DOI:
10.1039/C3PY00944K
(Paper)
Polym. Chem., 2014,
5, 234-240
Direct and indirect core–shell inversion of block copolymer micelles†
Received
17th July 2013
, Accepted 8th August 2013
First published on 9th August 2013
Abstract
The core–shell inversion of micelles of diblock copolymers under environmental stimuli in solution is a basic and important subject in macromolecular self-assembly and is also a basis for some of the applications of the assemblies. In literature, the inversion kinetic studies for diblock copolymers switched by pH or temperature found that such inversion always took place indirectly, comprising steps of dissociation and re-assembly. In such an indirect process, molecularly dispersed chains as intermediates are inevitable, because in the middle range of pH or temperature, both blocks are soluble. It is remarkable to see that another pathway, i.e. direct inversion without any intermediates, which is favourable for many applications, has not been achieved yet on a bench scale. In this paper, a novel block copolymer of PNIPAm-b-PBOB (PNIPAm: poly(N-isopropylacrylamide); BOB: benzoboroxole) was studied, where the hydrophobic PBOB block containing sugar-responsive benzoboroxole groups turned hydrophilic at neutral pH and the temperature-sensitive block PNIPAm underwent the coil–globule transition around its LCST. By deliberately adjusting the program of imposing the triggers, i.e. sugar addition and pH change for PBOB block and temperature change for PNIPAm, we succeeded in controlling the inversion kinetics of the micelle of PNIPAm-b-PBOB (M-BOB) in water. Both indirect and direct inversion pathways are realized as desired. The direct inversion may be accompanied by micellar fusion or fission, or more or less proceeds in situ. The direct inversion was realized by treating the copolymer micelle M-BOB with fructose at 25 °C and neutral pH for a certain period to make the PBOB core swell, followed by a temperature increase, in which collision and fusion sometimes maybe involved. The temperature increase induced agglomeration of the PNIPAm block and moved inward, which can match the further interaction of PBOB with fructose turning hydrophilic and moving outward. As far as we know, this is the first time that these indirect and direct micelle inversions for the same diblock copolymer have been achieved on a bench scale.
Introduction
Spherical micelles composed of a solvophobic core and a solvophilic shell are the most common self-assembled nanostructures in solution. Such structures are normally made of amphiphiles, such as surfactants, polypeptides, block copolymers, dendrimers, etc. Micelles of block copolymers have been extensively studied due to their great interest in theory as well as their promising and broad applications.1–3 Typically a diblock copolymer A-b-B, containing hydrophobic A block and hydrophilic B block, forms core–shell micelles in aqueous media, having A as the core and B as the shell. Changing the medium or imposing an environmental stimulus on the micelle solution may denature the blocks and then alter the roles of A and B, thus realizing micelle inversion. So micellar inversion is a basic and important subject in the studies of self-assembly of block copolymers and is a basis for some of the applications of the assemblies. Intuitively, there are two different pathways for this micelle inversion: (1) indirect inversion: the initial micelles dissociate into molecularly dispersed chains first and then the chains re-assemble to form the inverted micelles; (2) “direct” inversion: the initial micelles switch to the inverted ones directly without molecular dispersion, in which fusion or fission may be involved. An ideal direct process is “in situ” inversion where the process proceeds as the micelles keep their integration without significant polymeric chain loss.
In literature, almost all of the reported micelle inversions followed the first, indirect pathway. The important contributions in the field were from Liu and Armes.4,5 For example, schizophrenic diblock copolymers (A-b-B) underwent two opposite self-assembly directions to form core A–shell B in acidic conditions and core B–shell A in basic conditions. Inversion between the two states can be achieved by adjusting the solution pH constantly. However, when the pH reached the range close to neutrality, both A and B turned soluble, so the molecularly dispersed state became inevitable. This process was indicated by the “trough” shape of evolution of scattered light intensity against pH as in the middle pH range the dispersed unimer gave a very low scattered light intensity. A similar pathway was also observed for the diblock copolymer with blocks A and B having respective LCST (lower critical solution temperature) and UCST (upper critical solution temperature) behavior.6 Enlightened by this design, core–shell inversion has been achieved by using other synthetic block copolymers and block polypeptides.7,8 Recently, similar indirect inversion of micelles via precipitation was also reported by using a pH trigger.9
Searching the numerous data of self-assembly of block copolymers in solutions, we were surprised by the fact that only a limited amount of papers dealt with and succeeded in realizing this direct inversion. Eisenberg et al.10 achieved direct inside-out inversion of vesicles by using a triblock copolymer (PAA26-b-PS890-b-P4VP40), where the presence of the long and glassy middle block of polystyrene as the vesicular wall is the key point to hold the integration of the vesicles against the pH adjustment. Chen et al. of our group achieved micelle-to-vesicle inversion of a diblock copolymer by changing the medium but without detailed investigations on the pathway.11 Generally, the direct and in situ inversion of micelles is more favorable for many applications. For example, in drug delivery the molecularly dispersed state of polymeric drug carriers is detrimental for long circulation and metabolism as well as reducing the toxicity in vivo. The importance of such direct inversion of micelles has drawn some attention of theoreticians. Qiu et al. studied the micellar inversion of diblock copolymers by dissipative particle dynamics (DPD) simulation and concluded that the inversion mainly proceeded in situ without molecularly dispersed intermediates.12 The obvious discrepancy between this result and the most experimental reports mentioned above is understandable as in the simulation, complete and instantaneous denaturation of the blocks is assumed, which of course is very different from the practical experimental conditions. In fact, even in the stopped-flow studies of micellar inversion reported by Liu et al., where external stimuli, pH, temperature and salt concentration variations happened abruptly, the molecularly dispersed intermediates were not fully avoided.7b,13 However, the simulation work pictured the process of in situ inversion in which the speed of the outer blocks moving inward is comparable to that of the core blocks moving outward. In short, it is of great interest and a big challenge to explore the possibility of direct inversion of micelles of block copolymers in practical conditions. Herein we design a novel block copolymer A-b-B where denaturation of A and B is switched by independent stimuli. The stimuli are controllable to make the denaturation of A and B proceed in concord. Thus we are able to realize the core–shell inversion via different pathways, i.e. indirect via dissociation and re-assembly and direct, by just adjusting the program of imposing the stimuli. As far as we know, this is the first time that all these inversion pathways have been achieved in the same block copolymer on a bench scale.
Results and discussion
Polymer design and synthesis of monomer AABOB
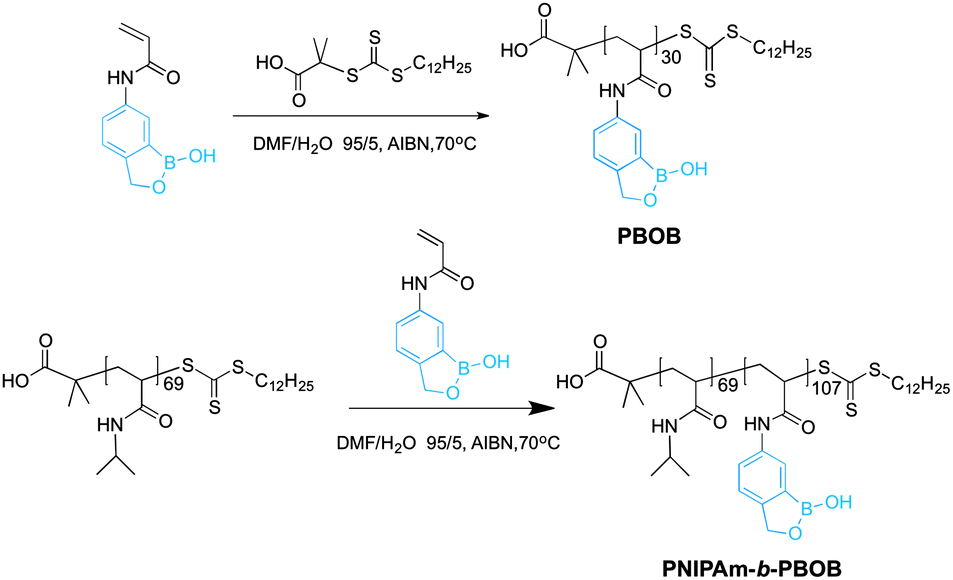
To achieve the indirect and direct core–shell inversion of micelles, a diblock copolymer PNIPAm-b-PBOB with multiple responses to environmental stimuli is designed. PNIPAm was chosen as one of the blocks because of its well-known thermo-induced coil–globule transition behavior. PBOB is special as its denaturation can be triggered by either addition of sugar or increasing pH. We synthesized a monomer containing a benzoboroxole (BOB) group (Scheme S1†), i.e. 1-hydroxy-1,3-dihydrobenzoxaborole, which is hydrophobic at room temperature and neutral pH, and turns to hydrophilic at high pH or after binding with sugars. It is well known that abundant sugar species often exist on cell surfaces, so the resultant micelles of PNIPAm-b-PBOB (M-BOB) are expected to be useful in our further bio-directed studies. BOB is a derivative of phenylboronic acid containing a very stable oxaborole ring and exhibits high stability. More importantly, the oxaborole structure enables BOB to interact with carbohydrates under favorable conditions, i.e. neutral pH,11,14–16 instead of the basic conditions required by traditional phenylboronic acid, although the latter has been introduced into polymers serving as drug carriers by Sumerlin and other research groups.17,18 Styreneboroxole as the first BOB-containing monomer was used for RAFT (Reversible Addition–Fragmentation chain Transfer) polymerization,19 as reported several months ago. Compared to styreneboroxole, the synthetic route of our AABOB was more efficient with a higher overall yield (24%). Briefly, BOB was prepared by treating o-bromobenzyl alcohol with butyllithium followed by addition of triisopropylborate.20 Nitro group was then introduced by fuming HNO3, followed by reduction and treatment with acryloyl chloride to give the desired monomer AABOB (experimental details and 1H NMR characterization (Fig. S1–S5) are in ESI†).
Preparation of homopolymer PBOB and block copolymer PNIPAm-b-PBOB
With AABOB in hand, RAFT was performed to produce homopolymer PBOB. The polymerization was conducted at 70 °C in a mixture of DMF/H2O (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 95:5) with RAFT chain transfer agent (CTA) and AIBN. For preparing the block copolymer, NIPAm was polymerized first at 60 °C in dioxane using DMP (2-dodecylsulfanylthiocarbonyl sulfanyl-2-methylpropionic acid) as the chain transfer agent and AIBN as the initiator (Scheme 1). The number-average molecular weight of the resulting PNIPAm was 8.1 kDa, i.e. DP = 69 from NMR, which served as a macro-CTA for sequential RAFT polymerization of AABOB to yield PNIPAm-b-PBOB block copolymer. The BOB-containing polymer could not be directly characterized by gel permeation chromatography (GPC) because of its limited solubility.9,17b The DP of BOB was calculated as 107 from the ratio of the peak area of CH2 in boroxole ring and that of CH(CH3)2 of NIPAm.
:v = 95:5) with RAFT chain transfer agent (CTA) and AIBN. For preparing the block copolymer, NIPAm was polymerized first at 60 °C in dioxane using DMP (2-dodecylsulfanylthiocarbonyl sulfanyl-2-methylpropionic acid) as the chain transfer agent and AIBN as the initiator (Scheme 1). The number-average molecular weight of the resulting PNIPAm was 8.1 kDa, i.e. DP = 69 from NMR, which served as a macro-CTA for sequential RAFT polymerization of AABOB to yield PNIPAm-b-PBOB block copolymer. The BOB-containing polymer could not be directly characterized by gel permeation chromatography (GPC) because of its limited solubility.9,17b The DP of BOB was calculated as 107 from the ratio of the peak area of CH2 in boroxole ring and that of CH(CH3)2 of NIPAm.
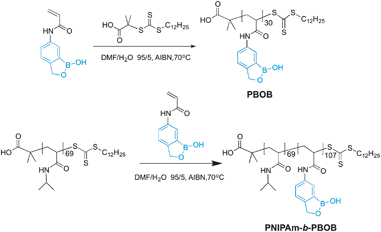 |
| | Scheme 1 Polymerization route of homopolymer PBOB and block copolymer PNIPAm-b-PBOB. | |
Sugar responses of PBOB homopolymer at neutral pH
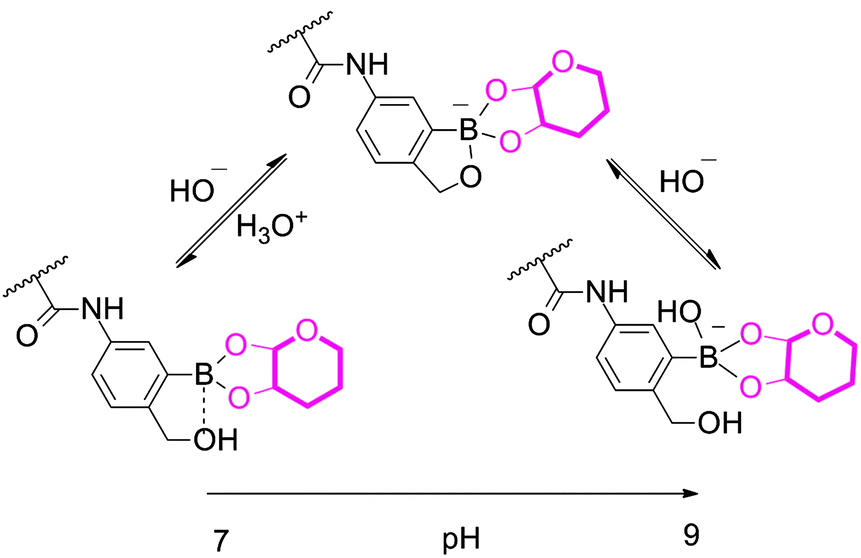
The sugar responsiveness of PBOB homopolymer was first checked by mixing the polymer with different monosaccharides. PBOB (DP = 30) was insoluble in PBS buffer (0.1 M, pH 7.4). To the suspension of PBOB (0.5 mg mL−1), each of four different monosaccharides was individually added. As shown in Fig. 1a, at pH 7.4, fructose (Fru, 50 mM), as the only hemiketal furanose among the four, made PBOB dissolved within 2 h, while glucose (Glc), galactose (Gal) and mannose (Man) did not even at a high concentration (100 mM), although Glc exhibited an obvious tendency to dissolve PBOB. When the pH of PBS buffer (0.1 M) was slightly adjusted from 7.4 to 7.6, PBOB became almost soluble with the three pyranose monosaccharides (Fig. 1b). When pH was further increased to 7.8, clear solutions in all the vials were observed. These results demonstrated the considerable binding effect of the monosaccharides to PBOB at neutral pH. Furthermore, the effect was quite sensitive to the slight adjustment of solution pH. This sugar induced hydrophobicity-to-hydrophilicity transition of PBOB is caused obviously by complexation between BOB and the diol structure of sugar, where the equilibrium between the neutral state and anionic state exists (Scheme 2).
 |
| | Fig. 1 Photographs of PBOB (0.5 mg mL−1) solutions with different monosaccharides in PBS buffer (0.1 M) at (a) pH 7.4, (b) pH 7.6, (c) pH 7.8. (Sample 0: control sample without sugars. Sugar concentration: Fru 50 mM, Glc, Man and Gal 100 mM.) | |
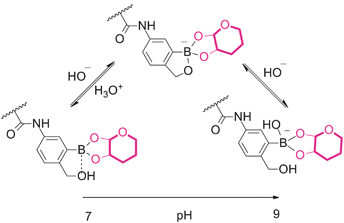 |
| | Scheme 2 Possible complexation equilibrium between BOB group and sugar (adapted from ref. 15). | |
Self-assembly of PNIPAm-b-PBOB in water
PNIPAm-b-PBOB is amphiphilic at room temperature at neutral pH as the block PBOB is hydrophobic under these conditions. Conventional self-assembly procedures were employed for this block copolymer. Briefly, deionized water was added into the polymer solution in DMF (3 mg mL−1, 4 mL) till the polymer concentration reached 1 mg mL−1. After stirring overnight, an equal volume of water was added to the DMF/water mixture followed by dialysis resulting in the stock solution of the micelles. For further characterization, an equal volume of PBS buffer was mixed with the stock solution leading to the final concentration of block copolymer of 0.25 mg mL−1 in PBS buffer (0.05 M, pH 7.4). TEM observations found that black and solid spheres of PNIPAm-b-PBOB have a diameter (DTEM) around 26 nm. This is close to the stretched length of PBOB block with DP of 107 (Fig. 2). However, as shown in Fig. 2, the Rh,app measured by DLS is too large (76 nm) to fit to those spherical micelles although a unimodal distribution (PDI < 0.1) is observed. It is worth mentioning that the pKa of BOB is rather low, around 7.2, so under the present conditions partially negatively charged BOB borate exists. It is known that the electrostatic interactions in polyelectrolyte micelles create an electrostatic field, which fluctuates with the Brownian motion of the micelles and reversely influences their motion dynamics by slowing it down.21 This results in so-called “slow mode” in the distribution of diffusion coefficients. As a consequence, the measured apparent Rh,app is always much larger than the true value for polyelectrolyte micelles.22,23
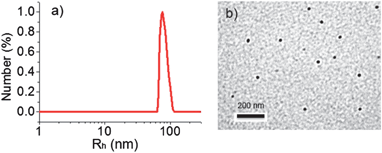 |
| | Fig. 2 (a) Size distribution and (b) TEM image of M-BOB micelles formed by PNIPAm-b-PBOB in pH 7.4 PBS buffer at room temperature (scale bar 200 nm). | |
Micelle response to pH
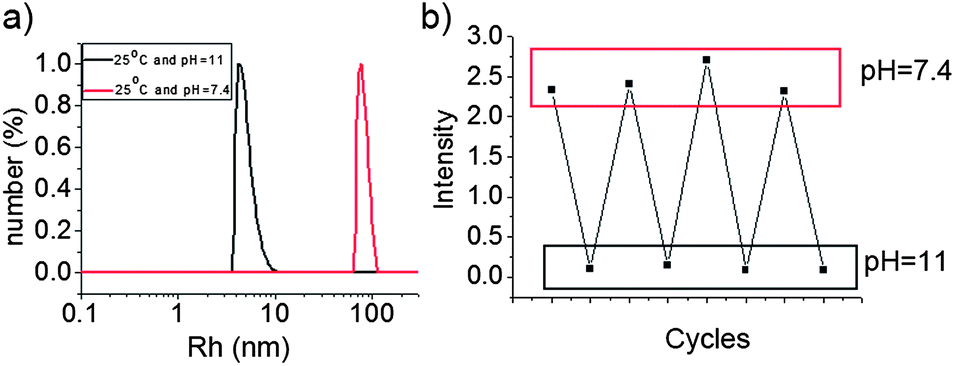
As BOB can be converted to its anionic form under high pH, the water-insoluble PBOB is expected to turn to anionic and then become water-soluble under basic conditions. M-BOB dissociation by pH increase was first performed as follows. When the pH of the M-BOB solution (0.5 mg mL−1) obtained in DMF/water was adjusted to pH 11 with addition of aqueous solution of NaOH (1 M), the hydrodynamic radius distribution of the final solution only exhibited a single peak with Rh,app = 5 nm (Fig. 3a), indicating the dissociation of M-BOB into unimers. Furthermore, this micelle dissociation was proved fully reversible by pH adjustment. As shown in Fig. 3b, addition of NaOH (aq.) to adjust the pH from 7.4 to 11 led to a drastic decrease of the scattered light intensity to close to zero. The light intensity then returned to the initial state when pH decreased to 7.4 by adding HCl (aq.) subsequently. In total, four cycles were performed showing satisfactory reversibility.
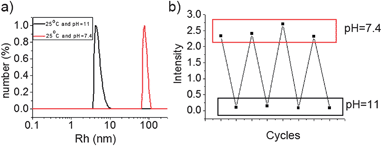 |
| | Fig. 3 (a) Size distribution of M-BOB at pH 7.4 and dissociated unimer at pH 11, (b) the corresponding scattered light intensity changes triggered by pH cycles at 25 °C. | |
Inverted micelle at high pH
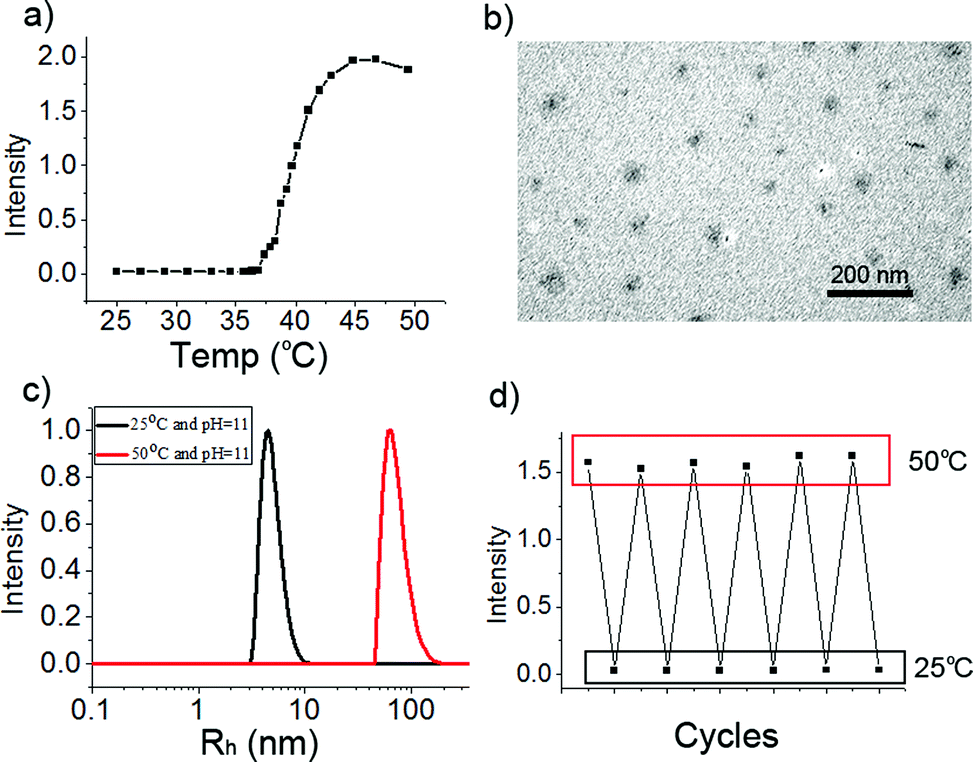
After M-BOB was dissociated in water at pH 11, heating to induce the well-known “coil to globule” transition of PNIPAm block was performed. As shown in Fig. 4a, with increasing temperature, the scattered light intensity kept constant below 37 °C followed by an abrupt increase between 37 °C and 45 °C, which was consistent with the typical phase transition behavior of PNIPAm. So micelles with PNIPAm as core and water-soluble anionic PBOB as shell at high pH were obtained, which were the base-inverted micelles. This inverted micelle was characterized by DLS to have Rh,app as 73 nm at pH 11 and 50 °C (Fig. 4c), and the micelle diameter around 20 nm was found (Fig. 4b) by TEM. Furthermore, reversible micelle formation and dissociation at pH 11 triggered by temperature was clearly demonstrated in several heating and cooling cycles (Fig. 4d).
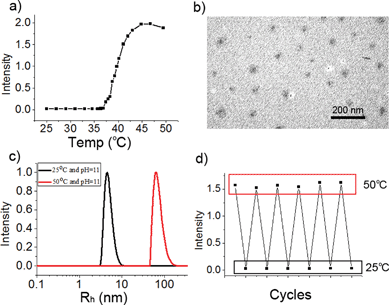 |
| | Fig. 4 (a) Relative scattered light intensity of PNIPAm-b-PBOB as a function of temperature, the intensity was recorded after 30 min standing of the solution (0.5 mg mL−1, pH 11) at a given temperature. (b) TEM image of the inverted micelles formed at pH 11 at 50 °C. (c) Size distribution of the inverted micelles at 50 °C (Rh,app = 73 nm) and the unimers at 25 °C (Rh,app = 5 nm) at pH 11 and (d) corresponding scattered light intensity changes triggered by heating–cooling cycles. | |
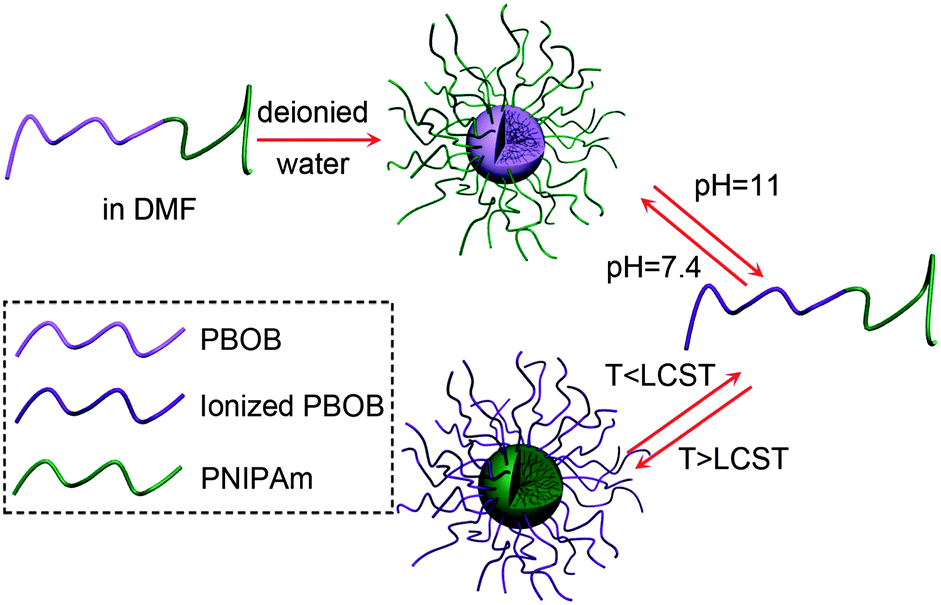
Scheme 3 illustrated the indirect micelle inversion we achieved so far from the observe micelle of PBOB core–PNIPAm shell to the inverted one of PNIPAm core–PBOB shell by sequential pH and temperature changes as well as that from the inverse to the obverse one via the opposite way. Both of the pathways are featured by dissociation into individual chains and then re-association to micelles. Although there are two triggers to use for the two blocks, direct inversion was not achieved because the denaturation of PBOB happens in a very small range of pH, thus it cannot be adjusted accurately to match the denaturation of PNIPAm responsive to temperature. So we need to seek help from sugar binding for PBOB denaturation.
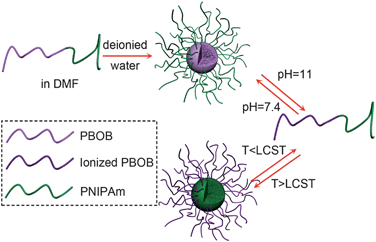 |
| | Scheme 3 Schematic illustration of indirect micelle inversion via molecularly dispersed chains as intermediate, trigged by pH and temperature. | |
Sugar response of block copolymer micelles and further inversion
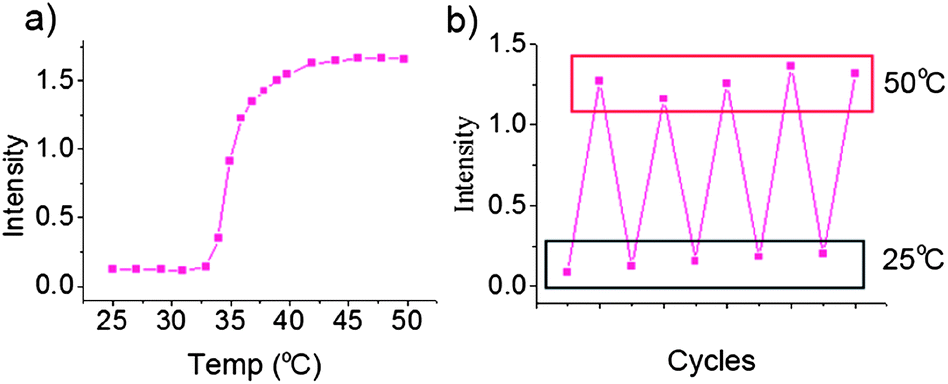
As discussed above, hydrophobic homopolymer PBOB turns to hydrophilic when BOB forms a complex with sugar. This complexation effect with sugars on the self-assembly behavior of the copolymers was then investigated. As shown in Fig. 5, after addition of monosaccharide Fru to the solution of M-BOB at 25 °C, a continuous decrease of the scattered light intensity was observed. Surprisingly, the process lasted as long as about 5 days to reach the final, stable and very low intensity, showing that the copolymer became molecularly dispersed as evidenced by a measured Rh,app as small as 8 nm (Fig. 5b). The process being much slower than that for sugar-homopolymer case, as we just discussed, could be attributed to the difficulty of diffusion of sugar into the hydrophobic core of PBOB of the micelle. The resultant sugar-bonded PNIPAm-b-PBOB unimer at pH 7.4 can further self-assemble into inverted micelles after heating to 32 °C (Fig. 6a) and the process was fully reversible (Fig. 6b). This is a new route to realize the indirect inversion.
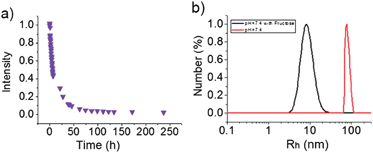 |
| | Fig. 5 (a) Decrease of scattered light intensity (Is/I0) of M-BOB prepared at 25 °C and pH 7.4 PBS buffer after addition of Fru (100 mM); (b) size distribution of M-BOB at 25 °C in 7.4 PBS at the beginning (red line) and the end (black line) of 100 mM Fru treatment. | |
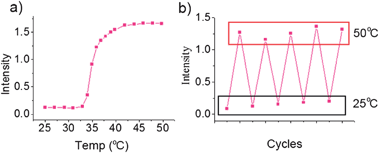 |
| | Fig. 6 (a) Relative scattered light intensity of Fru-bonded unimer PNIPAm-b-PBOB (0.25 mg mL−1 in PBS pH = 7.4 with 200 mM Fru) as a function of temperature indicating the formation of inverted micelles. (b) Corresponding scattered light scattering intensity changes triggered by heating–cooling cycles. | |
Direct micelle inversion above LCST
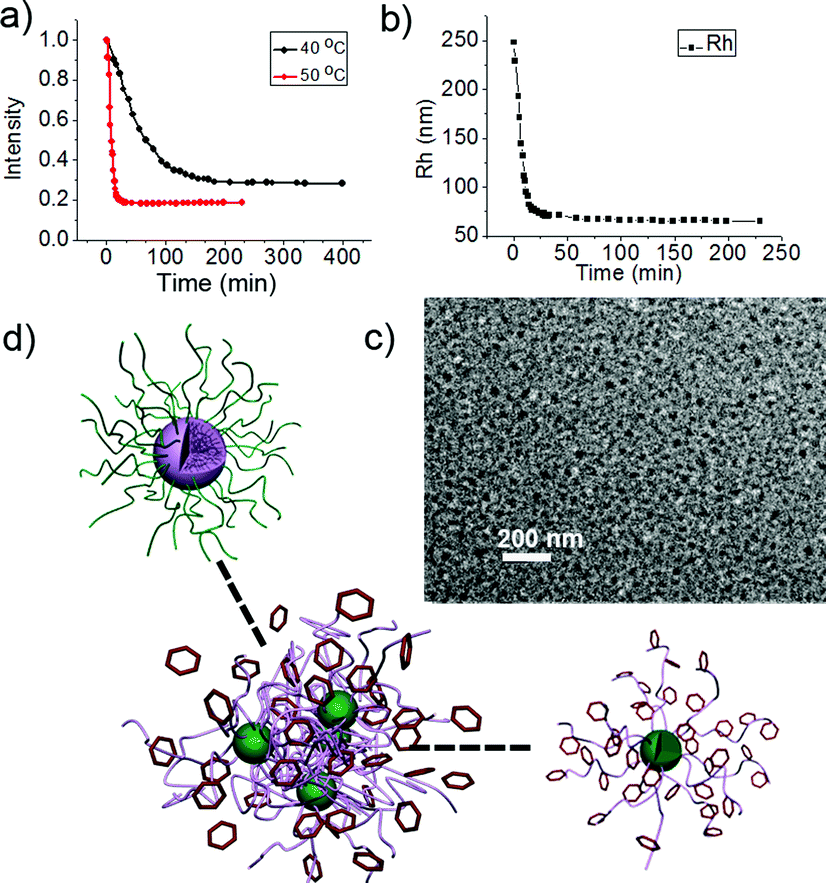
Here the two triggers, sugar and temperature, are used simultaneously to achieve direct micelle inversion. A solution of M-BOB in PBS (pH 7.4) was immediately placed into an incubator at 50 °C after addition of Fru, which was far above the LCST of PNIPAm. An abrupt decrease of the scattered light intensity was observed within the first 25 min prior to the intensity reaching a stable value around one fifth of the starting micelles (Fig. 7a, red line). Compared to the curve in Fig. 5a at 25 °C, the intensity decrease in Fig. 7a (red line) at 50 °C was so sharp and quick that the duration needed to reach equilibrium was shortened to 1/240. More importantly, the final normalized light intensity was stable at around 0.2 rather than the low value of single chains. Meanwhile a continuous quick decrease of Rh,app to a stable low value in this process was observed (Fig. 7b). So the changes in both light intensity and Rh,app indicate the formation of some small aggregates. The resultant aggregates were found to have irregular shape and a size about 10 nm under TEM (Fig. 7c). NMR measurements of the micelles before and after heating at 50 °C were performed. A decrease of the integrated area of CH(CH3)2 of PNIPAm and an increase of the phenyl group on oxaborole ring was clearly observed (Fig. S7†), which reflected the position exchange between the components in the core and shell. Furthermore, as shown in Fig. 8a, zeta-potential measurement monitoring the process displayed a drastic drop of the potential from −5 mV to −30 mV as a result of the PBOB block turning to ionic and moving to the surface of the small micelles. Combing all the results, we could conclude that under the simultaneous stimuli of interacting with sugar and quickly increasing temperature to 50 °C, direct inversion of M-BOB took place, which was accompanied by micellar fission (Fig. 7d).
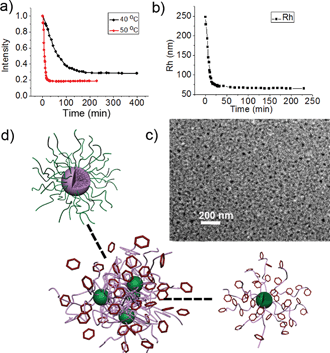 |
| | Fig. 7 (a) Scattered light intensity at 50 °C and 40 °C and (b) Rh,app of micelles at 50 °C for M-BOB in the presence of Fru as function of time. (c) TEM image of inverted micelles formed at the end of the process at 50 °C. (d) A scheme illustrating the direct inversion with micellar fission. | |
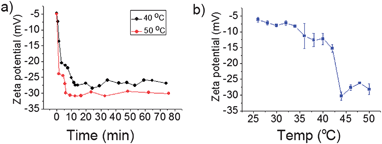 |
| | Fig. 8 Zeta potential (ζ) profiles of micelle M-BOBvia (a) directing heating at 50 °C and 40 °C; or (b) step-by-step temperature increase in the presence of Fru. | |
Direct inversion was performed at 40 °C as well. As shown in Fig. 7a (black line), a much slower scattered light intensity decrease compared to that at 50 °C was observed. As shown in Fig. 8a, at both 50 °C and 40 °C, the zeta potential went down abruptly in about 10 min before it became stable around −26 mV for 40 °C (black line) and −30 mV for 50 °C (red line). Such a high speed of zeta potential decrease implies that the diffusion of sugar into the core and the subsequent complexation and moving of the ionized blocks to the micellar surface are very quick. This process was probably promoted by the rapid agglomeration of PNIPAm to form microphase structure on the surface of the micelles. This idea was proved by an additional experiment shown in Fig. 8b where zeta potential was traced with a much slower increasing temperature program, i.e. one degree per 30 min. In this case, the potential varied a little until 40 °C and then a moderate and a drastic decrease took place subsequently in the range of 40–44 °C corresponding the coil–globule transition of the PNIPAm blocks. In other words, the complexation of PBOB was largely accelerated at the temperature above the LCST of PNIPAm block, which made micellar fission inevitable. Therefore, in order to realize an inversion without obvious fission, delicate temperature programming is necessary.
Direct inversion via programmed heating
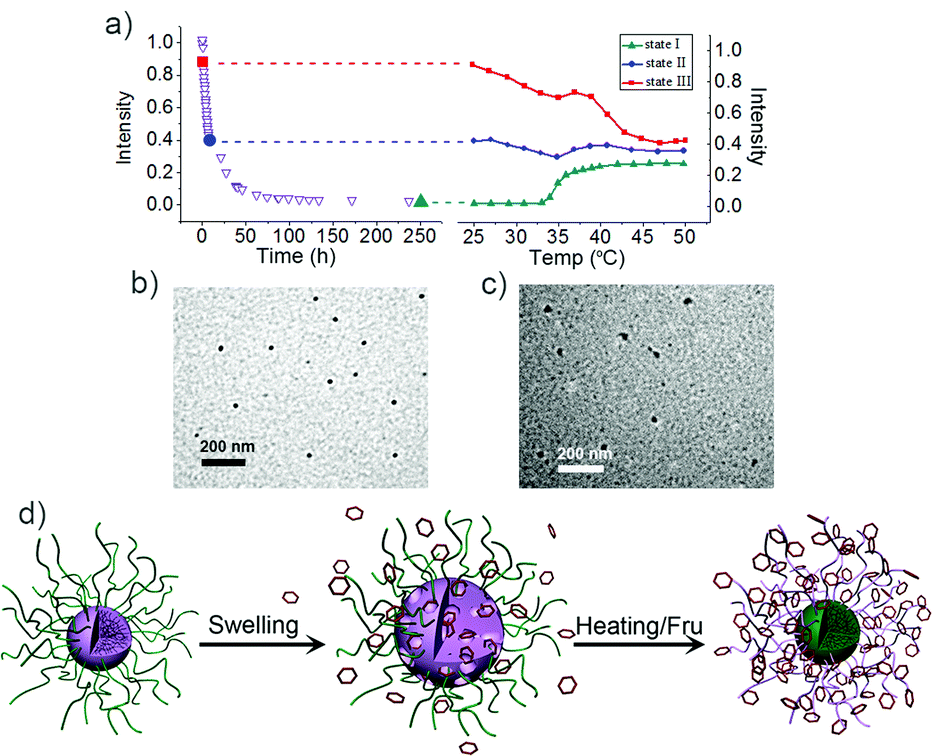
As mentioned above, M-BOB displayed a long continuous decrease of the scattered light intensity until it reached a very low single-chain featured value when the solution was treated with Fru at pH 7.4 and 25 °C. The corresponding change in the intensity–time correlation functions in this process is shown in Fig. S8† where the correlation function kept moving to longer decay time without any obvious indication of loss of polymer chains during the first 45 h, this meant increased swelling of the micelles due to the gradual increase of the hydrophilicity of the PBOB blocks. Similar tendency and explanation was reported in the literature.24 After 45 h the correlation function began to shift back, which indicated fission or even dissociation became dominant, as the PBOB block sufficiently complexed with Fru. This long duration of the complexation between the core of PBOB and Fru at 25 °C provides a good opportunity for us to design new procedures to control the inversion pathways: when heating is initiated from different states of the micelles, i.e. with different swelling degrees, which means different mobility of the core blocks, the swollen micelles may follow different ways to the final inversion. As shown in Fig. 9, three representatives, i.e. states I (green), II (blue) and III (red), produced after the copolymer interacted with Fru at 25 °C for 250 h, 6 h and 2 h respectively, were selected to perform the subsequent temperature-induced micelle inversion. The temperature was elevated by 2 °C per minute and kept constant for 30 min at each temperature for light scattering measurements.
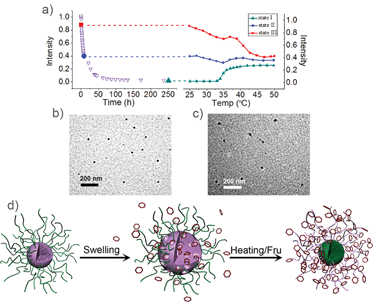 |
| | Fig. 9 (a) The evolution of scattered light intensity during the heating process started from state I–III as marked in the left curve, a copy of Fig. 5a. (b) TEM of M-BOB micelles formed by PNIPAm-b-PBOB in pH 7.4 PBS buffer at room temperature (scale bar 200 nm). (c) TEM of the inverted micelles via state of II. (d) Proposed schematic mechanism for in situ micelle inversion. | |
First, as a reference we discuss state I (green curve, Fig. 9), where heating started after 250 h treatment of the micelle with Fru. So it clearly showed the indirect inversion via the sugar-bonded individual chains of PNIPAm-b-PBOB (the same as that in Fig. 6a). For state III, the reaction with Fru at 25 °C was just initiated prior to heating. So at the beginning of heating the core is fully hydrophobic. The scattered light intensity over the whole temperature range generally showed a decreasing trend until it reached the plateau of the inverted micelle, reflecting the continuous swelling of the core due to PBOB becoming hydrophilic. However, in the temperature range 35 °C–40 °C, an apparent intensity increase was observed. In this temperature range around the LCST, PNIPAm blocks quickly agglomerated while PBOB blocks were still less hydrophilic so hardly moved towards the surface. Therefore, the intensity increase reflected collision and fusion of the micelles covered with the collapsed PNIPAm. In short, for the state III, the direct core–shell inversion took place accompanying micellar fusion. This direct micelle inversion process in such programmed heating was monitored by 1H NMR. As shown in Fig. 10a, at room temperature, for the solution of M-BOB in Fru solution (PBS pH 7.4), the methyl proton signals from the isopropyl groups of the PNIPAm block at 1.0 ppm can be clearly observed, whereas the signals for the aromatic protons of BOB on the PBOB blocks were not obvious although detectable. This result indicated that the PNIPAm block was solvated on the micelle shell and the hydrophobic PBOB block was embedded in the core and solvated poorly. As temperature was increased step by step, the integral area from PNIPAm steadily went down especially in the temperature range of 30 °C to 45 °C. Meanwhile, the integration of the PBOB block around 6.3–7.5 ppm increased significantly. In Fig. 10b, the molar ratio of PBOB vs. PNIPAm against temperature kept increasing from 35 °C, which further demonstrated the inversion process.
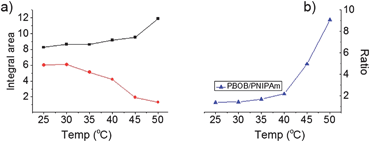 |
| | Fig. 10 Relative intensities of 1H NMR peaks normalized to the intensity at 25 °C at 1.05 ppm for M-BOB in buffer (0.25 mg mL−1) ((a) black dots: integrated area of phenyl group of PBOB, red dots: integrated area of CH(CH3)2 of PNIPAm, original 1H NMR spectra are in Fig. S9†). | |
State II shows an interesting result, for which in the whole heating process, except a slight fluctuation observed around 35 °C, the intensity was basically stable. When the temperature started increasing, the core of state II had been obviously swollen so the PBOB blocks were no longer tightly fixed but the core still had enough hydrophobicity to prevent fission. So in the subsequent heating to around LCST of PNIPAm, the speed of chain agglomeration and penetration into the core may match that of the further ionization of the PBOB block and movement to the surface. The TEM images in Fig. 9b and c also confirmed spherical micelle structures with a diameter around 30 nm obtained at the end of heating, close to the starting micelle M-BOB. In other words, in this case, the core–shell inversion took place without apparent fission or fusion, i.e. more close to so-called in situ inversion, as illustrated in Fig. 10d. In summary, thanks to the rather slow process of sugar Fru binding to the PBOB core of M-BOB at room temperature, different micelle inversion pathways, including those accompanied by fusion or fission or close to the in situ case, can be realized by starting different heating processes from micelles with different degrees of swelling.
Conclusions
In this paper, by using a novel block copolymer PNIPAm-b-PBOB, thanks to its multiple responses to pH and sugar for PBOB and to temperature for PNIPAM, different core–shell inversion kinetics of the micelle were realized. The indirect process with dispersed polymer chains as intermediate was performed by sequential pH and temperature change and the processes are demonstrated to be totally reversible. The direct inversion without single chains as intermediate on a bench scale, which had not been reported before, was successfully realized by adjusting the sugar interaction and heating program. The key point to realize such direct inversion, which is favorable for many applications of micelles, is to make the denaturation of the two blocks occur in concord. This success is expected to develop further to a general strategy to control the core–shell inversion kinetics for different block copolymer micelles with tunable time scales.
Acknowledgements
Ministry of Science and Technology of China (2009CB930402 and 2011CB932503), National Natural Science Foundation of China (No. 91227203 and 51322306) and the Shanghai Rising-Star Program (Grant 13QA1400600) are acknowledged for their financial support.
Notes and references
-
M. Jiang, A. Eisenberg, G. Liu and X. Zhang, Macromolecular Self-assembly (in Chinese), Science Press, Beijing, 2006 Search PubMed.
-
M. Lazzari, G. J. Liu and S. Lecommandoux, Block Copolymers in Nanoscience, Wiley-VCH, Weinheim, 2006 Search PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- S. Liu, N. C. Billingham and S. P. Armes, Angew. Chem., Int. Ed., 2001, 40, 2328 CrossRef CAS.
- S. Liu and S. P. Armes, Angew. Chem., Int. Ed., 2002, 41, 1413 CrossRef CAS.
- M. Arotçaréna, B. Heise, S. Ishaya and A. Laschewsky, J. Am. Chem. Soc., 2002, 124, 3787 CrossRef PubMed.
-
(a) J. Rodríguez-Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026 CrossRef PubMed;
(b) X. Jiang, G. Zhang, R. Narain and S. Liu, Langmuir, 2009, 25, 2046 CrossRef CAS PubMed.
-
(a) S. Guragain, B. P. Bastakoti, S.-I. Yusa and K. Nakashima, Polymer, 2010, 51, 3181 CrossRef CAS PubMed;
(b) X. J. Loh and J. Appl, Polym. Sci., 2013, 127, 992 CAS.
- J. Zou, S. Zhang, R. Shrestha, K. Seetho, C. L. Donley and K. L. Wooley, Polym. Chem., 2012, 3, 3146 RSC.
- F. Liu and A. Eisenberg, J. Am. Chem. Soc., 2003, 125, 15059 CrossRef CAS PubMed.
- G. Hou, L. Zhu, D. Chen and M. Jiang, Macromolecules, 2007, 40, 2134 CrossRef CAS.
- B. Hong, F. Qiu, H. Zhang and Y. Yang, J. Chem. Phys., 2010, 132, 244901 CrossRef PubMed.
- D. Wang, J. Yin, Z. Zhu, Z. Ge, H. Liu, S. P. Armes and S. Liu, Macromolecules, 2006, 39, 7378 CrossRef CAS.
- M. Dowlut and D. G. Hall, J. Am. Chem. Soc., 2006, 128, 4226 CrossRef CAS PubMed.
- M. Bérubé, M. Dowlut and D. G. Hall, J. Org. Chem., 2008, 73, 6471 CrossRef PubMed.
- S. Burroughs and B. Wang, ChemBioChem, 2010, 11, 2245 CrossRef CAS PubMed.
-
(a) P. De, S. R. Gondi, D. Roy and B. S. Sumerlin, Macromolecules, 2009, 42, 5614 CrossRef CAS;
(b) D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2008, 2477 RSC;
(c) D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106 RSC.
-
(a) B. Wang, R. Ma, G. Liu, Y. Li, X. Liu, Y. An and L. Shi, Langmuir, 2009, 25, 12522 CrossRef CAS PubMed;
(b) B. Wang, R. Ma, G. Liu, X. Liu, Y. Gao, J. Shen, Y. An and L. Shi, Macromol. Rapid Commun., 2010, 31, 1628 CrossRef CAS PubMed;
(c) R. Ma, H. Yang, Z. Li, G. Liu, X. Sun, X. Liu, Y. An and L. Shi, Biomacromolecules, 2012, 13, 3409 CrossRef CAS PubMed;
(d) G. Liu, R. Ma, J. Ren, Z. Li, H. Zhang, Z. Zhang, Y. An and L. Shi, Soft Matter, 2013, 9, 1636 RSC.
- H. Kim, Y. J. Kang, S. Kang and K. T. Kim, J. Am. Chem. Soc., 2012, 134, 4030 CrossRef CAS PubMed.
- V. V. Zhdankin, P. J. Persichini, L. Zhang, S. Fix and P. Kiprof, Tetrahedron Lett., 1999, 40, 6705 CrossRef CAS.
- O. Bertrand, C.-A. Fustin and J.-F. Gohy, ACS Macro Lett., 2012, 1, 949 CrossRef CAS.
- S. Förster, N. Hermsdorf, C. Böttcher and P. Lindner, Macromolecules, 2002, 35, 4096 CrossRef.
- M. Sedlák, Langmuir, 1999, 15, 4045 CrossRef.
- D. Roy and B. S. Sumerlin, ACS Macro Lett., 2012, 1, 529 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of PBOB and PNIPAm-b-PBOB including 1H NMR and GPC results, and details of DLS and zeta-potential measurements. See DOI: 10.1039/c3py00944k |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.