Selective O-acyl ring-opening of β-butyrolactone catalyzed by trifluoromethane sulfonic acid: application to the preparation of well-defined block copolymers†
Aline
Couffin
ab,
Blanca
Martín-Vaca
*ab,
Didier
Bourissou
*ab and
Christophe
Navarro
c
aUniversity of Toulouse, UPS, LHFA, 118 route de Narbonne, F-31062 Toulouse, France. E-mail: dbouriss@chimie.ups-tlse.fr; bmv@chimie.ups-tlse.fr; Fax: +33 561558204; Tel: +33 561557737
bCNRS, LHFA UMR 5069, F-31062 Toulouse, France
cArkema, Lacq Research Center, Po Box 34, 64170 Lacq, France
First published on 7th August 2013
Abstract
A detailed study of β-butyrolactone (β-BL) ring-opening polymerization (ROP) with methane and trifluoromethane sulfonic acids (MSA and HOTf, respectively) is reported. HOTf affords the best results in terms of activity and selectivity, leading to PBL of controlled molecular weights (Mn up to 8200 g mol−1) and narrow distributions (Đ < 1.25). Ring-opening of β-BL occurs selectively via O-acyl bond cleavage and crotonisation reactions do not occur to a significant extent. This leads to hydroxyl terminated PBL that is able to initiate efficiently ROP of ε-caprolactone (ε-CL). Using mono- and dihydroxylated initiators, including macroinitiators, a variety of well-defined block copolymers have been prepared upon successive monomer addition: PBL-b-PCL, PCL-b-PBL-b-PCL, PBL-b-PEG-b-PBL (PEG = polyethylene glycol), PCL-b-PBL-b-PEG-b-PBL-b-PCL and PCL-b-PBL-b-PBD-b-PBL-b-PCL (PBD = polybutadiene). 1H/13C NMR spectroscopy, Size Exclusion Chromatography (SEC) and MALDI-TOF mass spectrometry have been used to ascertain the structure of the obtained polymers.
Introduction
The past two decades have witnessed significant progress in Ring-Opening Polymerization (ROP) of lactones and cyclic carbonates. Nowadays, this unambiguously represents the most efficient and versatile method to prepare biodegradable polyesters and polycarbonates in a controlled manner.1–3 Thereby, polymers of desired properties are readily available starting from common monomers such as ε-caprolactone (ε-CL), lactide (LA), and trimethylene carbonate (TMC).β-Butyrolactone (β-BL) has been much less studied than the aforementioned monomers. In particular, it has only been scarcely involved in efficient copolymerization, to the best of our knowledge. This is probably due to the peculiar behaviour of β-BL, compared to the other monomers. Indeed, β-BL suffers from two main drawbacks (Fig. 1): (i) poor ring-opening selectivity, with inherent competition between O-acyl bond cleavage (leading to a propagating hydroxyl chain-end) and O-alkyl bond cleavage (leading to a propagating carboxylic acid chain-end); (ii) propensity to undergo crotonisation reactions yielding acrylic moieties (non-propagating chain-end).4 These two phenomena are often at the origin of some loss of polymerization control. They also represent obstacles to efficient copolymerization with other monomers, as propagation can only occur at hydroxyl chain-ends. Yet, copolymers attract increasing attention in materials science. Combining different monomers and varying their arrangement (random, block, gradient, alternated/linear, branched, comb, and star) enable us to achieve properties that are not accessible with homopolymers. It is thus highly desirable to identify catalysts that are able to efficiently promote copolymerization, in particular in the case of the challenging β-BL monomer. Much progress has been achieved in coordination–insertion ROP with metal complexes, and some systems have been shown to enable controlled ROP of β-BL,5 including in a few instances copolymerization with other cyclic monomers (ε-CL, LA, TMC, etc.).6
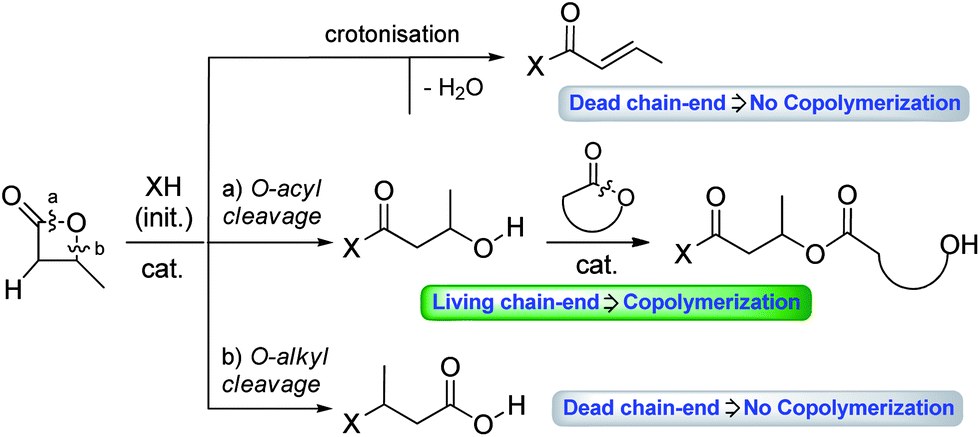 | ||
| Fig. 1 Selectivity issues in the ROP of β-BL and feasibility of copolymerization. | ||
During the past decade, organo-catalytic ROP has emerged as an attractive and versatile approach to the preparation of polyesters and polycarbonates of a well-defined structure under mild conditions.3 As for metal complexes, most studies have focused on the ROP of ε-CL, LA, TMC and variants thereof, but organo-catalysts have rarely been applied to the ROP of β-BL. Only N-heterocyclic carbenes NHC,7 and more recently, DBU, BEMP and TBD (Chart 1) have led to somewhat efficient polymerization,8,9 but none of these compounds yield selectively hydroxyl terminated PBL. When combined with tBuOH, 1,3,4-triphenyl triazol-5-ylidene allows for suppressing undesirable crotonisation reactions, but poor ring-opening selectivity results in the formation of PBL chains terminated by carboxylic acid moieties, hampering efficient copolymerization with other lactones. On the other hand, DBU, BEMP and TBD behave as polymerization initiators rather than catalysts. They are incorporated as chain-ends to the PBL, and under the relatively harsh conditions employed (60 °C in bulk), crotonisation reactions take place.10
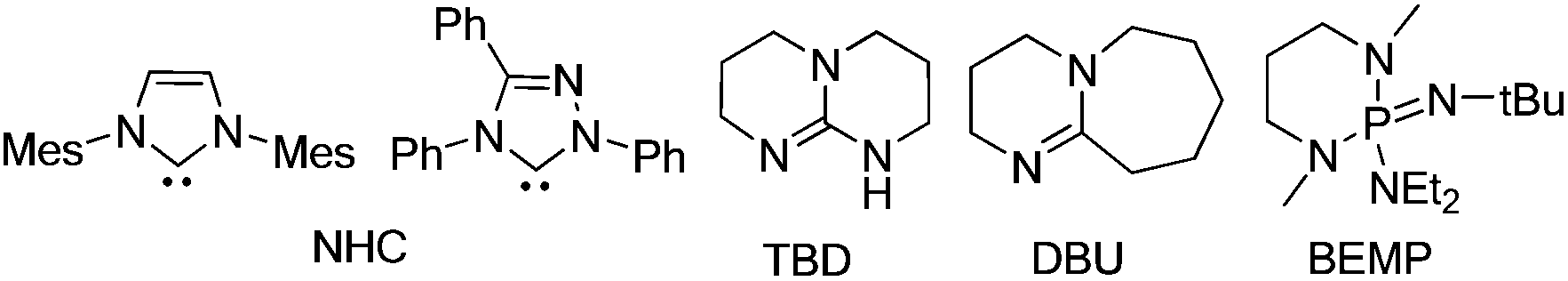 | ||
| Chart 1 Structure of the main organo-catalysts reported to date to promote efficient ROP of β-butyrolactone. | ||
At this stage, it is important to note that a broad variety of organo-catalysts has been developed for ROP and that different pathways are operating (basic/nucleophilic,11–14 acid15–20 or an association thereof21–23). Despite structural similarities, different monomers behave quite differently and in fact, each monomer/catalyst couple has its own specificities. Accordingly, basic/nucleophilic activation is particularly suited for LA and TMC but less efficient for ε-CL,13,14 whereas Brønsted acids perform better with ε-CL and TMC than with LA (only strong acids such as trifluoromethane sulfonic acid HOTf are active towards LA).17,18 In this respect, the diversity of available organo-catalysts is a noticeable advantage, and careful choice of the catalytic system has enabled achievement of very good results in the polymerization of functionalized monomers3d,24 as well as in the preparation of various types of copolymers (block, random, and gradient).3d,12c,17e,20c
In the past years, we and others have demonstrated the ability of sulfonic and phosphoric acids to promote homo- and co-polymerization of ε-CL, LA, TMC and related monomers.17,18,20 These acid catalysts are robust and versatile, and in some instances, they surpass the other organo-catalysts in terms of activity and/or selectivity. In this context, we decided to explore in detail the polymerization and copolymerization of β-BL promoted by sulfonic acids. A previous study by N. L. Pohl and co-workers suggests the feasibility of β-BL homopolymerization with HOTf,25 but the efficiency and controlled character of the process have not been evaluated and copolymerization with other monomers has not been investigated. In this study, we have considered both HOTf and methane sulfonic acid (MSA). HOTf is shown to be significantly more active and to enable good polymerization control (Mn up to 8200 g mol−1, Đ ≤ 1.25). β-BL selectively ring opens via O-acyl bond cleavage and crotonisation reactions do not occur to a significant extent. This leads to hydroxyl terminated PBL that are able to initiate efficiently ROP of ε-CL. A variety of block copolymers of well-defined structures (di-, tri- and penta-block, Mn up to 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) have thereby been prepared.
000 g mol−1) have thereby been prepared.
Experimental
Materials
All reactions were performed under an inert atmosphere of argon, using standard Schlenk techniques. Solvents were distilled prior to use over molecular sieves (C6D6 and CDCl3). β-Butyrolactone (β-BL) (98%, ALDRICH) and ε-caprolactone (ε-CL) (99.5%, ALDRICH) were purified by distillation over CaH2 and stored under argon. The sulfonic acids MSA (99%, ARKEMA) and HOTf (98%, ALDRICH) were used as received and stored in a glovebox. n-Pentanol (99%) and butan-1,4-diol (99%) were dried over sodium and distilled before use. Polyethylene glycol (FLUKA, 1500 g mol−1) was azeotropically distilled from toluene and dried under vacuum. Polybutadiene (Krazol® LBH-P 3000, kindly supplied by SARTOMER, 3000 g mol−1) was dried over phosphorus oxide before use.Characterization of the polymers
000000 g mol−1) and two STYRAGEL columns (HR1, 100–5000 g mol−1 and HR 4E, 50–100000 g mol−1) were used. Calibrations were performed using polystyrene standards (400–100000 g mol−1).
:1:1 respectively. The mixed solution was hand-spotted on a stainless steel MALDI target and left to dry. The spectrum was recorded in the reflectron mode. Baseline corrections and data analyses were performed using MassLynx version 4.1 and Polymerix Software, Sierra Analytics, Version 2.0.0.
Methods
Typical homo-polymerization procedure
β-Butyrolactone (β-BL) (200 μL, 2.5 mmol, 40 equiv.) was dissolved in deuterated benzene (0.6 mL, [β-BL]0 = 4 mol L−1). The initiator, n-pentanol (6.6 μL, 0.06 mmol, 1 equiv.), and trifluoromethane sulfonic acid (HOTf) (5.4 μL, 0.06 mmol, 1 equiv.) were successively added. The reaction mixture was stirred at 30 °C for 2 h until the complete consumption of β-BL, as determined by 1H NMR spectroscopy. An excess of diisopropylethylamine was added to neutralize the catalyst, and the solvent was evaporated under vacuum. The polymer was then dissolved in a minimum of dichloromethane and precipitated in cold methanol, then filtered and dried under vacuum. Conversion: >96%, yield: 90%. 1H NMR (C6D6, 300 MHz): 5.32 (m, 1H × 42, nOCH), 4.31 (m, 1H, CHOH), 3.97 (m, C4H9CH2O), 2.44 (m, 1H × 42, nCOCH2), 2.26 (m, 1H × 42, nCOCH2), 1.12 (m, 3H × 42, CH3CHO), 0.79 (t, 3H, J = 6.3 Hz, CH3); SEC (THF): Mn ∼ 3813 g mol−1, Đ ∼ 1.12.Typical block co-polymerization procedure
β-BL (200 μL, 2.5 mmol, 20 equiv.) was dissolved in C6D6 (0.6 mL, [β-BL]0 = 4 mol L−1). The initiator, n-pentanol (14 μL, 0.13 mmol, 1 equiv.), and HOTf (11 μL, 0.13 mmol, 1 equiv.) were successively added. The reaction mixture was stirred at 30 °C for 30 min. After the complete consumption of β-butyrolactone, as monitored by 1H NMR spectroscopy, ε-CL (260 μL, 2.5 mmol, 20 equiv.) was added. The reaction mixture was stirred at 30 °C for 30 min (until the complete consumption of ε-CL). An excess of diisopropylethylamine was added to neutralize the catalyst, and the solvent was evaporated under vacuum. The polymer was dissolved in a minimum of dichloromethane, precipitated in cold methanol, then filtered and dried under vacuum. Conversion: >96%, yield: 90%. 1H NMR (CDCl3, 300 MHz): 5.23 (m, 1H × 18, nOCH), 4.05 (t, 2H × 47, J = 6.6 Hz, nOCH2), 3.63 (t, 2H, J = 6.3 Hz, HOCH2), 2.58 (m, 1H × 18, nCOCH2), 2.48 (m, 1H × 18, nCOCH2), 2.29 (t, 2H × 47, J = 7.8 Hz, nCOCH2), 1.63 (m, 4H × 47 + 4H, CH2), 1.37 (m, 2H × 47 + 2H, CH2), 1.27 (m, 3H × 18, CH3CHO), 0.89 (t, 3H, J = 7.8 Hz, CH3); SEC (THF): Mn ∼ 6670 g mol−1, Đ ∼ 1.21.Results and discussion
ROP of β-BL promoted by sulfonic acids: MSA versus HOTf
Previous studies on LA, ε-CL and TMC have shown that the activity of sulfonic acids does not simply correlate with acidity,17a–c and we were thus keen to evaluate both MSA and HOTf towards β-BL. The reactions were carried out with rac-β-BL at 30 °C in C6D6 solution, using n-pentanol as an initiator. The monomer conversion was directly monitored by 1H NMR spectroscopy. MSA does promote the ROP of β-BL. High conversions (>96%) are achieved in reasonable reaction times (1 to 26 hours) provided 3 equiv. of catalyst per initiator and an initial monomer concentration of 8 mol L−1 are used (runs 1–4, Table 1). Under these reaction conditions, n-pentanol efficiently initiates the polymerization, ring-opening proceeds selectively by O-acyl bond cleavage and crotonisation reactions do not occur to a significant extent (see Fig. S1† for the 1H NMR spectrum of a representative PBL sample). However, MS MALDI-TOF analyses reveal the presence of several populations in addition to that of the n-pentyl ester end-capped PBL (Fig. S2†) and the Mn values of the obtained polymers do not increase linearly with the initial monomer to initiator ratio, even for rather low M/I values (Fig. 2, diamonds).|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Cat. | Run | [M]0/[I]0/[cat] | [M]0 | Timeb (h) | M n (g mol−1) | M n (g mol−1) | M n (th) (g mol−1) | Đ |
| a Polymerization of β-butyrolactone carried out at 30 °C in C6D6 solution. b Polymerization time necessary to achieve monomer conversion >96% according to 1H NMR spectroscopy. c Determined by 1H NMR spectroscopy. d Obtained from size exclusion chromatography analysis in tetrahydrofuran using polystyrene standards. e Calculated from the molar mass of β-BL (86 g mol−1) × the initial monomer/initiator ratio plus the molar mass of the initiator (88 g mol−1). f Monomer conversion = 93%. | ||||||||
| MSA | 1 | 20/1/3 | 8 | 1 | 1765 | 2050 | 1800 | 1.14 |
| 2 | 40/1/3 | 8 | 1.5 | 3790 | 3700 | 3528 | 1.16 | |
| 3 | 60/1/3 | 8 | 4.5 | 4730 | 5248 | 5248 | 1.20 | |
| 4 | 80/1/3 | 8 | 26 | 5070 | 5090 | 6960 | 1.28 | |
| HOTf | 5 | 20/1/1 | 8 | 0.25 | 1725 | 2260 | 1808 | 1.26 |
| 6 | 40/1/1 | 8 | 0.75 | 3790 | 4990 | 3528 | 1.25 | |
| 7 | 60/1/1 | 8 | 3.5 | 4900 | 6460 | 5248 | 1.31 | |
| 8 | 20/1/1 | 4 | 0.5 | 1810 | 2070 | 1800 | 1.13 | |
| 9 | 40/1/1 | 4 | 2 | 3700 | 3813 | 3528 | 1.12 | |
| 10 | 60/1/1 | 4 | 4.5 | 5500 | 5150 | 5248 | 1.20 | |
| 11 | 80/1/1 | 4 | 9 | 7055 | 7280 | 6960 | 1.21 | |
| 12 | 100/1/1 | 4 | 12f | 8090 | 8120 | 8690 | 1.25 | |
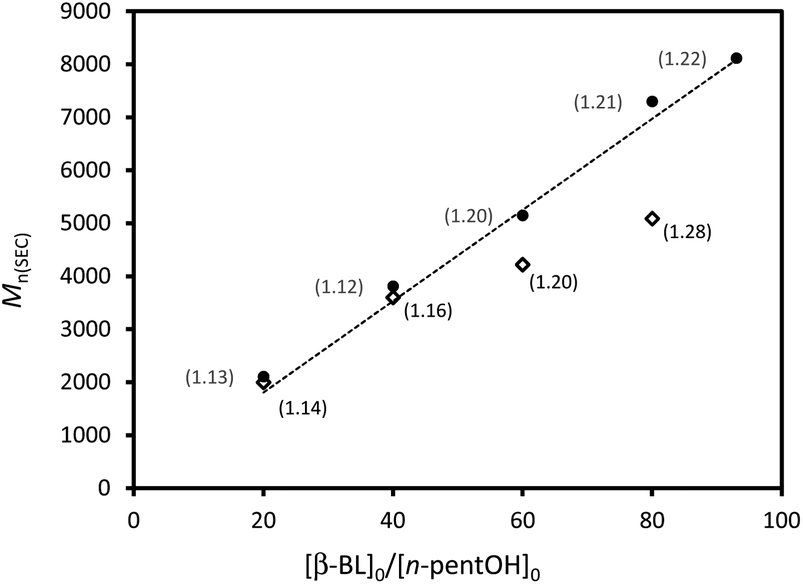 | ||
| Fig. 2 Plot of Mn(SEC) (estimated by SEC in THF with polystyrene standards) vs. β-BL/initiator ratio ([β-BL]0/[n-PentOH]0) using HOTf (◇) or MSA (●) as a catalyst. The dotted line represents the theoretical values of Mn. Đ in brackets. | ||
Under similar conditions, HOTf proved significantly more active than MSA and high monomer conversions were achieved much faster even using only one equiv. of catalyst per initiator. 20 equiv. of β-BL are converted in only 15 min with HOTf whereas 60 min are required with MSA (runs 1 and 5, Table 1). At [M]0 = 8 mol L−1, the Mn values of the polymers prepared with HOTf exceed substantially those expected from the initial monomer to initiator ratio and relatively large molecular weight distributions tend to be observed (Đ > 1.25). Better results were obtained at lower monomer concentration ([β-BL]0 = 4 mol L−1) and PBLs of well-controlled molecular weight (Mn up to 8200 g mol−1) and narrow distributions (Đ between 1.12 and 1.25) were prepared (runs 8–12, Table 1).
As illustrated in the plot of Mnvs. [M]0/[I]0 (Fig. 2, circles), the Mn values of the obtained polymers increase linearly with the monomer to initiator ratio and match well with the theoretical ones. HOTf also allows very good control over the chain-ends. The alcohol used as an initiator is quantitatively incorporated into the polymer chains as an ester chain-end. The two H atoms of the CH2OCO group are diastereotopic and appear in the 1H NMR spectrum as two overlapping triplets at δ ∼ 3.97 ppm (Fig. 3a, signal f). The spectrum also displays a broad multiplet signal at δ 4.37 ppm corresponding to the terminal CHOH moiety (signal a). The relative integration of these two signals (2/1) supports efficient initiation (all polymer chains have been initiated by n-pentanol) and selective O-acyl bond cleavage of the monomer. Crotonisation reactions occur to a very small extent if any at all (less than 0.2%, according to 1H NMR spectroscopy).26 Consistently, MALDI-TOF MS analysis (Fig. 3b) revealed the presence of a very major population of PBL with molar mass M = n × 86.09(Mβ-BL) + 88(MPentOH) + 23(MNa+) g mol−1, corresponding to the linear polymer chains initiated by n-pentanol and terminated by an hydroxyl group. No signals attributable to crotonisation reactions were detected, but at low m/z values (<800 g mol−1), a minor PBL population with molar mass M = n × 86.09(Mβ-BL) + 23(MNa+) g mol−1 was observed, indicating the formation of a small amount of macrocycles.
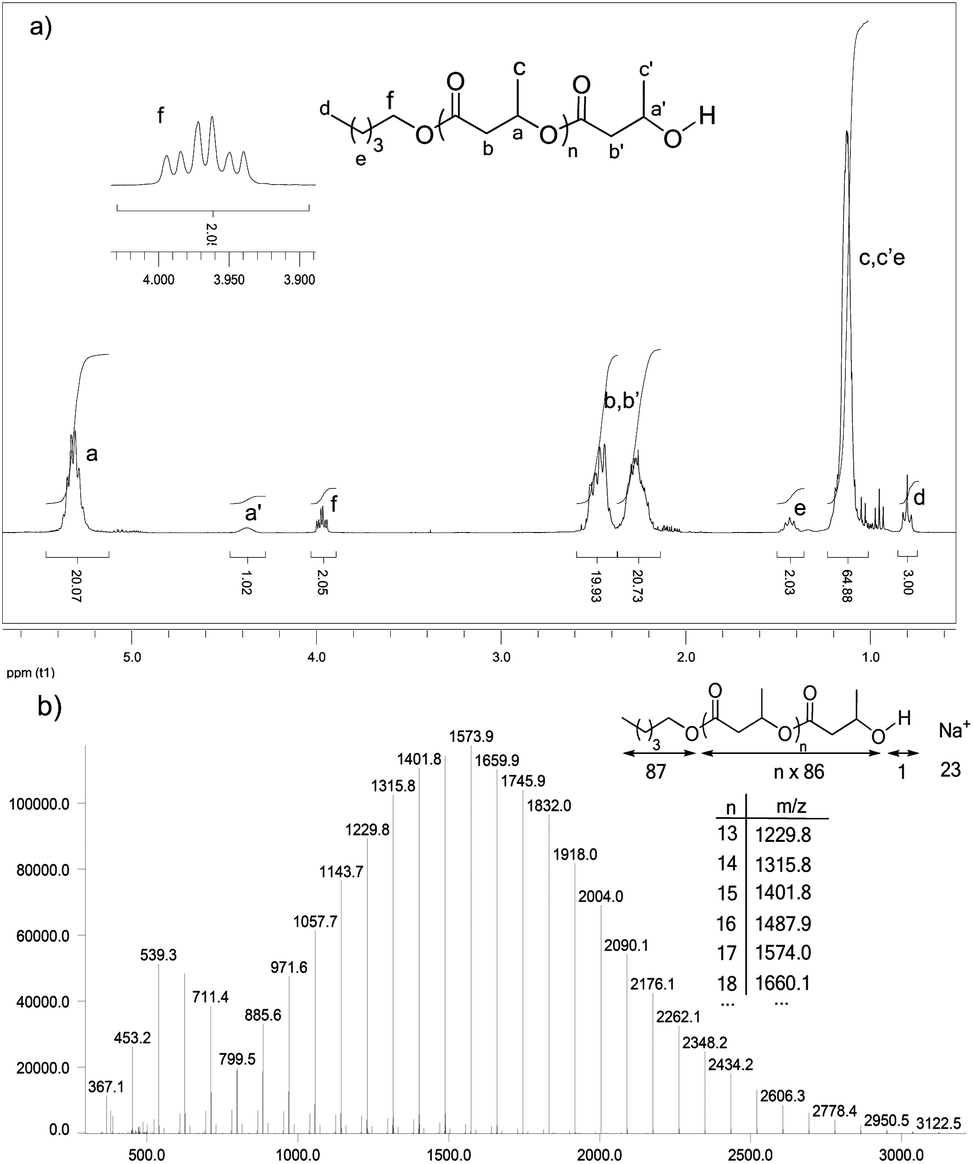 | ||
| Fig. 3 (a) 1H NMR spectrum (C6D6, 300 MHz) and (b) MALDI-TOF MS (region m/z 400 to 3200 g mol−1) of a PBL sample prepared with HOTf ([β-BL]0/[n-PentOH]0/[HOTf] = 20/1/1, toluene, 30 °C, [β-BL]0 = 4 mol L−1). | ||
The selectivity of the ring-opening of β-BL in favor of O-acyl bond cleavage and the absence of crotonisation reactions result in well-controlled propagation of the polymerization via hydroxyl chain ends. These features are quite unique for organo-catalyzed ROP of β-BL and make HOTf a good candidate to achieve its copolymerization with other lactones. In the following studies, we chose ε-CL as a case example and investigated the preparation of block copolymers by sequential monomer addition, using n-pentanol and butan-1,4-diol as well as hydroxyl-terminated polymers as initiators.
β-BL/ε-CL copolymerization catalysed by HOTf
The ability of HOTf to promote the formation of β-BL/ε-CL block copolymers was first evaluated with n-pentanol as an initiator and both orders of addition were considered. In line with previous studies,17b ε-CL was readily polymerized with HOTf and the ensuing n-pentyl ester end-capped PCL was found to efficiently initiate the polymerization of β-BL (representative 1H NMR spectra are depicted in Fig. S4†). The formation of β-BL/ε-CL block copolymers starting from β-BL is a priori more challenging (because of the side reactions associated with this monomer). β-BL was first polymerised under the reaction conditions described above ([M]0/[I]0/[cat] = 20/1/1, C6D6 solution, 30 °C, [β-BL]0 = 4 mol L−1). When β-BL was entirely consumed (15 min as deduced from 1H NMR spectroscopy monitoring), ε-CL (20 equiv.) was added. Complete conversion of ε-CL was achieved in 30 min. The catalyst was then quenched by addition of diisopropylethylamine (DIEA) and the formation of block copolymers was ascertained by NMR spectroscopy and SEC (Scheme 1a).27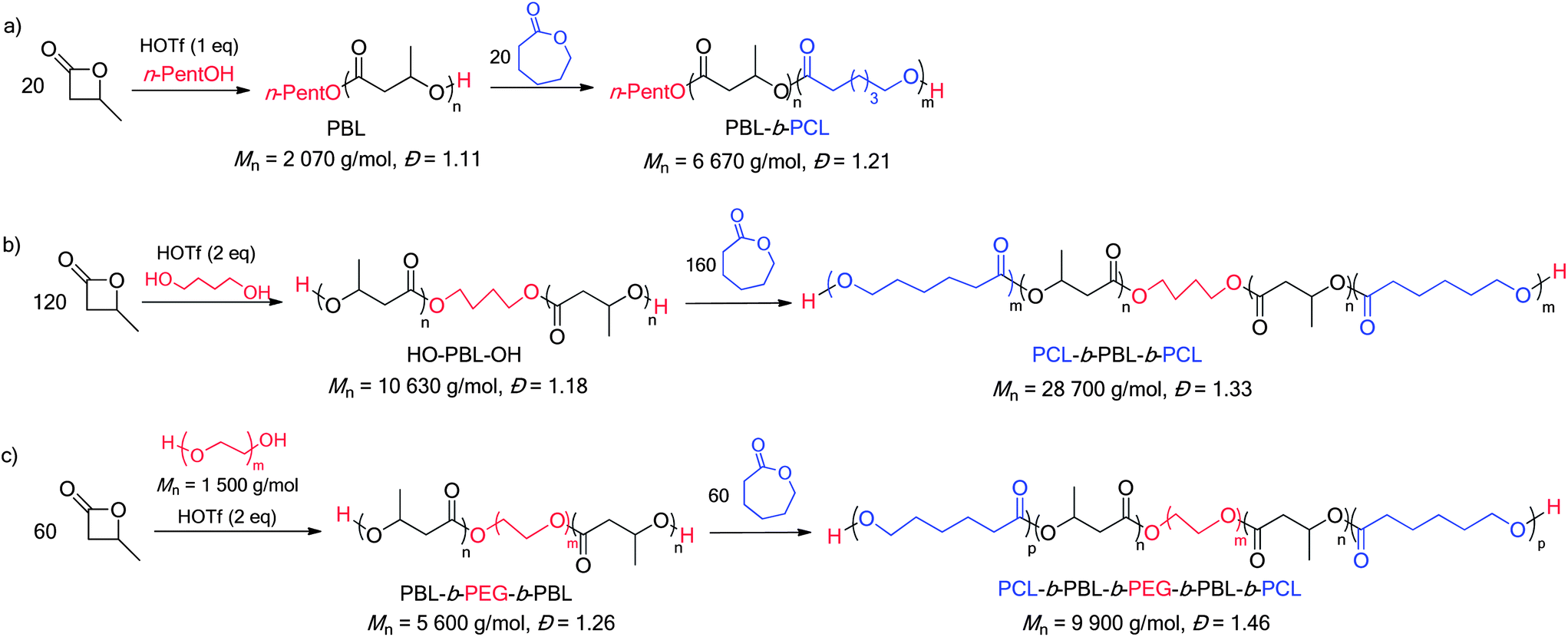 | ||
| Scheme 1 Preparation of PBL-b-PCL diblock (a), PCL-b-PBL-b-PCL triblock (b) and PCL-b-PBL-b-PEG-b-PBL-b-PCL pentablock copolymers. | ||
At the end of β-BL polymerization, the 1H NMR spectrum displays, as previously mentioned, a broad multiplet at δ 4.37 ppm for the terminal CHOH (Fig. 3a). This signal disappears upon addition and consumption of ε-CL, while a triplet, integrating for 2 protons, appears at δ 3.62 ppm for the CH2OH chain end of the formed PCL block (Fig. 4a).2813C NMR spectroscopy (Fig. 4b) further supports the formation of a block copolymer and the absence of redistribution reactions. In the carbonyl region, we observe only one signal at δ 173.3 ppm for the PCL block and two broad signals at δ 169.0 ppm for the atactic PBL block,29 but no signals are detected for β-BL/ε-CL heterodyads.30 SEC analysis (Fig. 4c) corroborates the formation of a block copolymer. The Mn value increases upon monomer feeding from 2070 to 6670 g mol−1, while narrow molar mass distributions are retained (Đ ∼ 1.21). These results demonstrate that the selectivity of HOTf for O-acyl bond cleavage of β-BL enables the preparation of block copolymers. Good control over the process is retained for PBL-b-PCL copolymers of higher molar masses. With a [β-BL]0/[ε-CL]0/[n-PentOH]0 ratio of 60/60/1, a block copolymer with Mn over 15000 g mol−1 (Đ = 1.30) was readily prepared (Table S1†).
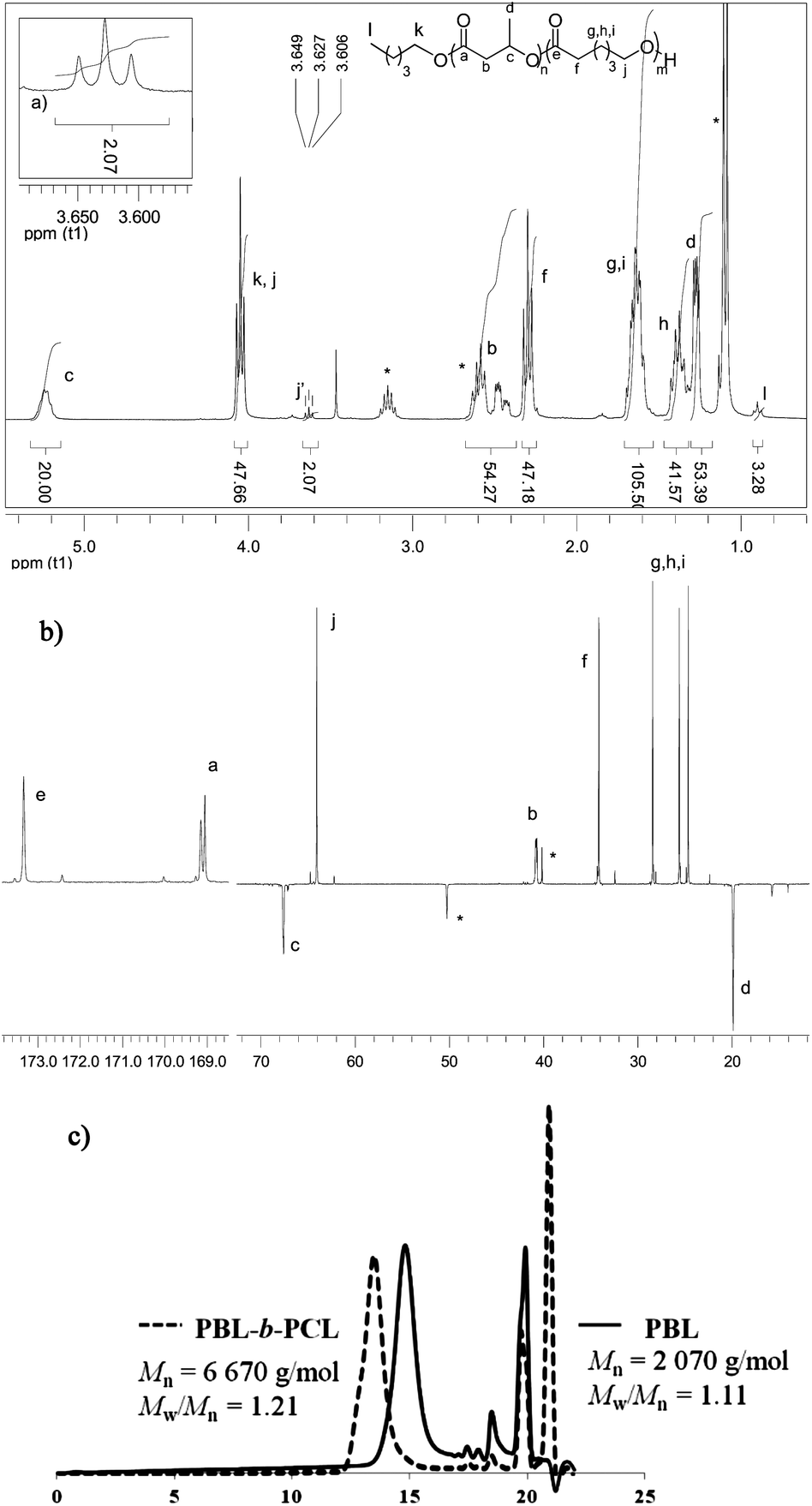 | ||
| Fig. 4 Analyses of a PBL20-b-PCL20 copolymer prepared by successive addition of β-BL and ε-CL ([β-BL]0/[ε-CL]0/[n-PentOH]0/[HOTf] = 20/20/1/1, toluene, 30 °C): (a) 1H NMR spectrum (CDCl3, 300 MHz). (b) 13C{1H} Jmod NMR spectrum (CDCl3, 75 MHz) (downward peaks refer to CH and CH3 groups). * refers to the DIEA·HOTf salt. (c) SEC traces of the first PBL20 block and final PBL20-b-PCL20 copolymer. Peaks at t > 17.5 min refer to low molar mass compounds (short oligomers, catalyst, monomer and solvent residues). | ||
We then became interested in using dihydroxylated initiators, to further extend the scope of such block copolymerizations and target block copolymers of higher orders (tri- and penta-block). To this end, a telechelic HO-PBL-OH of Mn = 10630 g mol−1 and Đ = 1.18 was initially prepared by ROP of β-BL (120 equiv.) using butan-1,4-diol as an initiator (Scheme 1b). Subsequent addition of ε-CL (160 equiv.) yielded a triblock PCL-b-PBL-b-PCL copolymer (Mn 28700 g mol−1 and Đ = 1.33) (see Fig S5 and S6† for the corresponding 1H NMR spectra and SEC traces). The synthetic methodology was then extrapolated to the preparation of polyester–polyether triblock copolymers. Dihydroxylated polyethylene glycol of Mn = 1500 and 10000 g mol−1 was used as a macroinitiator. Under these conditions, the rate of the β-BL polymerization decreases slightly, but very good control is retained and triblock copolymers of Mn = 5600 and 16730 g mol−1 (Đ = 1.26 and 1.23, respectively) were thereby obtained. The copolymerization process can be extended further and enables the preparation of pentablock copolymers. Accordingly, addition of ε-CL (60 equiv.) to the telechelic triblock PBL-b-PEG-b-PBL copolymer of Mn = 5600 g mol−1 (Scheme 1c) gives a pentablock PCL-b-PBL-b-PEG-b-PBL-b-PCL copolymer of Mn = 9900 g mol−1 and Đ = 1.46 (Fig. S7†). The 1H NMR chain end signals and the 13C NMR carbonyl signals (Fig. S8 and S9†) are diagnostic for a β-BL/ε-CL block copolymer and no redistribution of the monomer units along the polymer chain is detected. To illustrate the generality of the system, the nature of the macroinitiator was also varied and a pentablock copolymer was prepared starting from Krasol® LBH-P-3000, a hydrophobic hydroxyl terminated polybutadiene PBD (Mn = 5 570 g mol−1, Đ = 1.11).31 The copolymer was prepared by successive ROP of β-BL (60 equiv.) and ε-CL (60 equiv.) (Scheme S1†). It is noteworthy that HOTf shows good compatibility with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds of the polybutadiene block. The pentablock structure is consistently assessed by 1H and 13C NMR (Fig. S10 and S11†) and the extension of the polymer chain is substantiated by SEC analysis: from Mn = 9870 g mol−1, Đ = 1.21 for the triblock PBL-b-PBD-b-PBL copolymer, to Mn = 16790 g mol−1, Đ = 1.40 for the pentablock PCL-b-PBL-b-PBD-b-PBL-b-PCL copolymer (Fig. S12†).
C double bonds of the polybutadiene block. The pentablock structure is consistently assessed by 1H and 13C NMR (Fig. S10 and S11†) and the extension of the polymer chain is substantiated by SEC analysis: from Mn = 9870 g mol−1, Đ = 1.21 for the triblock PBL-b-PBD-b-PBL copolymer, to Mn = 16790 g mol−1, Đ = 1.40 for the pentablock PCL-b-PBL-b-PBD-b-PBL-b-PCL copolymer (Fig. S12†).
Conclusions
Trifluoromethane sulfonic acid was found to promote controlled ROP of β-butyrolactone, a rather challenging monomer, under mild conditions. The ring-opening of β-BL proceeds selectively via O-acyl bond cleavage and undesirable crotonisation reactions do not occur. These features make HOTf a quite unique organo-catalyst towards β-BL, and the propagating hydroxyl chain-end opens the way to efficient copolymerization. Accordingly, successive monomer addition was shown to enable the preparation of a variety of well-defined PBL/PCL block copolymers, including pentablock copolymers using dihydroxylated PEG and PBD macroinitiators.These results further substantiate the interest of simple sulfonic acids in ROP. HOTf displays unique features and in some instances surpasses the other organo-catalysts. The variety of organo-catalysts available for ROP and the diversity of the associated activation modes represent major advantages and variation/optimisation of the monomer/catalyst couples enable us to prepare efficiently an ever wider range of polymer structures.
Notes and references
- For selected reviews on metal-catalyzed ROP, see: (a) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215 RSC; (b) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef CAS PubMed; (c) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, in The Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH, Weinheim, 2009, p. 255 Search PubMed; (d) C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832 RSC; (e) A. K. Sutar, T. Maharana, S. Dutta, C. T. Chen and C. C. Lin, Chem. Soc. Rev., 2010, 39, 1724 RSC; (f) P. J. Dijkstra, H. Z. Du and J. Feijen, Polym. Chem., 2011, 2, 520 RSC; (g) S. Dagorne, M. Normand, E. Kirillov and J. F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869 CrossRef CAS PubMed.
- For selected reviews on enzymatic-catalyzed ROP, see: (a) S. Kobayashi, Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793 CrossRef CAS PubMed; (b) R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097 CrossRef CAS PubMed; (c) I. K. Varma, A.-C. Albertsson, R. Rajkhowa and R. K. Srivastava, Prog. Polym. Sci., 2005, 30, 949 CrossRef CAS PubMed; (d) S. Kobayashi, Macromol. Symp., 2006, 240, 178 CrossRef CAS; (e) S. Matsumura, Adv. Polym. Sci., 2006, 194, 95 CrossRef CAS; (f) A.-C. Albertsson and R. K. Srivastava, Adv. Drug Delivery Rev., 2008, 60, 1077 CrossRef CAS PubMed; (g) A. Heise, C. J. Duxbury and A. R. A. Palmans, in The Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH, Weinheim, 2009, p. 379 Search PubMed; (h) S. Kobayashi, Macromol. Rapid Commun., 2009, 30, 237 CrossRef CAS PubMed.
- For selected reviews on organo-catalyzed ROP, see: (a) N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813 CrossRef CAS PubMed; (b) D. Bourissou, S. Moebs-Sanchez and B. Martin Vaca, C. R. Chim., 2007, 10, 775 CrossRef CAS PubMed; (c) A. P. Dove, in The Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH, Weinheim, 2009, p. 357 Search PubMed; (d) M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093 CrossRef CAS; (e) A. P. Dove, ACS Macro Lett., 2012, 1, 1409 CrossRef CAS.
- O. Coulembier and P. Dubois, in The Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH, Weinheim, 2009, p. 227 Search PubMed.
- For selected references, see: (a) S. M. Guillaume, L. Annunziata, I. del Rosal, C. Iftner, L. Maron, P. W. Roesky and M. Schmid, Polym. Chem., 2013, 4, 3077 RSC; (b) J.-F. Carpentier, Macromol. Rapid Commun., 2010, 31, 1696 CrossRef CAS PubMed; (c) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165 RSC.
- For selected contributions, see: (a) Y. Hori, Y. Gonda, Y. Takahashi and T. Hagiwara, Macromolecules, 1996, 29, 804 CrossRef CAS; (b) Y. Hori, H. Hongo and T. Hagiwara, Macromolecules, 1999, 32, 3537 CrossRef CAS; (c) S. Li, M. Pignol, F. Gasc and M. Vert, Macromolecules, 2004, 37, 9798 CrossRef CAS; (d) Y.-C. Liu, C.-H. Lin, B.-T. Ko and R.-M. Ho, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5339 CrossRef CAS; (e) B. J. Jeffery, E. L. Whitelaw, D. Garcia-Vivo, J. A. Stewart, M. F. Mahon, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 12328 RSC; (f) D. C. Aluthge, C. Xu, N. Othman, N. Noroozi, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2013, 46, 3965 CrossRef CAS.
- O. Coulembier, X. Delva, J. L. Hedrick, R. M. Waymouth and P. Dubois, Macromolecules, 2007, 40, 8560 CrossRef CAS.
- (a) C. G. Jaffredo, J. F. Carpentier and S. M. Guillaume, Macromol. Rapid Commun., 2012, 1938 CrossRef CAS PubMed; (b) C. G. Jaffredo, J. F. Carpentier and S. M. Guillaume, Polym. Chem., 2013, 4, 3837 RSC.
- ROP of β-BL initiated by NHC–CO2 adducts has been reported recently, see: E. Brulé, V. Guérineau, P. Vermaut, F. Prima, J. Balogh, L. Maron, A. M. Z. Slawin, S. P. Nolan and C. M. Thomas, Polym. Chem., 2013, 4, 2414 RSC.
- According to ref. 4, DBU and TBD are inactive towards β-BL in solution at room temperature.
- F. Nederberg, E. F. Connor, M. Moller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712 CrossRef CAS.
- (a) A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, E. C. Hagberg, G. W. Nyce, R. M. Waymouth and J. L. Hedrick, Polymer, 2006, 47, 4018 CrossRef CAS PubMed; (b) A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, H. Li, E. C. Hagberg, R. M. Waymouth and J. L. Hedrick, in N–Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2006, p. 275 Search PubMed; (c) O. Coulembier, M. K. Kiesewetter, A. Mason, P. Dubois, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2007, 46, 4719 CrossRef CAS PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574 Search PubMed.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Macromolecules, 2007, 40, 4154 Search PubMed.
- For early work with acids in the absence of initiator, see: (a) H. R. Kricheldorf and R. Dunsing, Makromol. Chem., 1986, 187, 1611 Search PubMed; (b) H. R. Kricheldorf and I. Kreiser, Makromol. Chem., 1987, 188, 1861 Search PubMed.
- (a) Y. Shibasaki, H. Sanada, M. Yokoi, F. Sanda and T. Endo, Macromolecules, 2000, 33, 4316 Search PubMed; (b) F. Sanda, H. Sanada, Y. Shibasaki and T. Endo, Macromolecules, 2002, 33, 680 Search PubMed; (c) X. Lou, C. Detrembleur and R. Jérôme, Macromolecules, 2002, 35, 1190 Search PubMed.
- (a) D. Bourissou, B. Martin-Vaca, A. Dumitrescu, M. Graullier and F. Lacombe, Macromolecules, 2005, 38, 9993 Search PubMed; (b) S. Gazeau-Bureau, D. Delcroix, B. Martin-Vaca, D. Bourissou, C. Navarro and S. Magnet, Macromolecules, 2008, 41, 3782 Search PubMed; (c) D. Delcroix, B. Martin-Vaca, D. Bourissou and C. Navarro, Macromolecules, 2010, 43, 8828 Search PubMed; (d) N. Susperregui, D. Delcroix, B. Martin-Vaca, D. Bourissou and L. Maron, J. Org. Chem., 2010, 75, 6581 Search PubMed; (e) A. Couffin, Delcroix, C. Navarro, B. Martin-Vaca and D. Bourissou, Macromolecules, 2013, 46, 4354 Search PubMed.
- (a) M. Basko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 7071 Search PubMed; (b) M. Basko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3090 Search PubMed; (c) M. Basko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7919 Search PubMed; (d) M. Basko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2650 Search PubMed.
- (a) R. Kakuchi, Y. Tsuji, K. Chiba, K. Fuchise, R. Ryosuke, T. Satoh and T. Kakuchi, Macromolecules, 2010, 43, 7090 Search PubMed; (b) M. Oshimura, T. Tang and A. Takasu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1210 Search PubMed; (c) K. Makiguchi, S. Kikuchi, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2455 Search PubMed.
- (a) H. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999 Search PubMed; (b) D. Delcroix, A. Couffin, N. Susperregui, C. Navarro, L. Maron, B. Martin-Vaca and D. Bourissou, Polym. Chem., 2011, 2, 2249 Search PubMed; (c) K. Makiguchi, Y. Ogasawara, S. Kikuchi, T. Satoh and T. Kakuchi, Macromolecules, 2013, 46, 1772 Search PubMed; (d) S. Kan, Y. Jin, X. He, J. Chen, H. Wu, P. Ouyang, K. Guo and Z. Li, Polym. Chem., 2013 10.1039/c3py00667k.
- (a) R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556 Search PubMed; (b) A. Chuma, H. W. Horn, W. C. Swope, R. C. Pratt, L. Zhang, B. G. G. Lohmeijer, C. G. Wade, R. M. Waymouth, J. L. Hedrick and J. E. Rice, J. Am. Chem. Soc., 2008, 130, 6749 Search PubMed.
- (a) A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798 Search PubMed; (b) R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. B. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863 Search PubMed.
- For other hydrogen donor derivatives combined with a tertiary amine, see: (a) S. Koeller, J. Kadota, A. Deffieux, F. Peruch, S. Massip, J. M. Leger, J. P. Desvergne and B. Bibal, J. Am. Chem. Soc., 2009, 131, 15088 Search PubMed; (b) S. Koeller, J. Kadota, F. Peruch, A. Deffieux, N. Pinaud, I. Pianet, S. Massip, J. M. Leger, J. P. Desvergne and B. Bibal, Chem.–Eur. J., 2010, 16, 4196 Search PubMed; (c) A. Alba, A. Schopp, A. P. De Sousa Delgado, R. Cherif-Cheikh, B. Martin-Vaca and D. Bourissou, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 959 Search PubMed; (d) J. M. Becker, S. Tempelaar, M. J. Stanford, R. J. Pounder, J. A. Covington and A. P. Dove, Chem.–Eur. J., 2010, 16, 6099 Search PubMed.
- For selected contributions, see: (a) F. Jing and M. A. Hillmyer, J. Am. Chem. Soc., 2008, 130, 13826 Search PubMed; (b) O. T. du Boullay, N. Saffon, J.-P. Diehl, B. Martin-Vaca and D. Bourissou, Biomacromolecules, 2010, 11, 1921 Search PubMed; (c) A. Dove, Biomacromolecules, 2010, 11, 1930 Search PubMed; (d) J. A. Castillo, D. E. Borchmann, A. Y. Cheng, Y. Wang, C. Hu, A. J. García and M. Weck, Macromolecules, 2012, 45, 62 Search PubMed; (e) S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312 Search PubMed; (f) D. J. Coady, H. W. Horn, G. O. Jones, H. Sardon, A. C. Engler, R. M. Waymouth, J. E. Rice, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2013, 2, 306 Search PubMed; (g) A. C. Engler, J. M. W. Chan, K. Fukushima, D. J. Coady, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2013, 2, 332 Search PubMed.
- F. A. Jaipuri, B. D. Bower and N. L. Pohl, Tetrahedron: Asymmetry, 2003, 14, 3249 Search PubMed.
- Similar small amounts of crotonic acid are often detected within the β-BL monomer despite careful purification by distillation over CaH2 (Fig. S3†).
- Block copolymers of relatively low molecular weight (20 equiv. of each monomer with respect to the initiator) facilitate NMR characterization and enable us to follow precisely the advent and relative integration of the chain-ends.
- The signal corresponding to the CHO-CO-PBL-PCL linkages around δ 4.05 ppm cannot be observed due to overlapping with those of PCL CH2OCO ester groups.
- (a) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239 Search PubMed; (b) For data on PCL-b-PBL copolymers, see: H. Abe, Y. Doi and Y. Kumagai, Macromolecules, 1994, 27, 6012 Search PubMed.
- (a) H. Abe, Y. Doi, H. Aoki, T. Akehata, Y. Hori and A. Yamaguchi, Macromolecules, 1995, 28, 7630 Search PubMed; (b) G. Impallomeni, M. Giuffrida, T. Barbuzzi, G. Musumarra and A. Ballistreri, Biomacromolecules, 2002, 3, 835 Search PubMed.
- PCL-b-PBD-b-PCL copolymers have been prepared by ROP of ε-CL with Sn(Oct)2 as catalyst and Krazol as macroinitiator: F. Meng, S. Zheng, W. Zhang, H. Li and Q. Liang, Macromolecules, 2006, 39, 711 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, SEC and MALDI-TOF MS spectra of PBL and PBL-b-PCL. See DOI: 10.1039/c3py00935a |
| This journal is © The Royal Society of Chemistry 2014 |