1/2 poly(EHA) with Mn = 913100 and Mw/Mn = 1.21 (entry 3, Table 2) was obtained. When TFP containing 30% DMSO was employed for [EHA]0/[BPE]0/[Me6-TREN]0 = 5000/1/2 a lower monomer conversion (68%) was obtained compared to the TFE–DMSO solvent mixture resulting in poly(EHA) with Mn = 435100 and Mw/Mn = 1.35 (entry 4, Table 2).
Table 2 SET-LRP of EHA during catalysis with Cu(0)/Me6-TREN in binary mixtures of TFP or TFE with DMSO
| Entry |
[M]/[I]/[L] |
Solvent |
Temp. (°C) |
k
appp (min−1) |
Conv. (%) |
M
th
|
M
n (GPC) |
M
w/Mn |
| 1 |
5000/1/1 |
30% DMSO in TFE |
50 |
— |
66 |
617670 |
352900 |
1.23 |
| 2 |
5000/1/2 |
30% DMSO in TFE |
50 |
0.0024 |
76 |
700600 |
539500 |
1.20 |
| 3 |
10000/1/2 |
30% DMSO in TFE |
50 |
0.0021 |
67 |
1235010 |
913100 |
1.21 |
| 4 |
5000/1/2 |
30% DMSO in TFP |
50 |
— |
68 |
626880 |
435100 |
1.35 |
| 5 |
10000/1/2 |
30% DMSO in TFP |
50 |
— |
20 |
368560 |
210600 |
1.25 |
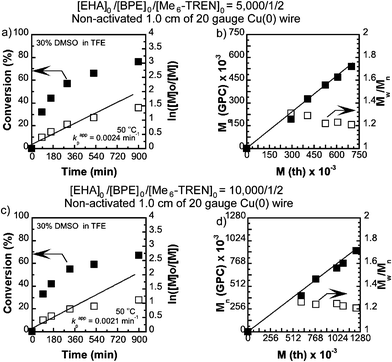 |
| | Fig. 3 Kinetic plots (left) and Mn and Mw/Mnvs. theoretical Mth (right) for SET-LRP reactions of 2-ethylhexyl acrylate (EHA) performed in a binary mixture of DMSO and trifluoroethanol (TFE). Conditions: (a and b) EHA = 1 mL, TFE containing 30% DMSO = 0.5 mL, and [EHA]0/[BPE]0/[Me6-TREN]0 = 5000/1/2, non-activated 1.0 cm Cu(0) wire, (c and d) EHA = 1 mL, TFE containing 30% DMSO = 0.5 mL, and [EHA]0/[BPE]0/[Me6-TREN]0 = 10000/1/2, non-activated 1.0 cm Cu(0) wire. | |
Conclusions
Excellent reaction media for SET-LRP of hydrophobic acrylates nBA and EHA were obtained when a combination of TFP or TFE with DMSO was used as a solvent. Very high monomer conversion and narrow molecular weight distribution with a relatively fast rate of reaction compared to pure TFP or TFE were obtained. Using these solvent mixtures for the first time, high molar mass poly(nBA) (Mn = 527700, Mw/Mn = 1.21, 12 h) and poly(EHA) (Mn = 913100, Mw/Mn = 1.20, 15 h) were produced. In both cases narrow molecular weight distribution of the polymers demonstrated that the polymerization reactions are living in nature and there is no significant impact of side reactions. Previously we reported that SET-LRP provided access for the first time to ultrahigh molar mass poly(MA) in DMSO (Mn = 1400000, Mw/Mn = 1.15, 10 h),3 in MeOH (Mn = 800000, Mw/Mn = 1.15, <3 h),41 poly(2-hydroxyethyl methacrylate) in DMSO (PHEMA, Mn = 1017900, Mw/Mn = 1.49, 38 h),17 and high molar mass poly(tBA) (Mn = 505120 and Mw/Mn = 1.30, 15 h) in TFE.45 In addition, high molecular weight linear polymers were synthesized by Single-Electron-Transfer Degenerative-chain-Transfer mediated Living Radical Polymerization (SET-DTLRP) initiated with iodoform, and catalyzed by sodium dithionite producing poly(nBA) with Mn = 85000, Mw/Mn = 1.9 in 5 h,55 and poly(tBA) with Mn = 823000, Mw/Mn = 1.15 in 71 h.56
Experimental
Materials
2-Ethylhexyl acrylate (99%, Acros) and n-butyl acrylate (99%, Sigma Aldrich) were passed through a short column of basic Al2O3 prior to use in order to remove the radical inhibitor. Dimethyl sulfoxide (DMSO) (Fisher, Certified ACS, 99.9) was used as received, TFP (SynQuest Laboratories) and TFE (AlfaAesar) were used as received. Hexamethylated tris(2-aminoethyl)amine (Me6-TREN) was synthesized as described in the literature.57 The bifunctional initiator BPE was synthesized by esterification of ethylene glycol with 2-bromopropionyl bromide in the presence of pyridine.42
Techniques
Gel Permeation Chromatography (GPC) analysis was performed on a Perkin-Elmer Series 10 high-performance liquid chromatograph, equipped with an LC-100 column oven at 30 °C, a Nelson Analytical 900 Series integration data station, a Perkin-Elmer 785 UV-vis detector (254 nm), a Varian star 4090 refractive index (RI) detector, and three AM gel columns (5 μm particle size and 500 Å, 1000 Å, and 10000 Å pore size). HPLC grade THF (Fisher) was used as an eluent at a flow rate of 1 mL min−1. The number-average molecular weight (Mn) and molecular weight distribution (Mw/Mn) were determined with poly(methyl methacrylate) (PMMA) standards purchased from American Polymer Standards.
Typical procedure for SET-LRP of nBA at [nBA]0/[BPE]0/[Me6-TREN]0 = 3110/1/1
A stock solution containing 1 mL nBA (1 mL, 6.89 mmol), initiator BPE (2.21 μmol, 0.7385 mg), ligand Me6-TREN (2.21 μmol, 0.510 mg), 0.5 mL of solvent and a stirring bar was charged to a Schlenk tube. The reaction mixture was then deoxygenated by six freeze-pump thaw cycles using liquid nitrogen as a freezing mixture. After these cycles the Schlenk tube was opened under a positive argon pressure to add the Cu(0) wire. It was further degassed and refilled by argon three times and transferred to a thermostatted oil bath operating at the desired temperature (50 °C). The introduction of the Cu(0) wire defines t = 0. Approximately one or two drops of samples were periodically removed from the reaction mixture by purging the side arm with argon for more than 2 min using a deoxygenated syringe and needle. The collected samples were then analyzed by 1H NMR in CDCl3 and subsequently by GPC to obtain the molecular weight and Mw/Mn data respectively.
Acknowledgements
Financial support by the National Science Foundation (DMR-1120901, DMR-1066116) and the P. Roy Vagelos Chair at Penn are gratefully acknowledged.
References
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS PubMed.
- V. Percec, A. V. Popov, E. Ramirez-Castillo and O. Weichold, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3283–3299 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- S. Zhai, B. Wang, C. Feng, Y. Li, D. Yang, J. Hu, G. Lu and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 647–655 CrossRef CAS.
- N. H. Nguyen, M. E. Levere and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 860–873 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4684–4695 CrossRef CAS.
- X. Leng, N. Nguyen, B. van Beusekom, D. A. Wilson and V. Percec, Polym. Chem., 2013, 4, 2995–3004 RSC.
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Macromolecules, 2009, 42, 2379–2386 CrossRef CAS.
- X. Jiang, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 403–409 CrossRef CAS.
- G. Chen, P. M. Wright, J. Geng, G. Mantovani and D. M. Haddleton, Chem. Commun., 2008, 1097–1099 RSC.
- M. W. Jones, M. I. Gibson, G. Mantovani and D. M. Haddleton, Polym. Chem., 2011, 2, 572–574 RSC.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4889–4893 CrossRef CAS.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2243–2250 CrossRef CAS.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2236–2242 CrossRef CAS.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4884–4888 CrossRef CAS.
- N. H. Nguyen, X. Leng and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2013, 4, 2760–2766 RSC.
- B. Barboiu and V. Percec, Macromolecules, 2001, 34, 8626–8636 CrossRef CAS.
- X.-H. Liu, G.-B. Zhang, B.-X. Li, Y.-G. Bai and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5439–5445 CrossRef CAS.
- N. H. Nguyen, J. Kulis, H.-J. Sun, Z. Jia, B. van Beusekom, M. E. Levere, D. A. Wilson, M. J. Monteiro and V. Percec, Polym. Chem., 2013, 4, 144–155 RSC.
- N. H. Nguyen, X. Jiang, S. Fleischmann, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5629–5638 CrossRef CAS.
- A. Anastasaki, C. Waldron, V. Nikolaou, P. Wilson, R. McHale, T. Smith and D. M. Haddleton, Polym.
Chem., 2013, 4, 4113–4119 RSC.
- Q. Zhang, J. Collins, A. Anastasaki, R. Wallis, D. A. Mitchell, C. R. Becer and D. M. Haddleton, Angew. Chem., Int. Ed., 2013, 52, 4435–4439 CrossRef CAS PubMed.
- J. Burns, C. Houben, C. Waldron, A. Anastasaki, A. Lapkin and D. Haddleton, Polym. Chem., 2013, 4, 4809–4813 RSC.
- Y.-N. Zhou and Z.-H. Luo, Polym. Chem., 2013, 4, 76–84 RSC.
- G. Lligadas, J. S. Ladislaw, T. Guliashvili and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 46, 278–288 CrossRef.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- C. Boyer, A. H. Soeriyadi, P. B. Zetterlund and M. R. Whittaker, Macromolecules, 2011, 44, 8028–8033 CrossRef CAS.
- N. H. Nguyen, M. E. Levere, J. Kulis, M. J. Monteiro and V. Percec, Macromolecules, 2012, 45, 4606–4622 CrossRef CAS.
- M. E. Levere, N. H. Nguyen, H.-J. Sun and V. Percec, Polym. Chem., 2013, 4, 686–694 RSC.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4227–4240 CAS.
- N. H. Nguyen, H.-J. Sun, M. E. Levere, S. Fleischmann and V. Percec, Polym. Chem., 2013, 4, 1328–1332 RSC.
- B. M. Rosen, X. Jiang, C. J. Wilson, N. H. Nguyen, M. J. Monteiro and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5606–5628 CrossRef CAS.
- M. J. Monteiro, T. Guliashvili and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1835–1847 CrossRef CAS.
- B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4950–4964 CrossRef CAS.
- A. Anastasaki, C. Waldron, P. Wilson, R. McHale and D. M. Haddleton, Polym. Chem., 2013, 4, 2672–2675 RSC.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- N. H. Nguyen, B. M. Rosen, X. Jiang, S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5577–5590 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1752–1763 CrossRef CAS.
- N. Nga Hang, C. Rodriguez-Emmenegger, E. Brynda, Z. Sedlakova and V. Percec, Polym. Chem., 2013, 4, 2424–2427 RSC.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2745–2754 CrossRef CAS.
- G. Lligadas, B. M. Rosen, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8360–8364 CrossRef CAS.
- X. Jiang, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2716–2721 CrossRef CAS.
- M. E. Levere, N. H. Nguyen, X. Leng and V. Percec, Polym. Chem., 2013, 4, 1635–1647 RSC.
- S. R. Samanta, M. E. Levere and V. Percec, Polym. Chem., 2013, 4, 3212–3224 RSC.
- X. Jiang, S. Fleischmann, N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5591–5605 CrossRef CAS.
- P. M. Wright, G. Mantovani and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7376–7385 CrossRef CAS.
- M. E. Levere, I. Willoughby, S. O'Donohue, P. M. Wright, A. J. Grice, C. Fidge, C. Remzi Becer and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1753–1763 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6880–6895 CrossRef CAS.
- C. Boyer, A. Atme, C. Waldron, A. Anastasaki, P. Wilson, P. B. Zetterlund, D. Haddleton and M. R. Whittaker, Polym. Chem., 2013, 4, 106–112 RSC.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013 10.1039/c3py00901g.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013 10.1039/c3py00902e.
- Y. Wang, H. Schroeder, J. Morick, M. Buback and K. Matyjaszewski, High-Pressure Atom Transfer Radical Polymerization of n-Butyl Acrylate, Macromol. Rapid Commun., 2013, 34, 604–609 CrossRef CAS PubMed.
- S. R. Samanta, H.-J. Sun, A. Anastasaki, D. M. Haddleton and V. Percec, Polym. Chem., 2013 10.1039/c3py01007d.
- J. F. J. Coelho, A. M. F. P. Silva, A. V. Popov, V. Percec, M. V. Abreu, P. M. O. F. Gonçalves and M. H. Gil, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2809–2825 CrossRef CAS.
- V. Percec, E. Ramirez-Castillo, A. V. Popov, L. A. Hinojosa-Falcon and T. Guliashvili, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2178–2184 CrossRef CAS.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41–44 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.