Reversible photocontrol of self-assembled peptide hydrogel viscoelasticity†
Todd M.
Doran
,
Derek M.
Ryan
and
Bradley L.
Nilsson
*
Department of Chemistry, University of Rochester, RC Box 270216, Rochester, NY 14627, USA. E-mail: nilsson@chem.rochester.edu; Fax: +1 585 276-0205; Tel: +1 585 276-3053
First published on 3rd September 2013
Abstract
Peptide hydrogels are promising biomaterials for applications ranging from drug delivery to tissue engineering. Peptide hydrogels that change their physical properties in response to an exogenous stimulus are advantageous as biomaterials that can be temporally controlled. Herein, we report the use of an azobenzene turn mimetic, [3-(3-aminomethylphenylazo)phenyl]acetic acid (AMPP), to engineer a light-responsive β-hairpin into the center of a hydrogel-forming peptide. In the trans state, AMPP exists in a β-arc conformation, and the peptide forms a rigid self-supporting gel. The peptide hydrogel rigidity is reduced upon trans–cis azobenzene isomerization, which promotes formation of putative β-hairpin assemblies. This process is reversible in that hydrogel viscoelasticity is restored upon reverse cis–trans photoisomerization. TEM imaging and spectroscopic data reveal that the loss in rigidity is a result of disruption of the well-ordered macromolecular structure and not due to disassembly of the constituent self-assembled β-sheet fibrils. These findings provide insight into the effect of β-arc and β-hairpin turns on the emergent properties of self-assembled peptide hydrogels and provide a basis for temporal control of hydrogel rigidity using near-UV light.
Introduction
Self-assembled peptide hydrogels have emerged as potential platforms for drug delivery, tissue engineering and biosensing.1–6 Peptide self-assembly into amyloid-like fibrils is driven by noncovalent hydrophobic, electrostatic and van der Waal's interactions. The resulting self-assembled fibrils entangle to form a network that immobilizes flow of water to provide self-supporting hydrogels that are strengthened by noncovalent cross-linking. Peptide-derived hydrogels are biocompatible, non-immunogenic, easily functionalized, and tunable in regard to mechanical rigidity.7 These properties make peptides ideal noncovalent hydrogel materials for biological applications.Hydrogels that respond to an exogenous stimulus to temporally modulate hydrogel rigidity and gel–sol/sol–gel transitions are desirable in the development of hydrogels for biomedical applications.2,8,9 Environmental pH,10 redox potential,11 temperature,12 ionic strength,13 enzyme activation,14,15 and light16,17 have been exploited to promote self-assembly and hydrogel formation. Specifically, designed amphipathic peptides containing β-hairpins have been employed to form well-ordered hydrogels that can be reversibly manipulated using pH,18 temperature,12,19 and ion complexation.20 In these cases, formation of the β-hairpin leads to desirable hydrogel properties through noncovalent cross-linking. Whereas these examples rely on β-hairpin folding for hydrogel formation, we envisaged using an azobenzene β-hairpin mimetic that could provide responsiveness by disrupting peptide secondary structure using light to introduce the β-hairpin turn.
Azobenzene has been used extensively to manipulate the macromolecular structure of peptide-based materials.21–31 Examples include the modulation of organo- and hydrogel rigidity with varying degrees of success.32–35 In general, azobenzene groups have been appended to the N-terminus of peptides in hydrogels due to synthetic convenience; cis–trans isomerization results in variance of hydrogel properties due to changes in the steric profile of the appended azobenzene.34,35 We have recently reported the use of turn nucleators, including azobenzene, to promote the self-assembly of amyloid-β and found that specific β-turn conformations have significant consequences on the self-assembly properties of the peptide. For example, when DPG, a type II′ β-hairpin mimetic, was incorporated into the putative turn region of Aβ, it self-assembled rapidly compared to the wild-type peptide.36 However, when the type I′ β-hairpin nucleator, cis-azobenzene, was incorporated into the same region of Aβ, self-assembly occurred, but not into well-ordered β-sheets.37 In a similar study, azobenzene located within the central hydrophobic core of Aβ disrupted fibrils upon trans–cis isomerization.38 Based on these findings, we hypothesized that in the context of a peptide hydrogel, cis–trans isomerization of a centrally located azobenzene backbone mimetic would result in a more global disruption of the overall hydrogel structure relative to the minor steric changes that occur when azobenzene is placed at a terminus.
We used an azobenzene backbone mimetic to modulate the secondary structure of a peptide known to form hydrogels in order to disrupt hydrogel order. [3-(3-aminomethylphenylazo)phenyl]acetic acid (AMPP) was incorporated into the (RADA)4 peptide. (RADA)4 readily forms hydrogels in saline solutions.39–43 We envisaged that trans-AMPP in a bent conformation would form a β arc, similar to those found in several naturally occurring peptide fibrils and thus leaving it competent to self-assemble into cross-β fibrils.44–50 In the cis conformation, however, the peptide would adopt a type I′ β-hairpin that would self-assemble into alternative β-hairpin fibrils. We found that AMPP in the trans conformation in (RADA)4 peptide assembled into a self-supporting hydrogel. When photoisomerized to the cis conformation, the (RADA)4 hydrogel weakened. Hydrogel rigidity was restored upon cis–trans photoisomerization. These results demonstrate that β-hairpin nucleation can be exploited to disrupt peptide quaternary structure leading to the disruption of the emergent properties of the resulting hydrogels.
Results
Peptide design and rationale
To assess our hypothesis that a centrally located photoswitch can perturb the viscoelastic properties of a hydrogel upon cis–trans isomerization, we incorporated the photochromic [3-(3-aminomethylphenylazo)]phenyl acetic acid (AMPP, Fig. 1A) into the (RADA)4 peptide. AMPP has been used previously as a β-hairpin nucleator in peptides.23,24,29,37,51 In cases where β-hairpin nucleation occurs in the context of an amyloid peptide, trans → cis isomerization leads to a decrease in the ordered structure of peptide fibrils.29,37 We hypothesized that inclusion of AMPP in a central location in (RADA)4 peptides would facilitate light-triggered structural transitions from extended or bent β-sheet in the trans AMPP configuration, (Fig. 1A, left and center) to β-hairpin in the cis configuration, (Fig. 1A, right). We also anticipated that this transition would perturb the hydrogel network, resulting in modulation of the emergent viscoelastic properties of the bulk material. This would be advantageous over terminally located azobenzene moieties because AMPP in a central location could lead to a more global disruption of higher-order structure. We also anticipated that the flexibility of the AMPP amino acid could lead to extended cross-linking, increasing hydrogel strength in the trans conformation (Fig. 1B). Another advantage of using AMPP is that trans → cis isomerization occurs rapidly at 365 nm, which is amenable to studies using the hydrogel for potential cell culture applications.52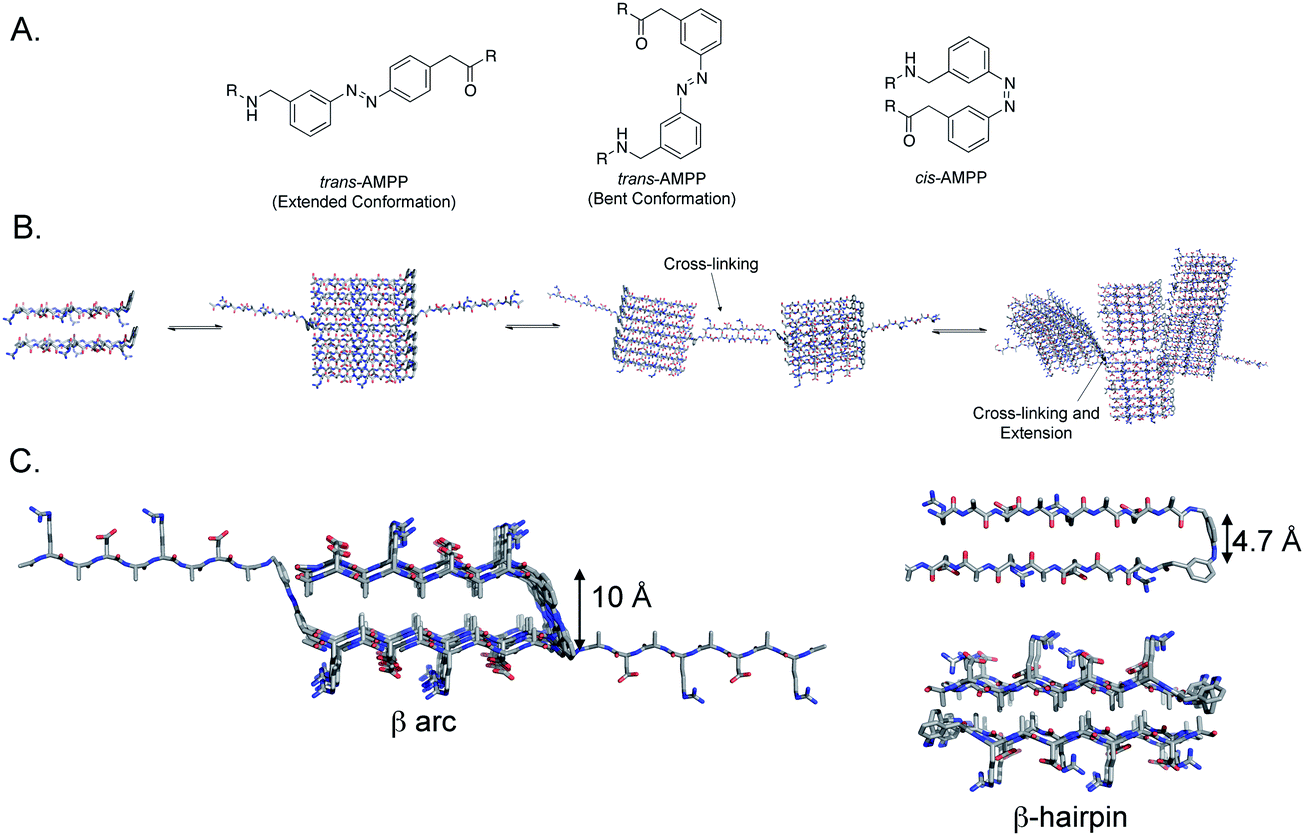 | ||
| Fig. 1 A. Chemical structures of the AMPP photoswitch in the extended-trans, bent-trans, and cis conformations. B. AMPP-containing (RADA)4 self-assembly scheme; cross-linking is shown as extended β-sheets nucleate interfibril interactions at branching points, leading to crosslinking and stabilization of the fibril hydrogel network. C. Model of peptide 1 folding in the trans (left) and cis (right) conformation. When azobenzene is in the trans conformation, the peptide adopts a strand-turn-strand motif resulting in a β arc. The flexible methylene units proximal to the azobenzene chromophore permit strand-swapping between pre-formed fibrils. In the cis conformation, a β-hairpin conformation results (upper right) that forms fibrils that are a bilayer of β-hairpin filaments that sequester the alanine side chain groups to the core of the bilayer (lower right). | ||
The Fmoc-AMPP-OH monomer was synthesized as described previously53 and incorporated into the center of the amphipathic Ac-(RADA)4-NH2 peptide in order to assess the effect of cis–trans isomerization on the self-assembly and hydrogelation behavior of this peptide.39,40,42 The Ac-(RADA)2-AMPP-(RADA)2-NH2 peptide (peptide 1, see ESI, Fig. S1†) was synthesized by standard Fmoc solid-phase peptide synthesis. Following HPLC purification, the peptide was isolated in the thermodynamically favored trans AMPP configuration (see ESI† for characterization and purification protocols).
Rheological properties of peptide 1
The self-assembly and hydrogelation behavior of peptide 1 was assessed. When dissolved in water (0.5 wt%, ∼3 mM) in the trans conformation (trans-1), the peptide self-assembled into β-sheet fibrils but failed to form an elastic hydrogel. At higher ionic strength (20 mM NaCl), trans-1 (0.5 wt%) formed an optically transparent, self-supporting gel (Fig. 2A). Irradiation at 365 nm for 15 min affected trans–cis isomerization of peptide 1. Photoisomerization of trans-1 hydrogel fibril assemblies to cis-1 decreased hydrogel viscoelasticity as evidenced by an apparent weakening of gel strength from a self-supporting gel to a free-flowing fluid (Fig. 2B). Hydrogel strength was regained upon cis–trans isomerization by irradiation at 434 nm for 15 min as evidenced by the reformation of a self-supporting gel (Fig. 2C). | ||
| Fig. 2 A. Digital photograph of 0.5 wt% self-supporting hydrogel of self-assembled trans-1. B. Digital photograph of trans-1 to cis-1 networks after irradiation for 15 min at 365 nm showing loss of rigidity. C. Digital photograph of 0.5 wt% solution of cis-1 to trans-1 after irradiation of cis-1 networks at 430 nm for 15 min exhibiting re-emergence of hydrogel stiffness. | ||
The rheological viscoelasticity of the hydrogels was measured using a dynamic frequency sweep from 0–50 rad s−1 with 0.2% strain (in the linear viscoelastic region). Trans-1 hydrogels had a G′ value of 260 ± 60 Pa and a G′′ of 30 ± 10 Pa; the hydrogels were stable up to oscillatory frequencies of 50 rad s−1 (Fig. 3A). Rheological analysis of cis-1-enriched assemblies showed a significant reduction of viscoelasticity compared to trans-1 networks with G′ value of 80 ± 20 and G′′ of 7 ± 2 (Fig. 3B). Upon regaining a population of trans-1 after irradiation at 434 nm, the viscoelastic strength of the gel was recovered (G′ = 310 ± 80, G′′ = 30 ± 10, Fig. 3C). The slight increase in G′ in trans-1 following re-isomerization was attributed to water evaporation during the photoisomerization step as well as evaporation during rheological measurements throughout the experiment. Nonetheless, the numbers fall within standard error.
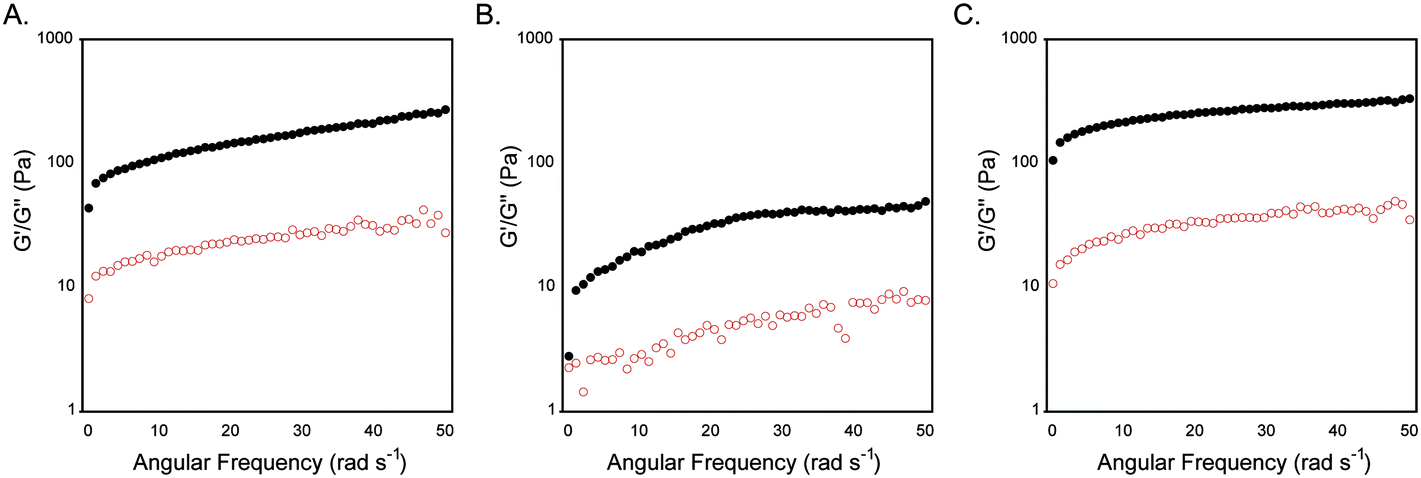 | ||
| Fig. 3 A. Dynamic frequency sweep of gel networks obtained from 0.5 wt% trans-1 in 20 mM NaCl. B. Dynamic frequency sweep of 1 after irradiation at 365 nm to affect trans–cis isomerization. C. Dynamic frequency sweep of hydrogel obtained after reisomerization to trans-1 after irradiation at 434 nm. | ||
Morphology of fibrils derived from peptide 1
It is well-established that photoisomerization of azobenzene groups between the cis and trans conformations does not proceed with complete fidelity in either direction.23 We therefore conducted studies to determine the degree of trans–cis and cis–trans isomerization within the self-assembled fibril network in order to understand how this influences the emergent viscoelastic properties of the network. Azobenzene molecules undergo a coincident change in absorbance properties upon cis–trans isomerisation. The extent of isomerization can be monitored, at least qualitatively, by UV spectroscopy. The thermodynamically favored trans-azobenzene conformation is characterized by a maximum at 330 nm; upon conversion to the cis orientation, the magnitude of this maximum is diminished and a shifted maximum at ∼430 nm is observed.23 As expected, the trans-1 fibril network exhibited a maximum intensity at 330 nm that diminished upon isomerization to cis-1 (Fig. 4B). A weak maximum was observed at 430 nm in the cis-enriched fibrils. Re-isomerization to trans-1 completely restored the expected UV signature (Fig. 4B). These data suggest that trans–cis photoisomerization does not go to completion in the context of self-assembled peptide fibrils and that the trans orientation is favored. The degree of isomerization was quantified by disaggregation of the fibril network after trans–cis conversion by dissolution in 40% acetonitrile/water (v/v) and determination of cis and trans ratios via HPLC analysis. It was found that ∼70% of trans-1 is converted to cis-1 upon irradiation at 365 nm for 15 min (Fig. S2 and S3†). It should be noted that the obligatory disaggregation step likely facilitates some trans–cis conversion to shift back to the thermodynamically favored trans conformation, although we expect that this shift does not occur beyond ∼5% more trans isomer; however, with this knowledge, the percent conversion reported is likely an underrepresentation.54,55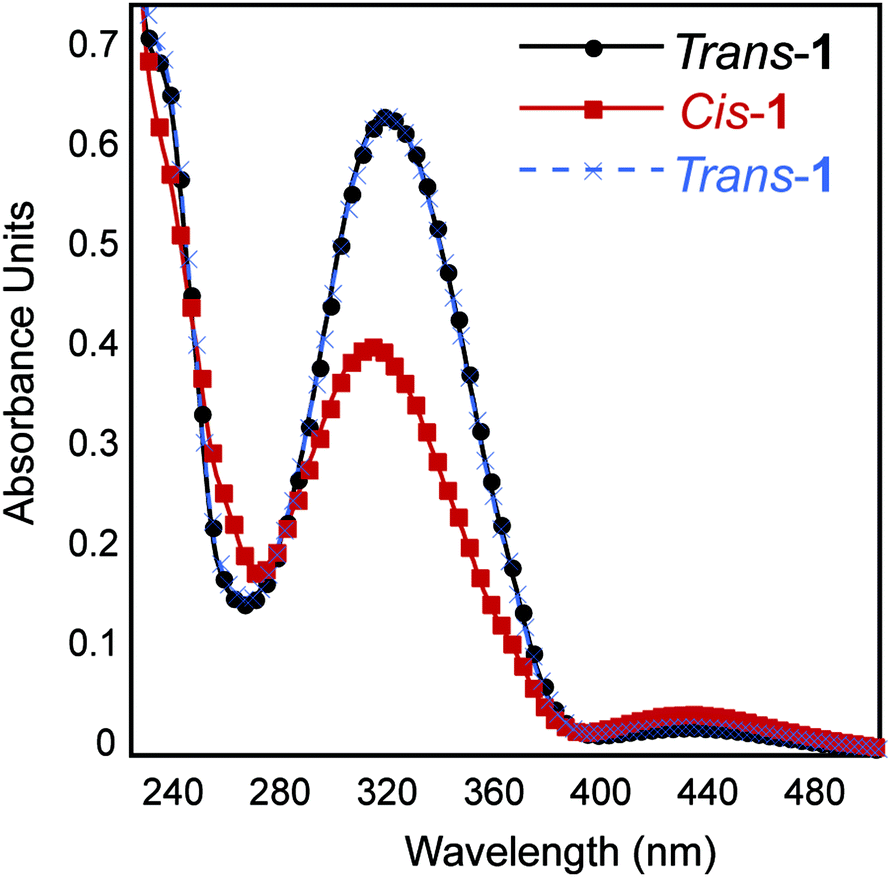 | ||
| Fig. 4 UV-Vis absorbance spectra of 0.5 wt% assembled trans-1 and cis-1 in 20 mM NaCl. | ||
The secondary structure of the hydrogel was examined using circular dichroism (CD) spectroscopy. The CD spectrum of a 0.5 wt% trans-1 solution displayed a minimum at 218 nm, characteristic of β-sheet structure (Fig. 5). The CD spectrum also showed a local minimum at ∼330 nm, which is evidence that the trans-azobenzene is held in a chiral framework within the fibril network. The CD spectrum of cis-1-enriched fibrils showed a minimum at 218 nm, but with reduced intensity compared to trans-1 fibrils, indicating the presence of β-sheet, albeit at lower concentrations (Fig. 5). In the cis conformation, while β-sheet secondary structure exists, it is unlikely that fibrils are stacked in a registry that would produce π–π stacking. It is surprising that although the trans–cis conversion is incomplete, the CD spectrum exhibits a complete loss of signal at 330 nm. One explanation is that while only 70% conversion is recorded, this is an underestimate due to the stringent conditions imposed to disaggregate the fibrils before injection onto an HPLC column. Alternatively trans–cis conversion probably leads to an overall disruption of the well-ordered trans-1 fibrils. Therefore, while trans-conformers remain, the macromolecular arrangement that lead to the chiral trans-1 fibrils has been largely disrupted.
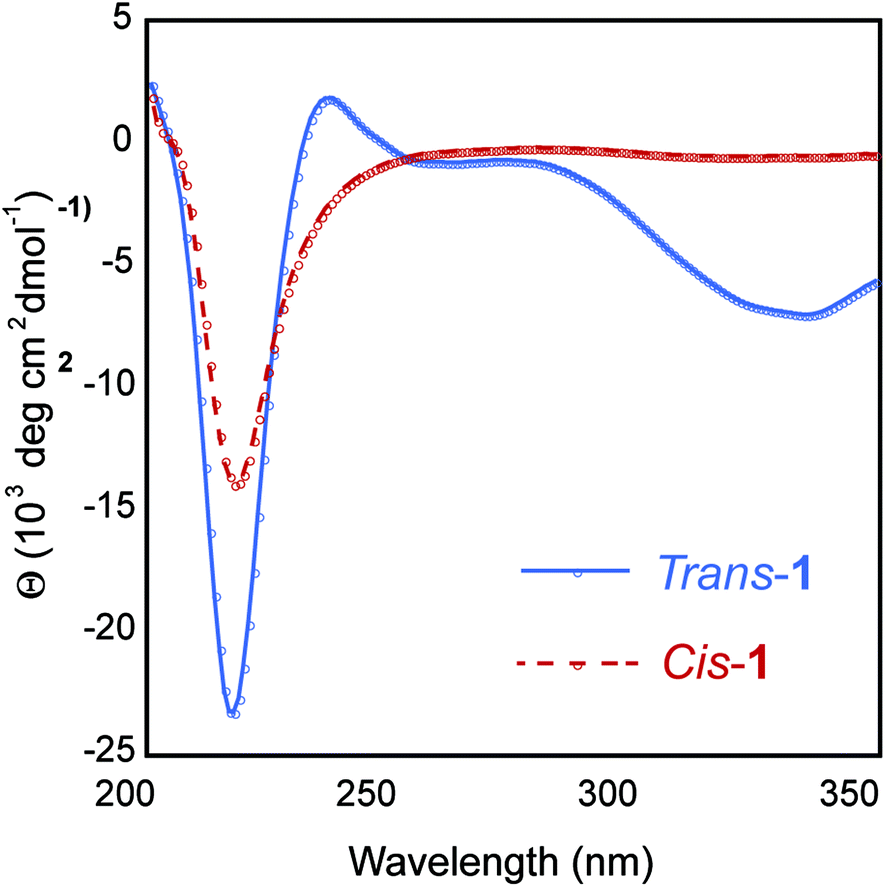 | ||
| Fig. 5 CD spectra of 0.5 wt% assembled trans-1 and cis-1 in 20 mM NaCl. | ||
The β-sheet character of peptide 1 in the cis and trans conformation was examined using Fourier-transform infrared (FT-IR) spectroscopy. Peptide β-sheets are characterized by an amide stretch of ∼1630 cm−1.56 In the trans-conformation, assemblies of peptide 1 exhibited a characteristic 1630 cm−1 stretch, indicating β-sheet secondary structure (Fig. 6). After trans–cis isomerization within the CaF2 IR plates, the cis-conformer also exhibited a stretching frequency of 1630 cm−1. The absence of a peak at 1695 cm−1 may support a parallel packing arrangement, consistent with our models (Fig. 1C). This parallel packing arrangement would appear to be electrostatically unfavourable because it results in like-charges being in close proximity in a cross-strand orientation (Fig. 1B and C). This potentially explains the requirement for salt for gelation to occur, as has been seen in similar amphipathic, self-assembling peptides.57
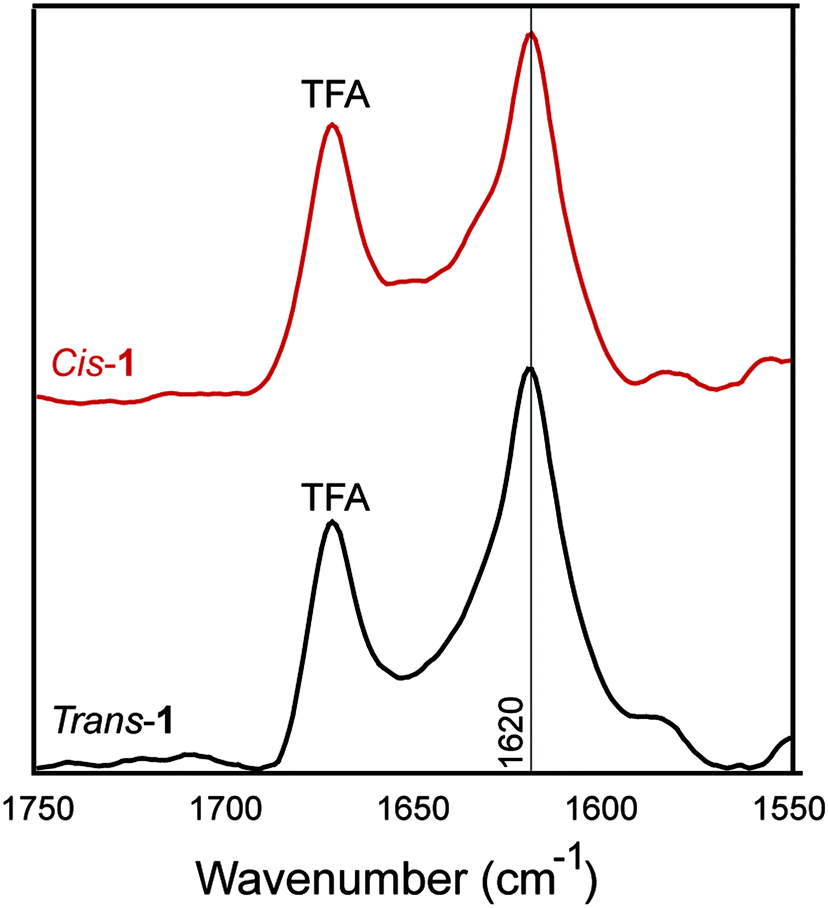 | ||
| Fig. 6 IR spectra of 0.5 wt% assembled trans-1 and cis-1 in 20 mM NaCl. | ||
Negative stain transmission electron microscopy (TEM) confirmed that the trans-1 hydrogels were composed of self-assembled fibrils. Fibrils were found to have a 3.4 ± 0.7 nm diameter and were several micrometers in length (Fig. 7). The diameter of the trans-1 fibrils is consistent with the fibril structure proposed in Fig. 1. Negative stain TEM of cis-1 hydrogels showed densely packed filaments with diameters of 2.4 ± 0.5 nm, which indicates that isomerization has a minor effect on the macromolecular morphology of the fibrils. The cis-1 networks also contained 3.5 ± 0.6 nm diameter fibrils, which are likely derived from residual, unconverted trans-1 peptide (Fig. 7). While there was a reduction in the quantity of fibrils in the cis-1 sample compared with the trans-1 sample when viewed at lower magnifications (Fig. 7C and D), this decrease in fibril density was minor. Collectively, these data suggest that the reduction in hydrogel rigidity upon trans–cis isomerization is primarily due to a disruption of the well-ordered β arch structures rather than fibril disassembly when in the cis conformation. Presumably, the increase in β-hairpin conformations in the cis-conformation reduces the degree of peptide in an open conformation that is available for fibril–fibril cross-linking (as shown in Fig. 1B) that strengthens the macromolecular network of the hydrogel.
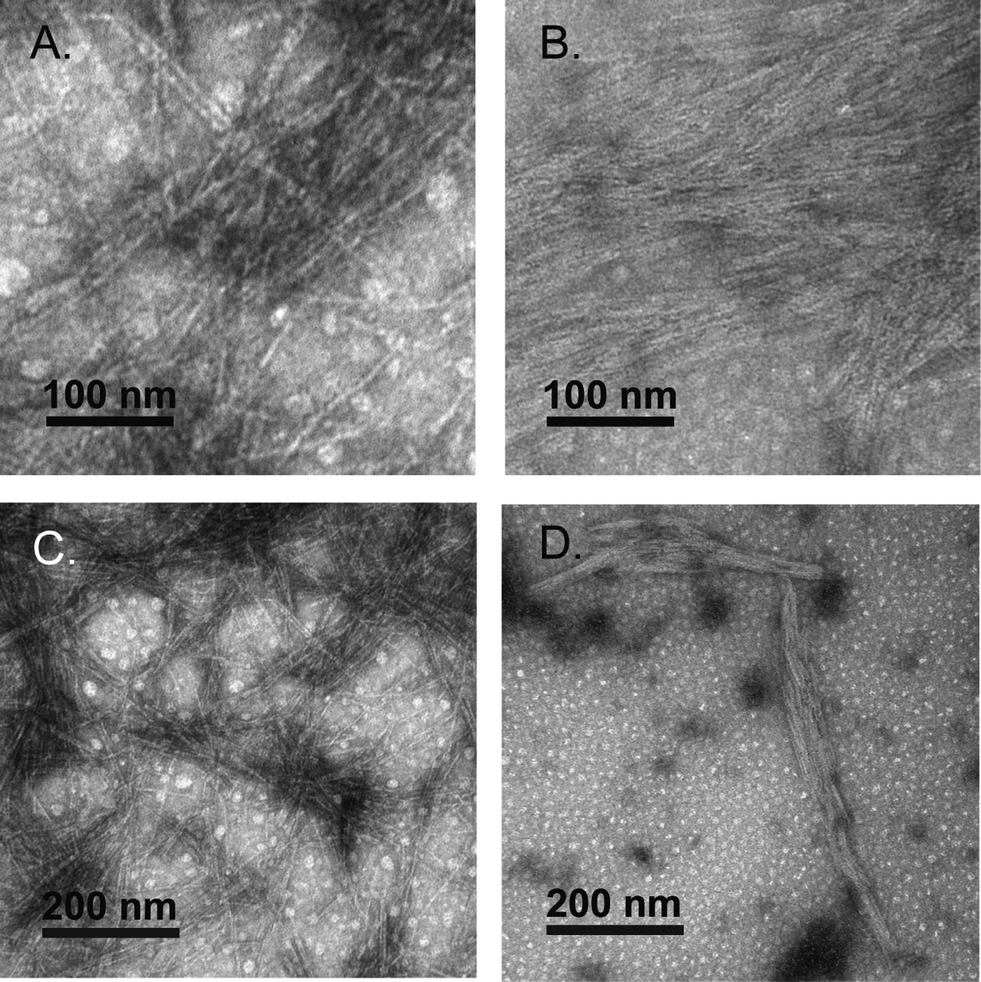 | ||
Fig. 7 A. Negative-stain TEM images of trans-1 at high magnification (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×). B. Negative-stain TEM images of cis-1 at high magnification (200000×). C. Negative-stain TEM images of trans-1 at high magnification (100000×). D. Negative-stain TEM images of cis-1 at high magnification (100000×). 000×). B. Negative-stain TEM images of cis-1 at high magnification (200000×). C. Negative-stain TEM images of trans-1 at high magnification (100000×). D. Negative-stain TEM images of cis-1 at high magnification (100000×). | ||
Discussion
Peptide hydrogel formation requires self-assembly of a peptide into well-ordered fibrils, followed by noncovalent cross-linking of the resulting fibrils.19 Therefore, perturbing self-assembly propensity of peptides prone to forming hydrogels is a logical strategy for hydrogel rigidity modulation. Studies in which photoreversible β-turns have been incorporated into peptide sequences to perturb peptide self-assembly reveal that the conformation of the turn has significant consequences on self-assembly properties.29,37 Zinth and coworkers showed that in amyloid-forming peptides, trans–cis isomerization of a centrally located azobenzene moiety leads to fibril destabilization and disassembly.29,30 When in the trans conformation, the β-sheets are able to stack, presumably in a linear ensemble; however, in the cis conformation, the β-hairpin leads to fibril destabilization and disassembly.29 Similarly, we and others have shown that an azobenzene turn mimetic in the trans-conformation leads to efficient self-assembly in the Aβ42 peptide, but the cis-AMPP conformer fails to self-assemble into fibrils and instead forms distinctive nontoxic aggregates.37,38 Conversely, DProGly β-hairpin turns in Aβ40 lead to effective fibril formation, often at accelerated rates.36 One proposed explanation for this disparity is that cis-azobenzene forms a type I′ β-turn, while DProGly adopts a type II′ conformation.36,51,58 Other studies reveal that turn conformation can influence the emergent viscoelastic properties of hydrogels in addition to self-assembly properties alone. Schneider et al. have incorporated a DProLPro type II′ β-turn into the MAX peptides to create stimulus responsive hydrogels.10,12,13,18–20,59 The type II′ turn creates an intramolecular sheet-turn-sheet motif that extends laterally to self-assemble and readily cross-link to form a hydrogel.10,12,13,18–20 In these systems, self-assembly occurs when triggered by salt, pH, or temperature to minimize charge.10,12,13,18,59 Therefore, it is important to characterize the consequences of specific turn conformation to establish rules for turn formation in both peptide self-assembly and hydrogel formation.In the system discussed herein, the azobenzene turn mimetic provides insight into the role of β-arc vs. β-hairpin formation in peptide self-assembly.49 Many amyloid assemblies, including Aβ, IAPP, and others, feature a so-called β-arc turn in a sheet-turn-sheet motif. Steven and Kajava et al. have articulated that β-arc strand-turn-strand motifs in which side chains are rotated 90° with respect to a β-hairpin represent a common motif in peptide fibrils known as β-arches.49 The β-arc consists of β-sheets in which the side chain groups are oriented perpendicular to the fibril axis; β-strands are thus separated by an approximate 10 Å distance (Fig. 1C, left hand structure). The primary intramolecular mode of interaction between β-strands that flank the β-arc turn is via side chain-side chain contact. β-Arch assemblies can be differentiated from β-hairpins in that all hydrogen bond formation occurs intermolecularly. Self-assembled peptides have also been engineered that feature a β-hairpin orientation of the constituent peptides.10,12,13,16,18–20 In the β-hairpin conformation, the peptide strands are rotated 90° relative to the β-arc conformation (Fig. 1C, right hand structures). β-Hairpins are thus stabilized by an intramolecular hydrogen bond network between the β-strands that flank the β-turn, providing a strand separation of 4.7 Å. Rigorous comparisons between the emergent properties of peptide assemblies that feature either β-arc or β-hairpin turns have been problematic until the recent observation that trans-AMPP adopts β-arc-like conformations and cis-AMPP adopts β-hairpin conformations. Thus, AMPP becomes a light-sensitive mimetic of either turn type.
Peptide assemblies of the trans-1 peptide most likely feature a β-arc architecture. As stated in the previous paragraph, we concluded that trans-AMPP adopts a bent arrangement in the context of Aβ-derived systems, leading to a stable β-arc-like turn.37 Models of assemblies of the trans-1 conformer reveal that the intra-strand distances are ∼10 Å, consistent with the formation of a β-arc structure (Fig. 1C). The β-arc formed by trans-1 is not structurally rigid, since AMPP contains two methylene units on either side of the azobenzene moiety that provide added flexibility to the turn.23 This facilitates the adoption of an extended conformation of the flanking β-strand segments in which the β-arc motif is interrupted. TEM images of the fibrils of trans-1 support a packing structure that predominately features β-arch stacks (based on the fibril diameter which approximately half the total length of the extended peptide). However, the occasional incorporation of trans-1 units into fibrils in which the peptide is extended provides an extrafibrillar extension that facilitates interfibril cross-linking with other fibrils (Fig. 1B and C). One possible explanation for hydrogel weakening upon trans → cis isomerization could be that the cis AMPP conformer does not contain the same flexibility as the trans conformer; the extrafibrillar extensions that exist in trans-1 assemblies are thus eliminated in cis-1 assemblies, reducing the density of fibril–fibril crosslinks.
Conversion of trans-1 assemblies to cis-1 assemblies results in formation of morphologically similar fibrils. Questions remain regarding the molecular structure of these resulting fibrils and the molecular mechanisms by which they interconvert. As shown in Fig. 1C, trans-1 fibrils are proposed to be β-arc-containing structures that stack into classical amyloid cross-β morphologies. Conversely, the cis-1 fibrils are proposed to be a bilayer of β-hairpin fibrils in which the alanine side chains are organized to one face of each constituent β-sheet of the bilayer and are thus segregated to a hydrophobic core of the resulting bilayer.18,19,42,60,61 The hydrophilic arginine and aspartic acid side chains thus comprise the solvent exposed exterior faces of the proposed bilayer. Each of these fibril-packing architectures is consistent with the available data for trans-1 and cis-1 fibrils respectively. However, confirmation of these structures requires challenging higher resolution structural studies exploiting techniques such as solid state NMR spectroscopy. Future studies in this vein will enable additional insight into how the fibrils derived from trans-1 and cis-1 differ and how they interconvert.
Conclusion
We have reported the development of photosensitive, self-assembled peptide hydrogels in which reversible transitions in hydrogel rigidity can be induced. Reversible, stimulus-responsive hydrogelation of self-assembled peptides will facilitate the creation of enhanced materials for ex vivo tissue engineering for the growth of cells in 3D matrices that can be selectively disassembled to allow harvesting of the cultured cells. In addition, these materials could be designed to allow light-triggered release of encapsulated cargo, including pharmaceutical agents. The use of azobenzene to affect reversible conformational transitions in the hydrogel network that translate to changes in viscoelasticity provides critical proof of principle and applications for these materials are currently under investigation.Experimental section
Peptide synthesis
Peptides were synthesized on a CEM Liberty peptide synthesizer with a Discover microwave (CEM) utilizing an Fmoc/tBu protection scheme. Fmoc-AMPP-OH was prepared as previously described.53 Couplings were facilitated through HBTU/HOBt activation. Peptides were cleaved from the resin by suspension in TFA/TIS/H2O (98:1:1, v/v/v), purified by reverse-phase HPLC and characterized by MALDI-TOF MS ([M + H]+m/z calculated 1967.3, observed 1968.4). Peptide concentration was determined by HPLC injection and correlation of the integrated peak area to a concentration curve calibrated by amino acid analysis (AIBiotech, Richmond, VA) (Fig. S2 and S5†).
Hydrogel preparation and photoisomerization
Peptide hydrogels were prepared by dissolving the lyophilized peptide (∼3 mM) in a solution of 20 mM NaCl and sonicating for 5 min; self-assembly and gelation occurred over 30 min. Photoisomerization to cis-1 was performed by irradiating with a 500 W mercury lamp (Oriel) with a 365 nm bandpass filter for 15 min. Isomerization to the trans-1 was achieved by irradiation using a 434 nm bandpass filter for 15 min. Digital images were obtained after sonication and gelation. Gels were gently disturbed prior to imaging.Rheology
Rheological measurements were conducted on a TA Instruments AR-G2 rheometer operating in oscillatory mode, using a 20 mm parallel plate geometry equipped with a solvent trap filled with silicon oil to prevent evaporation. Samples of each gel were prepared as described above and were applied to the rheometer stage immediately following photoisomerization. The stage was then covered with a solvent trap in order to prevent sample evaporation. Following application of the sample a dynamic frequency sweep was obtained from 0.1–50 rad s−1 with 0.2% strain at 25 °C. All reported values for G′ and G′′ are an average of at least three runs. All measurements were performed in the linear viscoelastic region for each gel.UV-Vis absorbance
Absorbance measurements were recorded on a Lambda 35 UV/Vis spectrophotometer (Perkin-Elmer). Spectra were obtained from 200–600 nm with a 1.0 nm step, and 1.0 nm bandwidth at 25 °C in a 1 mm quartz cuvette (Helma) at 0.5 wt% 1 (∼3 mM).Circular dichroism spectroscopy
CD spectra were recorded on an AVIV 202 CD spectrometer. Spectra were obtained from 260–190 nm with a 1.0 nm step, 1.0 nm bandwidth, and a 3 s collection time per step at 25 °C in a 0.1 cm path length quartz cuvette (Hellma) at 0.5 wt% 1 (∼3 mM). The AVIV software was used for background subtraction, conversion to molar ellipticity, and data smoothing with a least-squares fit.FT-IR spectroscopy
IR spectra were obtained on a Shimadzu FTIR 8400S spectrometer. Spectra were recorded at a resolution of 2 cm−1 on a 0.5 wt% solution of prepared gel in 20 mM NaCl placed between CaF2 plates. Trans–cis isomerization was carried out within the CaF2 plate. Raw data was smoothed and baseline corrected using IRSolution software.Negative stain transmission electron microscopy
A solution of trans-1 fibrils (10 μL) was applied to 200 mesh, carbon-coated copper grids for 30 s. Excess fluid was removed by capillary action. Residual salts and buffer were washed with water (10 μL) for 10 s followed by removal by capillary action (repeated twice). The grids were stained with 5% uranyl acetate (10 μL) for 5 min and the excess staining solution was removed via capillary action. The grid was washed twice with water (10 μL) to remove excess staining reagent. The stained grids were allowed to air dry for at least 10 minutes prior to imaging. For cis-1, the trans-1 fibril solution was photoisomerized at 365 nm for 15 min and the resulting solution was spotted onto carbon-coated copper grids as described above. Electron microscope imaging was performed on a Hitachi 7650 transmission electron microscope in high contrast mode with an accelerating voltage of 80 kV. Fibril width was determined by performing at least 100 measurements on unique fibrils for each peptide using the program ImageJ (http://rsbweb.nih.gov/ij/).62Acknowledgements
We gratefully acknowledge Chris Willoughby and TA Instruments for use of an AR-G2 rheometer. We thank Karen Bentley of the University of Rochester Medical Center Electron Microscope Research Core for assistance with transmission electron microscopy experiments. We also thank Prof. Joseph P. Dinnocenzo for helpful discussions regarding photoisomerization of azobenzene. This work was supported by a DuPont Young Professor Award to B.L.N., by the Alzheimer's Association (NIRG-08-90797), and by a Provost's Multidisciplinary Award (University of Rochester). Mass spectroscopy instrumentation was partially supported by a grant from the U.S. National Science Foundation (CHE-0840410).References
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS PubMed.
- S. Kyle, A. Aggeli, E. Ingham and M. J. McPherson, Trends Biotechnol., 2009, 27, 423–433 CrossRef CAS PubMed.
- D. W. P. M. Lowik, E. H. P. Leunissen, M. v. d. Heuvel, M. B. Hansen and J. C. M. v. Hest, Chem. Soc. Rev., 2010, 39, 3394–3412 RSC.
- J. P. Jung, J. Z. Gasiorowski and J. H. Collier, Biopolymers, 2010, 94, 49–59 CrossRef CAS PubMed.
- S. Cavalli, F. Albericio and A. Kros, Chem. Soc. Rev., 2010, 39, 241–263 RSC.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822–835 RSC.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
- K. Rajagopal, M. S. Lamm, L. A. Haines-Butterick, D. J. Pochan and J. P. Schneider, Biomacromolecules, 2009, 10, 2619–2625 CrossRef CAS PubMed.
- C. J. Bowerman and B. L. Nilsson, J. Am. Chem. Soc., 2010, 132, 9526–9527 CrossRef CAS PubMed.
- D. J. Pochan, J. P. Schneider, J. Kretsinger, B. Ozbas, K. Rajagopal and L. Haines, J. Am. Chem. Soc., 2003, 125, 11802–11803 CrossRef CAS PubMed.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 CrossRef CAS.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 2006, 1070–1071 CrossRef PubMed.
- R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19–24 CrossRef CAS PubMed.
- L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025–17029 CrossRef CAS PubMed.
- T. J. Measey, B. N. Markiewicz and F. Gai, Chem. Phys. Lett., 2013, 580, 135–140 CrossRef CAS PubMed.
- J. P. Schneider, D. J. Pochan, B. OZbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- R. P. Nagarkar, R. A. Hule, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2008, 130, 4466–4474 CrossRef CAS PubMed.
- B. Ozbas, K. Rajagopal, L. Haines-Butterick, J. P. Schneider and D. J. Pochan, J. Phys. Chem. B, 2007, 111, 13901–13908 CrossRef CAS PubMed.
- M. S. Vollmer, T. D. Clark, C. Steinem and M. R. Ghadiri, Angew. Chem., Int. Ed., 1999, 38, 1598–1601 CrossRef CAS.
- G. A. Woolley, Acc. Chem. Res., 2005, 38, 486–493 CrossRef CAS PubMed.
- A. Aemissegger, V. Kräutler, W. F. v. Gunsteren and D. Hilvert, J. Am. Chem. Soc., 2005, 127, 2929–2936 CrossRef CAS PubMed.
- V. Kräutler, A. Aemissegger, P. H. Hünenberger, D. Hilvert, T. Hansson and W. F. v. Gunsteren, J. Am. Chem. Soc., 2005, 127, 4935–4942 CrossRef PubMed.
- L.-s. Li, H. Jiang, B. W. Messmore, S. R. Bull and S. I. Stupp, Angew. Chem., Int. Ed., 2007, 46, 5873–5876 CrossRef CAS PubMed.
- F. Zhang, A. Zarrine-Afsar, M. S. Al-Abdul-Wahid, R. S. Prosser, A. R. Davidson and G. A. Woolley, J. Am. Chem. Soc., 2009, 131, 2283–2289 CrossRef CAS PubMed.
- F. Zhang, K. A. Timm, K. M. Arndt and G. A. Woolley, Angew. Chem., Int. Ed., 2010, 49, 3943–3946 CrossRef CAS PubMed.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- A. A. Deeg, T. E. Schrader, S. Kempter, J. Pfizer, L. Moroder and W. Zinth, ChemPhysChem, 2011, 12, 559–562 CrossRef CAS PubMed.
- A. A. Deeg, T. Schrader, H. Strzalka, J. Pfizer, L. Moroder and W. Zinth, Spectroscopy, 2012, 27, 387–391 CrossRef CAS.
- S. A. Waldauer, S. Hassan, B. Paoli, P. M. Donaldson, R. Pfister, P. Hamm, A. Caflisch and R. Pellarin, J. Phys. Chem. B, 2012, 116, 8961–8973 CrossRef CAS PubMed.
- X. Li, Y. Gao, Y. Kuang and B. Xu, Chem. Commun., 2010, 46, 5364–5366 RSC.
- W. Li, I.-s. Park, S.-K. Kang and M. Lee, Chem. Commun., 2012, 48, 8796–8798 RSC.
- Y. Matsuzawa and N. Tamaoki, J. Phys. Chem. B, 2010, 114, 1586–1590 CrossRef CAS PubMed.
- Y. Huang, Z. Qiu, Y. Xu, J. Shi, H. Lin and Y. Zhang, Org. Biomol. Chem., 2011, 9, 2149–2155 CAS.
- T. M. Doran, E. A. Anderson, S. E. Latchney, L. A. Opanashuk and B. L. Nilsson, J. Mol. Biol., 2012, 421, 315–328 CrossRef CAS PubMed.
- T. M. Doran, E. A. Anderson, S. E. Latchney, L. A. Opanashuk and B. L. Nilsson, ACS Chem. Neurosci., 2012, 3, 211–220 CrossRef CAS PubMed.
- C. Hoppmann, C. Barucker, D. Lorenz, G. Multhaup and M. Beyermann, ChemBioChem, 2012, 13, 2657–2660 CrossRef CAS PubMed.
- S. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS.
- S. Zhang, T. C. Holmes, C. M. DiPersio, R. O. Hynes, X. Su and A. Rich, Biomaterials, 1995, 16, 1385–1393 CrossRef CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- H. Yokoi, T. Kinoshita and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8414–8419 CrossRef CAS PubMed.
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. E. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Döbeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef PubMed.
- A. T. Petkova, W.-M. Yau and R. Tycko, Biochemistry, 2006, 45, 498–512 CrossRef CAS PubMed.
- J. Hennetin, B. r. Jullian, A. C. Steven and A. V. Kajava, J. Mol. Biol., 2006, 358, 1094–1105 CrossRef CAS PubMed.
- S. Luca, W.-M. Yau, R. Leapman and R. Tycko, Biochemistry, 2007, 46, 13505–13522 CrossRef CAS PubMed.
- A. K. Paravastu, R. D. Leapman, W.-M. Yau and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18349–18354 CrossRef CAS PubMed.
- A. V. Kajava, U. Baxa and A. C. Steven, FASEB J., 2010, 24, 1311–1319 CrossRef CAS PubMed.
- R. Tycko, Annu. Rev. Phys. Chem., 2011, 62, 279–299 CrossRef CAS PubMed.
- S.-L. Dong, M. Loweneck, T. E. Schrader, W. J. Schreier, L. Moroder and C. Renner, Chem.–Eur. J., 2006, 12, 1114–1120 CrossRef CAS PubMed.
- J.-i. Edahiro, K. Sumaru, Y. Tada, K. Ohi, T. Takagi, M. Kameda, T. Shinbo, T. Kanamori and Y. Yoshimi, Biomacromolecules, 2005, 6, 970–974 CrossRef CAS PubMed.
- A. Aemissegger and D. Hilvert, Nat. Protocols, 2007, 2, 161–167 CAS.
- N. Nishimura, T. Sueyoshi, H. Yamanada, E. Imai, S. Yamamoto and S. Hasegawa, Bull. Chem. Soc. Jpn., 1976, 49, 1381–1387 CrossRef CAS.
- S. Samanta, H. I. Qureshi and G. A. Woolley, Beilstein J. Org. Chem., 2012, 8, 2184–2190 CrossRef CAS PubMed.
- W. K. Surewicz, H. H. Mantsch and D. Chapman, Biochemistry, 1993, 32, 389–394 CrossRef CAS.
- C. J. Bowerman, W. Liyanage, A. J. Federation and B. L. Nilsson, Biomacromolecules, 2011, 12, 2735–2745 CrossRef CAS PubMed.
- H. E. Stanger and S. H. Gellman, J. Am. Chem. Soc., 1998, 120, 4236–4237 CrossRef CAS.
- K. Rajagopal, B. Ozbas, D. J. Pochan and J. P. Schneider, Eur. Biophys. J., 2006, 35, 162–169 CrossRef CAS PubMed.
- D. M. Marini, W. Hwang, D. A. Lauffenburger, S. Zhang and R. D. Kamm, Nano Lett., 2002, 2, 295–299 CrossRef CAS.
- C. J. Bowerman and B. L. Nilsson, Biopolymers, 2012, 98, 169–184 CrossRef CAS PubMed.
- M. D. Abramoff, P. J. Magelhaes and S. J. Ram, Biophotonics International, 2004, 11, 36–42 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Peptide characterization data and peptide concentration curve. See DOI: 10.1039/c3py00903c |
| This journal is © The Royal Society of Chemistry 2014 |