The synthesis and characterization of supramolecular elastomers based on linear carboxyl-terminated polydimethylsiloxane oligomers†
Lin
Yang
a,
Yaling
Lin
*b,
Lianshi
Wang
a and
Anqiang
Zhang
*a
aCollege of Material Science and Engineering, South China University of Technology, 381 Wushan Rd, Guangzhou 510641, Guangdong, China. E-mail: aqzhang@scut.edu.cn; Fax: +86-20-87112466; Tel: +86-20-87112466
bCollege of Resource and Environment, South China Agriculture University, 483 Wushan Rd, Guangzhou 510642, Guangdong, China. E-mail: linyaling@scau.edu.cn
First published on 7th August 2013
Abstract
Supramolecular elastomers obtained through a two-step reaction of linear carboxyl-terminated polydimethylsiloxane oligomers (PDMS–COOH2) with diethylenetriamine (DETA) and urea show reasonable hysteresis and acceptable self-healing properties. The results of temperature-dependent infrared analysis suggest the existence of hydrogen bonding interactions with good thermal reversibility in the matrix. Differential scanning calorimetry (DSC) and X-ray diffraction (XRD) analyses demonstrate that the supramolecular network structure is totally amorphous at room temperature. The rheological, mechanical and self-healing properties are closely related to the PDMS chain length, whereas the stability seems to be independent of the PDMS chain length. The viscoelastic properties of these materials are believed to be the result of entangled chains in the amorphous matrix. Only the hydrogen bonds formed by 1,1-dialkylurea groups and imidazolidone derivatives serve as effective crosslinks for contributing to the stability of the supramolecular network.
1 Introduction
The concept of using multiple hydrogen bonds to prepare supramolecular elastomers is similar to that used to prepare traditional thermoplastic elastomers (TPEs) and can be traced back to the late 1980s.1 The introduction of hydrogen bonding groups can significantly increase the viscoelastic properties of the related polymer chains. With respect to the functionalities of urazoylphenyl benzoic acid groups,2 urea/urethane-modified ureidopyridinone units (–U–UPy–/–T–UPy–)3,4 and bis-ureas,5 the presence of strong hydrogen bonding interactions between the bonded groups can result in microphase aggregates or crystalline domains in the polymer matrix, which can act as highly robust crosslinks that are comparable to the covalent bonds in conventional rubbers or the glassy and semicrystalline domains in TPEs.Recently, another hydrogen-bonded supramolecular elastomer based on the model of traditional TPEs was obtained by Chen and coworkers.6 In this system, the covalently connected soft segments were replaced by a hydrogen-bonded supramolecular soft matrix. In contrast to the network mentioned above, this new system combines the mechanical stiffness with dynamic healing properties.
Instead of functionalizing linear or star-shaped polymers, the use of multifunctional molecules with an average functionality higher than 2 also forms a supramolecular network.7 However, obtaining supramolecular elastic materials from this route presents a significant challenge because the interactions between the molecules induce crystallization. It was reported that unique supramolecular elastomers capable of self-healing were obtained through a two-step reaction of a dimer fatty acid with diethylenetriamine (DETA) and urea.8–10 According to the analysis of Montarnal and coworkers, the high functionality (>2) of the fatty acid and the wide distribution of the shapes and lengths of the modified oligomers, equipped with a variety of strongly associating groups, are two critical requirements for the formation of such supramolecular networks while effectively avoiding crystallization. By using a mixture of single, di-, and tri-carboxylic acid-terminated polydimethylsiloxane (PDMS–COOHx, x = 1–3), we have successfully obtained a similar supramolecular elastomer capable of self-healing that also exhibits rubber-like properties at room temperature.11 Although the mechanical strength of these materials is relatively low, they are regarded as suitable medical materials for wound dressing because of their acceptable water vapor transmission rate and water absorption rate at room temperature.12
The multi-step reaction process is accompanied by the increase in both the hydrogen bonding group fractions and chain length; accordingly, a high functionality (>2) can be obtained through a single component of the linear bifunctional molecules, provided the hydrogen bonding groups are treated as new functional groups. Thus, we have attempted to prepare a similar supramolecular elastic material based on the multi-step reaction process mentioned above using linear bifunctional oligomers. To afford the material with a low glass transition temperature (Tg) and to avoid crystallization, linear PDMS–COOH2 oligomers with distributed repeat units were used. Compared with using a mixture of single, di-, and triacids, the use of a single bifunctional diacid is a more tunable and reasonable route in the preparation of supramolecular networks with certain structures, which could aid in carrying out a more accurate evaluation of the crosslinking mechanisms for this type of material.
This report describes the synthesis of supramolecular elastic materials based on siloxane oligomers (SESi). A detailed discussion on the crosslinking mechanisms is given according to the changes in structure–property relationships during the reaction process. The dynamic mechanical properties and self-healing properties of several SESi samples with different PDMS chain lengths are also evaluated.
2 Experimental section
2.1 Materials
Octamethylcyclotetrasiloxane (D4, PMX-0244, 99.9% purity) was supplied by Dow Corning Silicones. 1,1,3,3-Tetramethyldisiloxane (HMM, 98% purity) was supplied by Kaihua Taicheng Silicone Co., Ltd (Quzhou, China). Tert-butyl methacrylate (tBMA, 98% purity) was supplied by Chemlin Chemical Industry Co., Ltd. (Nanjing, China). Other reagents, such as DETA (98% purity), urea (99% purity) and the solvents, were used as received.2.2 Synthesis of the PDMS–COOH2 oligomers
PDMS–COOH2 was synthesized through the three-step process presented in Scheme 1; hydrogen-terminated polydimethylsiloxane (PDMS–H2) oligomers were synthesized through the cationic ring-opening polymerization of D4,13 followed by a hydrosilylation reaction between PDMS–H2 and tBMA. PDMS–COOH2 was finally obtained after acidic hydrolysis of the t-butyl ester terminated polydimethylsiloxane (PDMS–tBMA2) intermediate product.14 (A detailed description can be found in the ESI.†)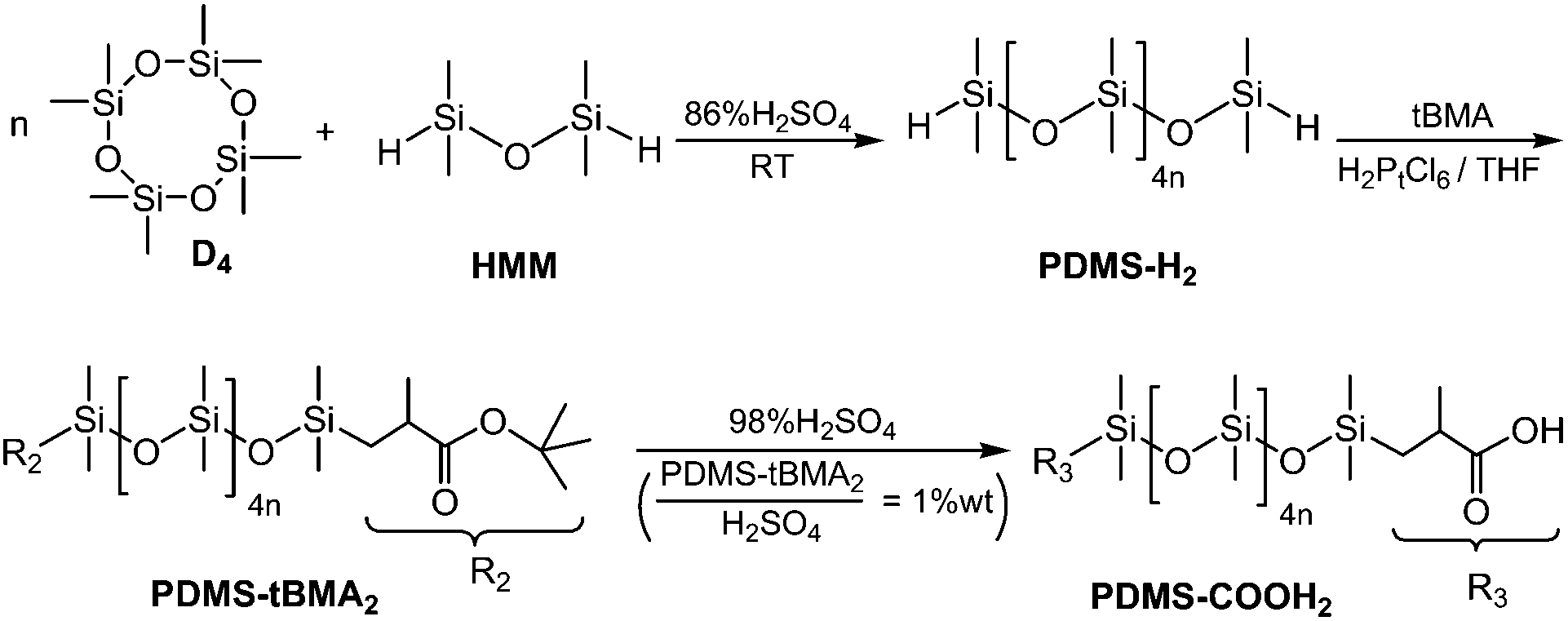 | ||
| Scheme 1 The synthesis and structure of PDMS–COOH2. | ||
2.3 Synthesis of SESi
The SESi materials were prepared through the two-step process presented in Scheme 2 and Table 1, which is a similar route to that used for the dimer fatty acid system reported by Leibler.8 First, we prepared a series of linear PDMS oligomers with distributed chain lengths, all bearing the NH–CH2–CH2–NH2 moiety at their extremities. This series was realized by reacting PDMS–COOH2 with excess DETA at 135 °C for several hours. Second, the oligomers were functionalized with urea and imidazolidone groups through the reaction between amine functions and urea at 135 to 160 °C (see the ESI†).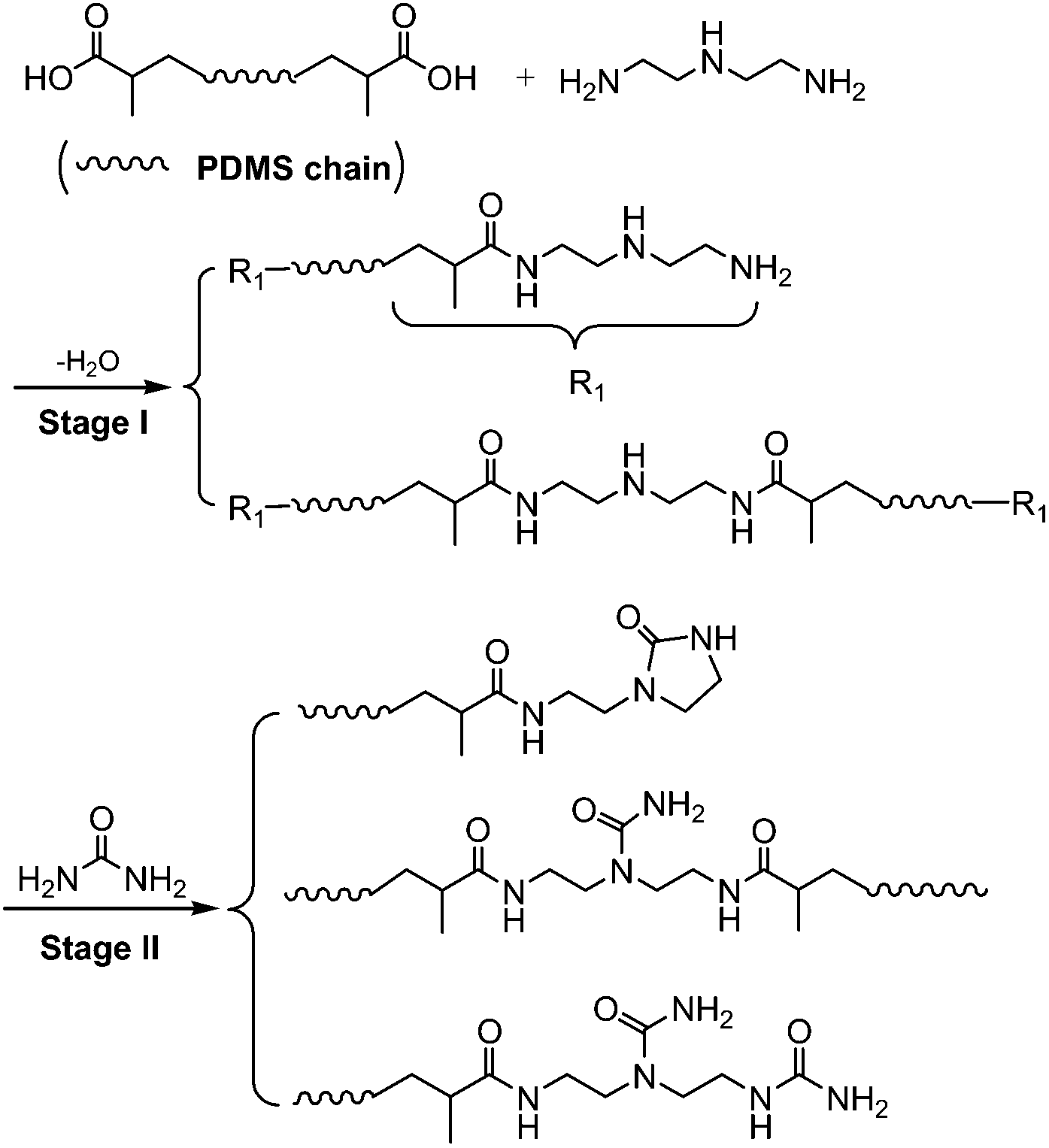 | ||
| Scheme 2 The synthesis and structure of the SESi materials. | ||
| Material | Molecular weight of PDMS–COOH2 (g mol−1) | DETA/PDMS–COOH2 (mol mol−1) | Urea/PDMS–COOH2 (mol mol−1) | ||
|---|---|---|---|---|---|
| M n | M n | PDIb | |||
| a Calculated from the 1H-NMR technique, n represents the number of siloxane repeat units. b Measured from the GPC analysis. | |||||
| SESi1 | 1.0 × 103 (n = 10) | 0.94 × 103 | 1.38 | 2.3/1 | 1/1 |
| SESi2 | 4.0 × 103 (n = 44) | 4.15 × 103 | 1.88 | 2.3/1 | 1/1 |
| SESi3 | 1.1 × 104 (n = 147) | 1.13 × 104 | 2.98 | 2.3/1 | 1/1 |
2.4 Characterization
Infrared spectra were recorded using a Bruker Vertex 70 Fourier transform-infrared spectrometer equipped with a Harrick ATC-024 temperature controller.1H-NMR spectra were collected with a Bruker AVANCE III-400 (400 MHz) spectrometer using CDCl3 as the solvent.
Gel permeation chromatography (GPC) was performed on Elite EC2000 GPC apparatus (Dalian, China) equipped with a Shodex K-G guard column and a Shodex K-804L chromatographic column. Detection was achieved using a refractive index detector, and the samples were analyzed at 30 °C using CHCl3 as the eluent at a flow rate of 1 mL min−1. The instrument was calibrated using low polydisperse polystyrene standards.
Thermal analyses were performed at a heating and cooling rate of 10 K min−1 in a nitrogen atmosphere with Netzsch DSC 204C apparatus. The SESi specimens were first heated and held at 150 °C for 5 min and were then cooled and held at −150 °C for 5 min. The thermal transitions of these materials were evaluated from thermograms collected during the second heating process.
Dynamic mechanical measurements were performed at 10 Hz in tensile mode using Netzsch DMA-242 apparatus. The tests were carried out at a heating rate of 5 K min−1 from −170 °C to 100 °C.
XRD analyses were performed using a PANalytical X'Pert-Pro X-ray diffractometer with filtered monochromatic Cu Kα radiation in the 2θ range of 5° to 90°.
An RPA 2000 rubber process analyzer was used for rheological temperature sweeps (30 °C to 160 °C) and frequency sweeps (0.05 rad s−1 to 200 rad s−1). The dynamic temperature sweep was performed at a strain amplitude of 3% and a frequency of 1 Hz. The dynamic frequency sweep was performed at a strain amplitude of 3% with the temperature range of 40 °C to 100 °C.
Tensile tests were performed on rectangular specimens (50 mm × 10 mm × 1 mm) using a Zwick Z010 tensile testing machine at a stretching rate of 500 mm min−1. Stress relaxation tests and creep tests were also performed on the Z010 tensile testing machine. The applied strain for the stress relaxation tests was 100%. The applied loads for the creep tests were 0.05 MPa, 0.025 MPa and 0.0125 MPa for SESi1, SESi2 and SESi3, respectively. Cyclic tensile tests were conducted at an elongation rate of 20 mm min−1 with an applied strain of 20%. The internal friction (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) of the SESi samples was calculated from the equation below:
δ) of the SESi samples was calculated from the equation below:
| ΔW = πσoεosinδ | (1) |
3 Results and discussion
3.1 Reaction mechanisms
The reaction progress of PDMS–COOH2 with DETA and urea was monitored with 1H-NMR spectra of samples collected from the mixture (Fig. S2†). First, an acylation reaction between PDMS–COOH2 and DETA was efficiently carried out at 135 °C with the evolution of water. A small amount of imidazoline by-product was also formed through a second dehydration reaction between the amide groups and the lateral –NH– groups (Scheme 3). Second, the –NH2 groups reacted with the partial urea groups to form monoalkyl-urea derivatives. The –NH– groups then reacted with the remaining urea to form 1,1-dialkylurea derivatives, while imidazolidone derivatives also formed with longer reaction times (Fig. S2†). Therefore, the reaction process in the second step is illustrated in Scheme 4. According to the GPC analysis (Fig. S3†), the transformation of monoalkyl-urea into biuret or 1,3-dialkylurea derivatives is comparatively minimal (Scheme 5).15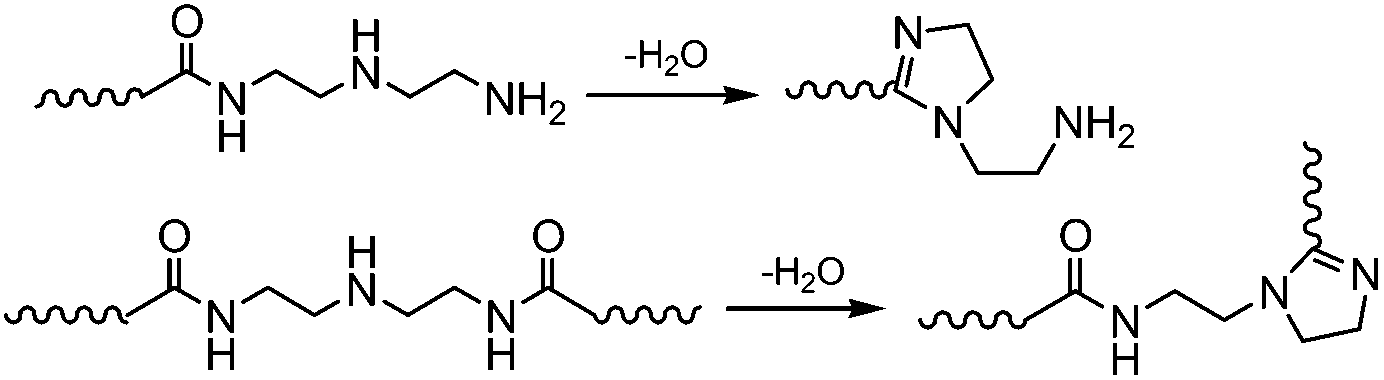 | ||
| Scheme 3 The formation of imidazoline by-products in the first step. | ||
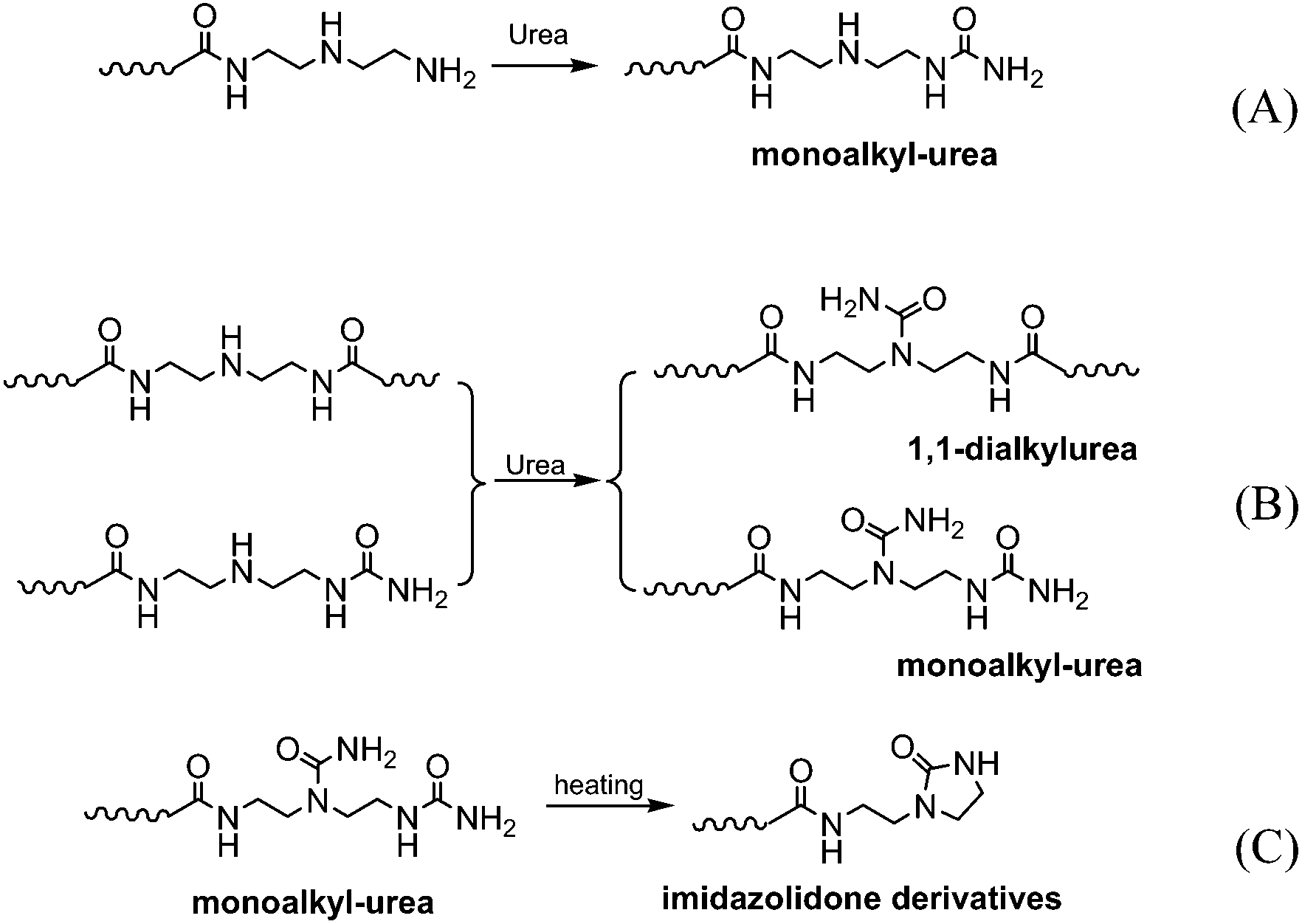 | ||
| Scheme 4 The evolutions of the reaction process in the second step. | ||
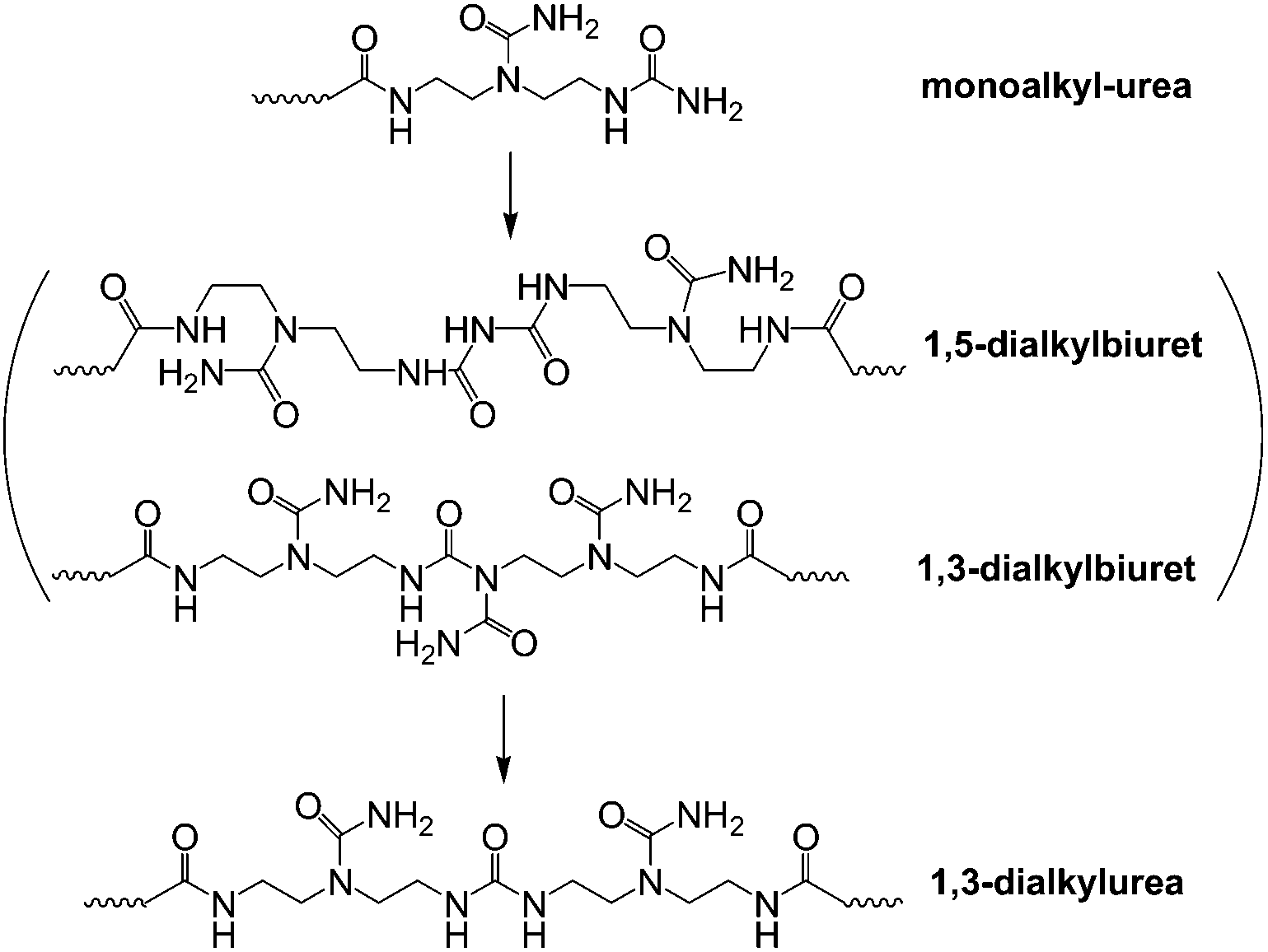 | ||
| Scheme 5 The transformation of monoalkyl-urea into biuret or 1,3-dialkylurea derivatives. | ||
3.2 Identification of the hydrogen bonding interactions
The existence of hydrogen bonding interactions in the SESi matrix and its related thermal properties were studied using temperature-dependent infrared spectroscopy. The sample film was cast from chloroform solution on a potassium bromide pellet, and the spectra were recorded in transmission mode.
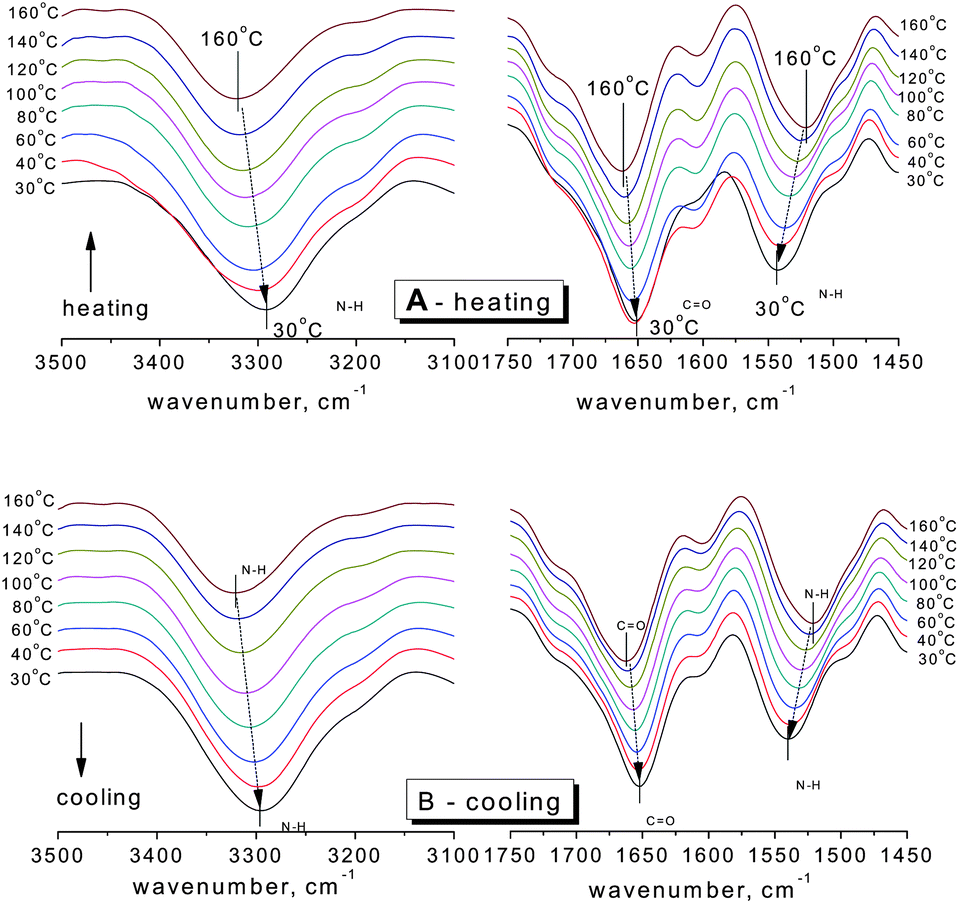
Fig. 1 shows the changes of the N–H stretching vibration (3500–3100 cm−1) and amide vibration (amide I and amide II vibrations, 1750–1450 cm−1) of SESi1 during the heating and cooling processes at different temperatures (30/40/60/80/100/120/140/160 °C). The spectra were recorded after maintaining the sample at the designated temperature for 5 minutes. During the heating process, the frequency of the hydrogen bonded N–H stretching vibration shifts systematically from 3293 cm−1 to 3321 cm−1 over the temperature range of 30 °C to 160 °C. A significant decrease in the area of the hydrogen bonded N–H stretching vibration absorption peak can also be observed. This reduction in area is likely related to the strong dependence of the absorptivity coefficient on the hydrogen bond strength in the N–H stretching band.16 When the temperature was increased, both the strength of the hydrogen bond and the absorptivity coefficient of the N–H stretching band decreased. The amide I mode (mainly the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration) also shifts to higher frequency (1651 cm−1 to 1663 cm−1 from 30 °C to 160 °C) with increasing temperature, and there is increasing evidence of a shoulder at approximately 1710 cm−1, which can be interpreted as the transformation of hydrogen bonded CO (in the urea functionalities) groups to free CO groups.17 In contrast, the amide II mode (mainly the N–H in-plane bending vibration) seems to shift to a lower frequency (1543 cm−1 to 1522 cm−1 from 30 to 160 °C) with increasing temperature. During the cooling process, a complete inverse shifting behavior was observed in the frequency of the bonded bands. Once the temperature was decreased to 30 °C, the peak positions and peak areas of the bonded groups remained almost unchanged, compared with the original state (Fig. 2 and Table 2). Therefore, the thermal reversibility of the hydrogen bonding interactions in the SESi matrix is confirmed.
O stretching vibration) also shifts to higher frequency (1651 cm−1 to 1663 cm−1 from 30 °C to 160 °C) with increasing temperature, and there is increasing evidence of a shoulder at approximately 1710 cm−1, which can be interpreted as the transformation of hydrogen bonded CO (in the urea functionalities) groups to free CO groups.17 In contrast, the amide II mode (mainly the N–H in-plane bending vibration) seems to shift to a lower frequency (1543 cm−1 to 1522 cm−1 from 30 to 160 °C) with increasing temperature. During the cooling process, a complete inverse shifting behavior was observed in the frequency of the bonded bands. Once the temperature was decreased to 30 °C, the peak positions and peak areas of the bonded groups remained almost unchanged, compared with the original state (Fig. 2 and Table 2). Therefore, the thermal reversibility of the hydrogen bonding interactions in the SESi matrix is confirmed.
 | ||
| Fig. 1 The FT-IR spectra of SESi1 in the temperature range of 30 °C to 160 °C (A) during the heating process and (B) during the cooling process. | ||
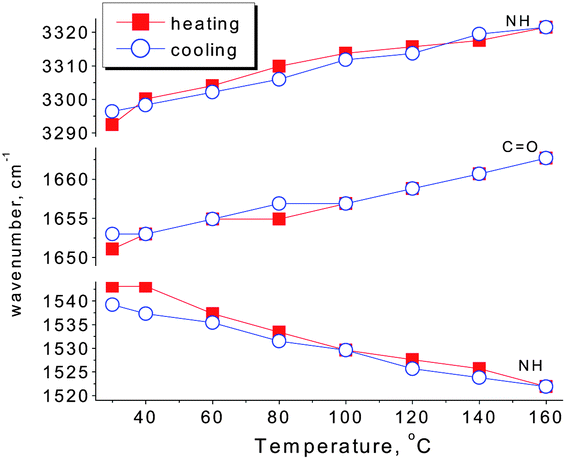 | ||
| Fig. 2 Changes in the peak positions of the N–H stretching vibration (νN–H), CO stretching vibration (νCO) and N–H in-plane bending vibration (δN–H). | ||
| Temperature | ν N–H |
ν
CO
|
δ N–H | ||||
|---|---|---|---|---|---|---|---|
| S | W 1/2 | S | W 1/2 | S | W 1/2 | ||
| a Compared to the peak area at the original state. | |||||||
| Heating | 30 °C | 1.00 | 297 | 1.00 | 127 | 1.00 | 110 |
| 160 °C | 0.62 | 282 | 1.15 | 154 | 1.10 | 122 | |
| Cooling | 160 °C | 0.60 | 282 | 1.11 | 152 | 1.09 | 102 |
| 30 °C | 1.02 | 290 | 0.94 | 135 | 0.98 | 107 | |
However, such a reversible process can only be conducted below a certain temperature. The use of higher temperatures can bring about further reactions between the alkyl-urea derivatives.10,15 The degradation of urea groups at 200 °C was also reported by Versteegen and coworkers.18 All of the above reactions led to a decrease in the strength of the hydrogen bonding interactions. Therefore, temperatures higher than 160 °C are not favorable for maintaining the structural stability and reversibility of the hydrogen bonding interactions (Fig. S4†).
3.3 Thermal transition analyses
Differential scanning calorimetry (DSC) and dynamic mechanical analysis were used to evaluate the thermal transitions of these SESi materials. The second heating and cooling traces of SESi1, SESi2 and SESi3 are depicted in Fig. 3, while the DMA results are presented in Fig. 4.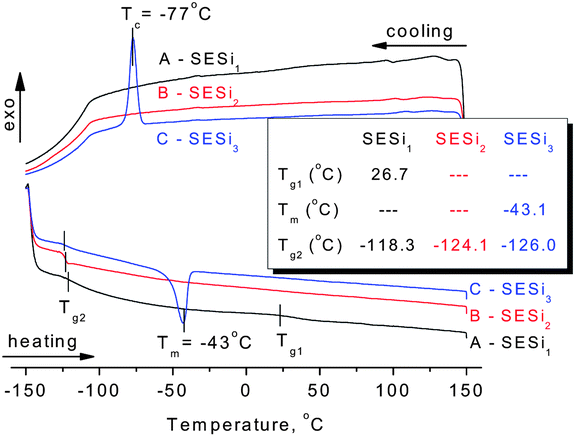 | ||
| Fig. 3 The DSC traces of (A) SESi1, (B) SESi2 and (C) SESi3. (Inset: the thermal transition temperatures of materials SESi1, SESi2 and SESi3; Tg1, Tg2 and Tm denote the glass transition temperature of the carbon chains, the melting point of the crystalline PDMS chains and the glass transition temperature of the siloxane chains, respectively.) | ||
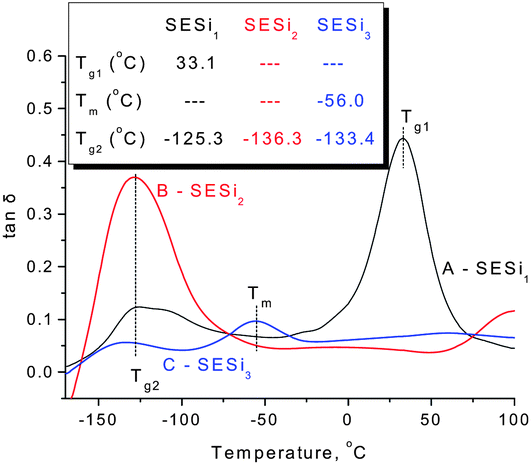 | ||
| Fig. 4 The DMA curves of (A) SESi1, (B) SESi2 and (C) SESi3. (Inset: the thermal transition temperatures of materials SESi1, SESi2 and SESi3; Tg1, Tg2 and Tm have the same meaning as in Fig. 3.) | ||
For all of the SESi materials, a clear glass transition temperature (Tg) attributed to the siloxane chains (Tg2) was observed between −126 °C and −118 °C, as shown in Fig. 3. The slight increase in Tg2 with a concurrent significant decrease in the PDMS chain length indicates a poor interaction between the carbon chains and the PDMS chains. This is advantageous for the low-temperature properties of the materials.18 The crystallization of PDMS chains in the SESi matrix is closely related to the molecular weight of PDMS–COOH2. For SESi1 and SESi2, which were prepared from low-molecular-weight PDMS–COOH2, no melting or crystallization peak is observed. However, for SESi3, clear melting/crystallization peaks are apparent at −43/−77 °C, attributed to the thermal transition temperatures of the crystalline phase of the PDMS chains.19 This phenomenon can be explained by the decrease in chain mobility with increasing hydrogen bonding interactions between the PDMS chains, induced by decreasing the molecular weight of PDMS–COOH2.20 The proportion of carbon chains also increases with the decreasing PDMS–COOH2 molecular weight, which could act as a specific diluent with respect to preventing siloxane unit aggregation. Compared with the Tg2 of PDMS, Tg1 attributed to the carbon chains is significantly affected by the molecular weight of PDMS–COOH2. The use of low-molecular-weight PDMS–COOH2 leads to higher components of carbon units and thus a clearer and elevated Tg1. According to the results of the DSC analysis, the Tg1 of SESi1 (which has the highest component of carbon units) is nearly 26.7 °C; this value already reaches the Tg of supramolecular elastomers fabricated based on dimer fatty acids (28 °C).10
Poor compatibility with the PDMS chains may lead to stacking of the hydrogen-bonded groups,21 possibly yielding a material similar to TPEs. However, no crystalline structure was detected above the room temperature, according to the DSC and XRD tests (Fig. 3 and S5†). This finding indicates the poor stacking behavior of the hydrogen-bonded groups in the SESi matrix, which may result from a diverse distribution of the hydrogen bond types. This is acceptable because a series of different bonded groups, such as amide groups, urea groups, and imidazolidone groups, coexist in the SESi matrix.
3.4 Viscoelastic properties
Dynamic temperature sweeps were performed to evaluate the evolution of the storage modulus (G′) and loss modulus (G′′) with increasing temperature (Fig. 5). For SESi1, a clear glass transition behavior was observed at low temperatures; in contrast, at high temperatures, a highly elastic rheological response with G′ > G′′ was detected. A wide temperature-independent rubbery plateau in G′ was detected in the temperature range of 80 °C to 140 °C, indicating the formation of a well-defined network structure that does not evolve significantly with the temperature.22 In SESi2 and SESi3, glass transition behaviors were not observed, and the rubbery plateaus expand to lower temperatures, indicating enhanced rubbery properties at low temperatures with increasing PDMS chain lengths. Additionally, a sharp decrease in G′ and a dispersion maximum in G′′ were observed in the range of 160 °C to 200 °C for all SESi specimens. This phenomenon was apparent as soon as the experimental time exceeded the lifetime of the physical crosslinks in the thermoreversible networks.23 This observation reflects a significant decrease in the hydrogen bond strength of the test samples at higher temperatures, which is consistent with the results from the FT-IR analysis.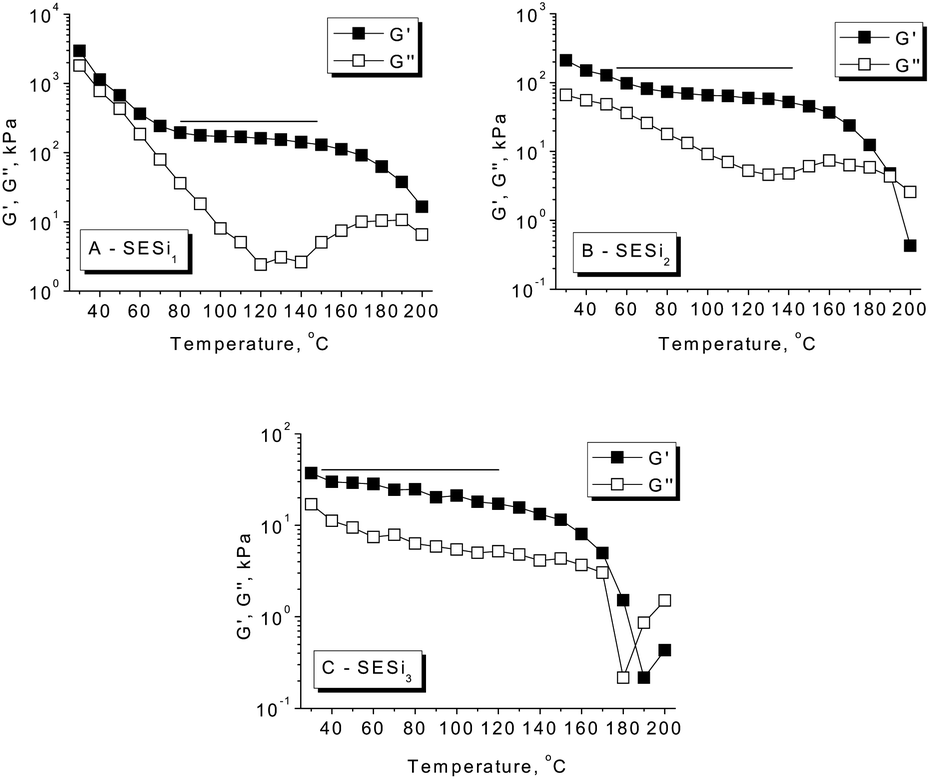 | ||
| Fig. 5 The temperature dependence of the dynamic storage modulus (G′) and loss modulus (G′′) for the different SESi materials: (A) SESi1, (B) SESi2 and (C) SESi3. | ||
For each SESi sample, the time–temperature superposition (TTS) principle was applied to construct a master curve by shifting the isothermal dynamic frequency sweep data collected at various temperatures along the frequency axis. Fig. 6 shows the TTS master curves in which G′ and G′′ are plotted against the reduced frequency (ωaT) in a double-logarithmic format. The behavior of SESi1 at high frequencies indicates the proximity of the glass transition; however, at low frequencies, the elastic rubbery character dominates. Similar rheological behaviors were also found for SESi2 and SESi3, with the rubbery plateaus shifting to higher frequencies with increasing PDMS chain lengths. That is, although an increase in the PDMS chain length decreases the hydrogen bonding strength, better chain mobility behaviors can be obtained, which could afford the material with better rubbery properties at low temperatures.
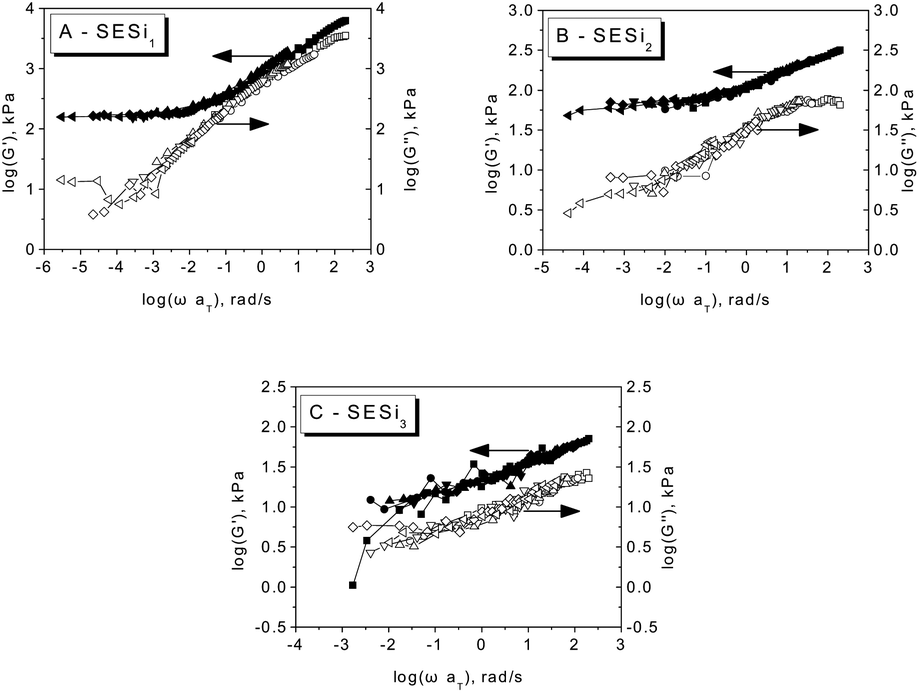 | ||
| Fig. 6 The master curves of the dynamic storage modulus (G′) and loss modulus (G′′) for different SESi materials referenced to 30 °C for (A) SESi1, (B) SESi2 and (C) SESi3. | ||
In Fig. 7, the shift factors logaT were plotted as a function of the inverse temperature, in which differences in the temperature dependence of the shift factors for the three SESi samples can be seen. For samples with short PDMS chain lengths, a large absolute value of logaT was obtained, accompanied by a high temperature dependence of the shift factor. Short PDMS chain lengths impart significant constraint on the chain mobility, induced by the hydrogen bonding interactions. The strong temperature dependence of the hydrogen bonding interactions results in a significant change of the chain mobility with variations in temperature, leading to a large absolute value of logaT. Generally, strong interactions between the polymer chains lead to a strong temperature dependence of the shift factor.1
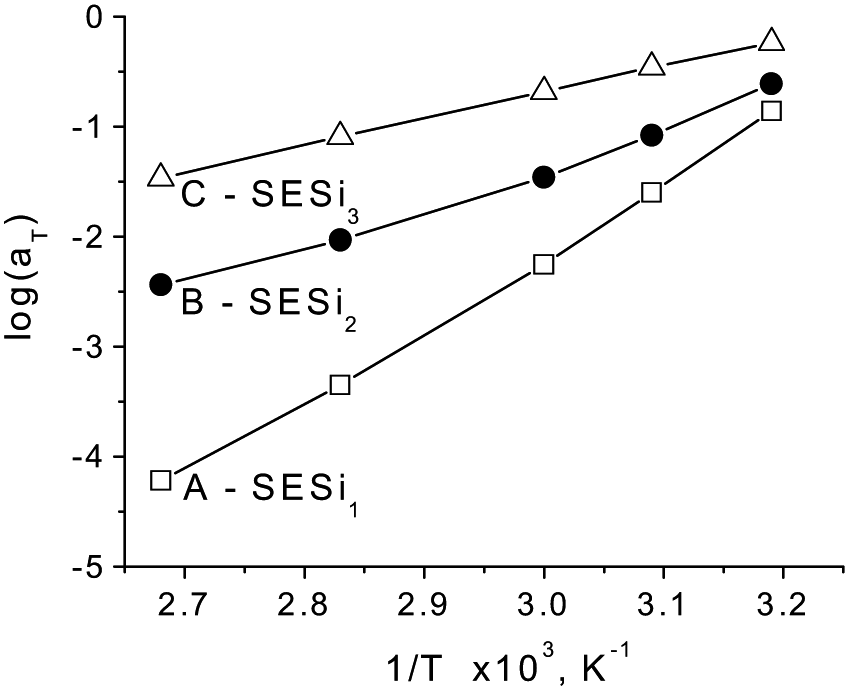 | ||
| Fig. 7 The shift factors aT as a function of 1/T for different SESi materials: (A) SESi1, (B) SESi2 and (C) SESi3. | ||
The rubber-like behavior of the SESi materials was also confirmed through creep tests and stress relaxation tests (Fig. S6 and 7†). From these results, we can conclude that interactions between the PDMS chains afford the SESi materials with stable crosslinking structures, which are independent of the PDMS chain length.
As is known, hysteresis is mainly determined by the chain mobility of the polymer materials, which is usually reflected by the internal friction, evaluated by cyclic tensile tests (Fig. S8†). For a pure, crosslinked silicone rubber such as SR1, hysteresis is negligible because the PDMS chains move almost freely at room temperature. The addition of fillers increases the internal friction of silicone rubbers (SR2), which originates from the strong interaction between the rubber matrix and the fillers. For the SESi materials investigated here, hydrogen bonding interactions between the PDMS chains act like a specific type of reinforcing filler, increasing the internal friction; as a result, hysteresis can be observed for all of the SESi samples. For SES1, a significant residual strain and relatively high internal friction were observed at room temperature due to the high Tg (26.7 °C) of this material. Compared with SR2, close values of the residual strain and internal friction were observed for SESi2 and SESi3, as shown in Table S1†. Hence, SESi elastomers also show acceptable hysteresis properties.
3.5 Self-healing properties
In general, supramolecular elastomers based on hydrogen bonding interactions can be processed like conventional thermoplastics (Fig. S9†). Moreover, a high fraction of hydrogen-bonded groups in the matrix is favorable for obtaining acceptable self-healing properties.6,10,24 Based on the above considerations, we have mainly focused our investigation on the self-healing abilities of the SESi1 and SESi2 materials. Rectangular specimens of SESi1 and SESi2 were first cut into two pieces and were brought into contact immediately at room temperature and then healed at various temperatures for 16 hours. It seems that the necessary healing temperatures for the different SESi samples are closely related to the PDMS chain lengths. For specimens with short PDMS chains, a high temperature is required (Fig. 8). This observation is reasonable because the self-healing ability of these hydrogen-bonded materials is mainly dependent on the mobility of the non-associated groups on the fracture surface.10 As mentioned above, shorter PDMS chains lead to a lower degree of chain mobility; hence, the use of a higher temperature is favorable, helping the non-associated groups to pair more efficiently. Additionally, to achieve complete healing, longer healing times are necessary for SESi samples with shorter PDMS chains, for the same reason (Fig. 9).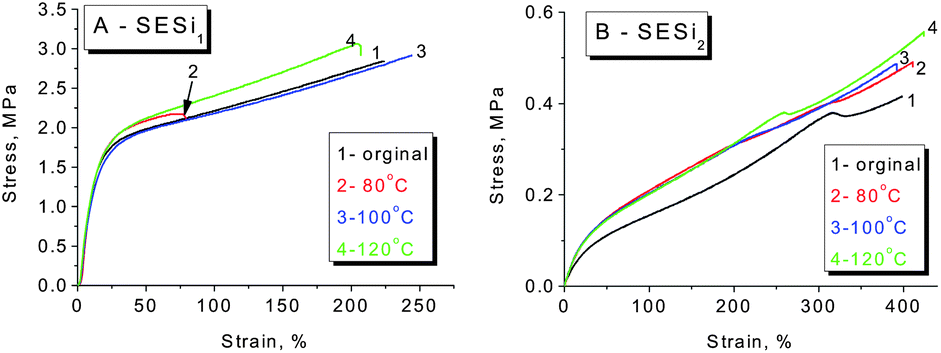 | ||
| Fig. 8 The self-healing properties of different SESi materials, (A) SESi1 and (B) SESi2. The samples were cut into two pieces and were then brought into contact immediately at room temperature, followed by healing at various temperatures for 16 hours. | ||
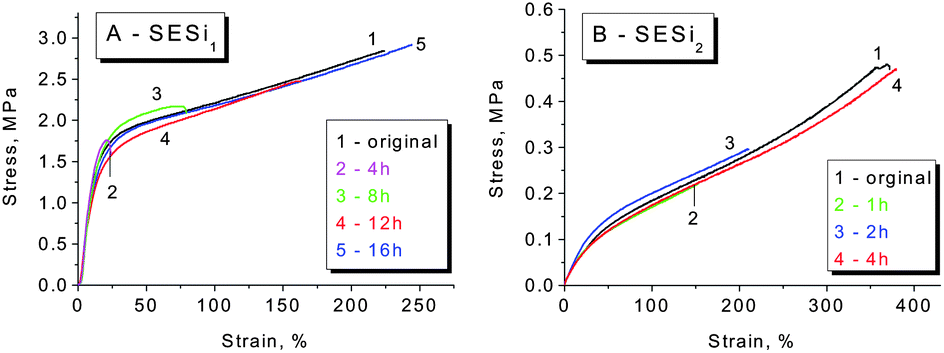 | ||
| Fig. 9 The self-healing properties of different SESi materials: (A) SESi1 and (B) SESi2, at 100 °C. The samples were cut into two pieces and then were brought into contact immediately at room temperature, followed by healing at 100 °C for various healing times. | ||
3.6 Crosslinking mechanism
A specific type of elastic material based on hydrogen bonding interactions was successfully obtained through a single component of linear bifunctional molecules. The presence of a broad rubber plateau in the modulus of the linear polymer is either due to the presence of entanglements or physical cross-links in microcrystalline domains.20 The DSC and XRD results indicate that microcrystalline domains did not exist in the SESi matrix at room temperature. Consequently, we interpreted the observed viscoelastic properties of the SESi materials as being the result of the entanglements of associated chains in an amorphous matrix. Moreover, we believe that the high functionality (>2) of the polymer chains was obtained after the two-step reaction because stable supramolecular networks exist in the SESi matrix. This could be observed by modifying the PDMS chains with several bonded groups. However, we cannot treat all bonded groups as effective sites for contributing to the increase of functionality; only the groups capable of forming hydrogen bonds are strong enough for consideration.According to the evolution of the reaction process, at least four stages can be included in the synthesis of SESi materials. (1) Stage A denotes the formation of amide groups, and in this stage, an associated liquid is obtained; (2) Stage B denotes the formation of monoalkyl-urea groups, and in this stage, a viscoelastic fluid is obtained; (3) Stage C denotes the formation of 1,1-dialkylurea groups, and in this stage, an unstable viscoelastic solid is obtained; (4) Stage D denotes the formation of imidazolidone derivatives, and in this stage, the stable elastic material is finally obtained (Fig. 10). The physical properties combined with the related content of the bonded groups and the mechanical strength at different stages are illustrated in Fig. 10. Throughout the reaction process, the mechanical strength of the system gradually increased. The content of bonded groups increases during the first three stages; in contrast, a decrease in this fraction was observed during the last stage. This result was caused by the formation of imidazolidone derivatives through the reaction between the monoalkyl-urea groups and 1,1-dialkylurea groups with a molar ratio of 1:1 (Scheme 4). From Fig. 10, we can conclude that imidazolidone derivatives are able to form strong hydrogen bonding interactions, which can act as effective crosslinks. Thus, stable rubbers are formed at this stage. However, it is difficult to evaluate the differences in strength among the hydrogen bonds formed by the remaining three groups, although a significant increase in the mechanical strength was observed during the first three stages. One may consider the increase of mechanical strength during the first three stages as the result of increased contents of bonded groups. In fact, the evolution of the physical properties also reflects a gradual increase in the stability of the structure, which was found to be independent of the PDMS chain length. Hence, we believe that the structural stability of the SESi system is mainly determined by the bond strength of different hydrogen bonds. That is, the hydrogen bonds formed by groups generated from the latter stage should have higher bond strengths (at least, at the overall level). Because imidazolidone derivatives only exist at chain ends, 1,1-dialkylurea groups are likely the groups that effectively increase the functionality of the polymer chains.
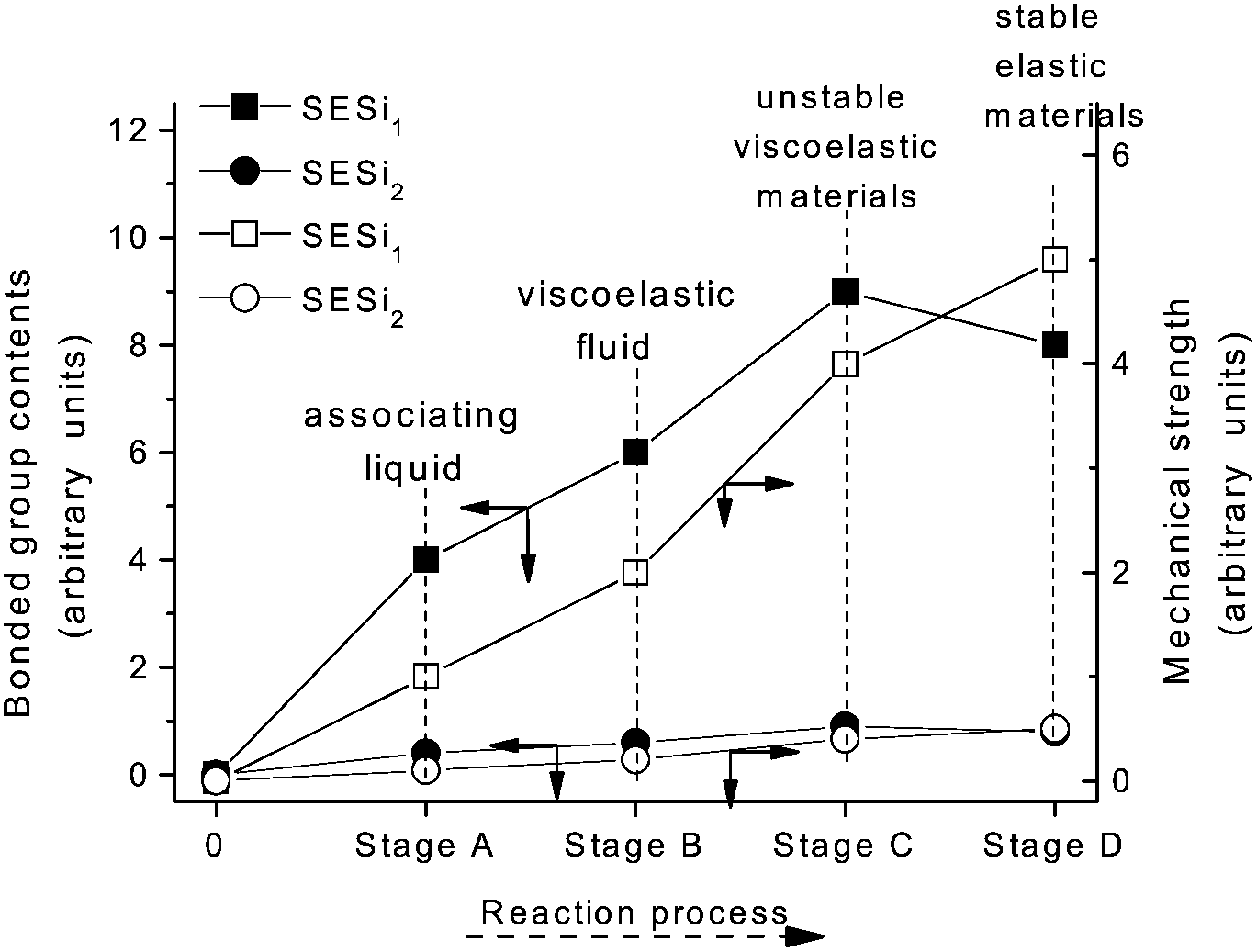 | ||
| Fig. 10 The evolution of the physical properties, mechanical strength (hollow) and bonded group contents (solid) during the reaction progress of the SESi materials. | ||
Although all bonded groups can contribute to the mechanical strength of SESi materials, only the groups forming strong hydrogen bonds that can act as stable crosslinks are recognized to increase the functionality of the polymer chains. Fig. 11 shows the possible supramolecular network of SESi, based on the associations of the bonded groups mentioned above in which the effective cross-links are mainly formed by 1,1-dialkylurea groups and imidazolidone derivatives. A further study is required to compare the bond strengths of different hydrogen bonds in detail, which could help in the construction of a more accurate model.
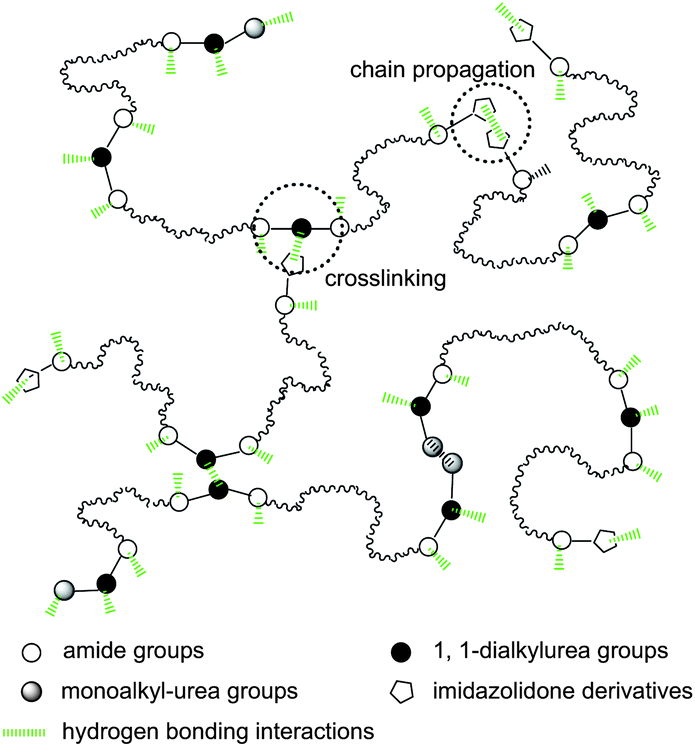 | ||
| Fig. 11 A schematic representation of a supramolecular network cross-linked by hydrogen bonding interactions from 1,1-dialkylurea groups and imidazolidone derivatives. | ||
4 Conclusions
The detailed study presented above shows that the SESi materials obtained through the two-step reaction of PDMS–COOH2 with DETA and urea are supramolecular elastomers with reasonable hysteresis and acceptable self-healing properties. We have testified that the rheological, mechanical and self-healing properties of these materials are closely related to the PDMS chain length; thus, a series of SESi materials with different properties can be achieved by altering the PDMS chain length. However, the stability of the structure seems to be independent of the PDMS chain length. Because no crystalline structure was detected at room temperature, we have interpreted the viscoelastic properties of these materials as the result of the entanglement of associated chains in the amorphous matrix. Moreover, we believe that only the hydrogen bonds formed by 1,1-dialkylurea groups and imidazolidone derivatives can be treated as effective cross-links with respect to contributing to the stability of the supramolecular networks.Finally, we underline the fact that high functionality (>2) is favored but is not necessary in the preparation of supramolecular networks because the functionality of the polymer chains can be increased during the reaction. That is, a supramolecular network can be obtained through the modification of bifunctional molecules under specific conditions. Only the bonded groups that form strong hydrogen bonds that can act as stable cross-links are considered as contributing to the increased functionality of the polymer chains.
Acknowledgements
The authors acknowledge the financial support through the NSFC (National Natural Science Foundation of China) grant 51003032, 31201552, and the “Fundamental Research Funds for the Central Universities, SCUT” grant 2009ZM0263, 2012ZZ0016.Notes and references
- R. Stadler and L. Freitas, Prog. Colloid Polym. Sci., 1986, 264, 773 CrossRef CAS.
- C. Hilger, M. Drager and R. Stadler, Macromolecules, 1992, 25, 2498 CrossRef CAS.
- H. Kautz, D. J. M. Van Beek, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 4265 CrossRef CAS.
- D. J. M. Van Beek, A. J. H. Speiering, G. W. M. Peters, K. Nijenhuis and R. P. Sijbesma, Macromolecules, 2007, 40, 8464 CrossRef CAS.
- O. Colombani, C. Barioz, L. Bouteiller, C. Chaneac, L. Fomperie, F. Lortie and H. Montes, Macromolecules, 2005, 38, 1752 CrossRef CAS.
- Y. L. Chen, A. M. Kushner, G. A. Williams and Z. B. Guan, Nat. Chem., 2012, 4, 467 CrossRef CAS PubMed.
- L. Bouteiller, Adv. Polym. Sci., 2007, 207, 79 CrossRef CAS.
- D. Montarnal, P. Cordier, C. Soulie-Ziakovic, F. Tournilhac and L. Leibler, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7925 CrossRef CAS.
- D. Montarnal, F. Tournilhac, M. Hidalgo, J. L. Couturier and L. Leibler, J. Am. Chem. Soc., 2009, 131, 7966 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS PubMed.
- A. Q. Zhang, L. Yang, Y. L. Lin, L. S. Yan, H. C. Lu and L. S. Wang, J. Appl. Polym. Sci., 2013, 129, 2435 CrossRef CAS.
- A. Q. Zhang, L. Yang, Y. L. Lin, L. S. Yan, H. C. Lu, Y. H. Qiu and Y. L. Su, J. Biomater. Sci., Polym. Ed., 2013 DOI:10.1080/09205063.2013.808151.
- O. Masaya, JPN Pat., 6,100,684A, 1994.
- A. Knebelkamp, P. Lersch and C. Weitemeyer, US Pat., 5,637,746, 1997.
- J. G. Erickson, J. Am. Chem. Soc., 1954, 76, 3977 CrossRef CAS.
- D. J. Skrovanek, P. C. Painter and M. M. Coleman, Macromolecules, 1986, 19, 669 CrossRef.
- M. M. Coleman, M. Sobkowiak, G. J. Pehlert and P. C. Painter, Macromol. Chem. Phys., 1997, 198, 117 CrossRef CAS.
- R. M. Versteegen, R. Kleppinger, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 772 CrossRef CAS.
- (a) N. E. Botterhuis, D. J. M. Van Beek, G. M. L. Van Gemert, A. W. Bosman and R. P. Sijbesma, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3877 CrossRef CAS; (b) A. Tiwari, A. K. Nema, C. K. Das and S. K. Nema, Thermochim. Acta, 2004, 417, 133 CrossRef CAS PubMed; (c) T. Dollase, H. W. Spiess, M. Gottlieb and R. Yerushalmi-Rozen, Europhys. Lett., 2002, 60, 390 CrossRef CAS.
- J. H. K. Ky Hirschberg, F. H. Beijer, H. A. Van Aert, P. C. M. M. Magusin, R. P. Sijbesma and E. W. Meijer, Macromolecules, 1999, 32, 2696 CrossRef CAS.
- E. Yilgor, E. Burgaz, E. Yurtsever and I. Yilgor, Polymer, 2000, 41, 849 CrossRef CAS.
- Y. Lei and T. P. Lodge, Soft Matter, 2012, 8, 2110 RSC.
- M. Muller, A. Dardin, U. Seidel, V. Balsamo, B. Ivan, H. W. Spiess and R. Stadler, Macromolecules, 1996, 29, 2577 CrossRef.
- A. Phadke, C. Zhang, B. Arman, C. C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. USA, 2012, 109, 4383 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis and characterization, including 1H-NMR, GPC, XRD, FTIR, and tensile properties of SESi materials. See DOI: 10.1039/c3py01005h |
| This journal is © The Royal Society of Chemistry 2014 |