Comparative kinetics of damage to the plasma and mitochondrial membranes by intra-cellularly synthesized and externally-provided photosensitizers using multi-color FACS
Sara
Haupt
a,
Zvi
Malik
b and
Benjamin
Ehrenberg
*a
aDepartment of Physics and Institute of Nanotechnology and Advanced Materials, Bar Ilan University, Ramat Gan, Israel
bFaculty of Life Sciences, Bar Ilan University, Ramat Gan, Israel
First published on 3rd October 2013
Abstract
Photodynamic therapy (PDT) of cancer involves inflicting lethal damage to the cells of malignant tumors, primarily by singlet oxygen that is generated following light-absorption in a photosensitizer molecule. Dysfunction of cells is manifested in many ways, including peroxidation of cellular components, membrane rupture, depolarization of electric potentials, termination of mitochondrial activity, onset of apoptosis and necrosis and eventually cell lysis. These events do not necessarily occur in linear fashion and different types of damage to cell components occur, most probably, in parallel. In this report we measured the relative rates of damage to two cellular membranes: the plasma membrane and the mitochondrial membrane. We employed photosensitizers of diverse hydrophobicities and used different incubation procedures, which lead to their different intra-cellular localizations. We monitored the damage that was inflicted on these membranes, by employing optical probes of membrane integrity, in a multi-color FACS experiment. The potentiometric indicator JC-1 monitored the electric cross-membrane potential of the mitochondria and the fluorometric indicator Draq7 monitored the rupture of the plasma membrane. We show that the electric depolarization of the mitochondrial membrane and the damage to the enveloping plasma membrane proceed with different kinetics that reflect the molecular character and intracellular location of the sensitizer: PpIX that is synthesized in the cells from ALA causes rapid mitochondrial damage and very slow damage to the plasma membrane, while externally added PpIX has an opposite effect. The hydrophilic sensitizer HypS4 can be taken up by the cells by different incubation conditions, and these affect its intracellular location, and as a consequence either the plasma membrane or the mitochondria is damaged first. A similar correlation was found for additional extracellularly-provided photosensitizers HP and PpIX.
Introduction
Biological photodynamic action is currently an accepted method for damaging cells. It involves the use of light-triggered reactions that commence with the absorption of light by a photosensitizer molecule. Photodynamic action is usually conducted with tetra-pyrrole molecules and their analogs. The application of photodynamic action for eradication of cancer cells is based on two important properties of photosensitizers: their preferential uptake by solid tumors and their efficient production of electronically excited singlet oxygen (1O2), via energy transfer from the sensitizer's long-lived excited triplet state to molecular oxygen. Different types of tumors (including skin, lung, breast and bladder) have been treated by the intravenous application of a sensitizer followed by illumination with a low-intensity light. Malignant cells undergo a necrotic or apoptotic reaction with minimal damage to surrounding tissue. PDT has therefore become a clinical protocol for treating malignancies as well as for antibacterial and antiviral treatments. Some review articles on photodynamic action's properties and on pre-clinical tests are cited.1–9There is evidence to indicate that amphiphilic sensitizers, which tend to be localized in lipid membrane environments, cause damage, upon photodynamic treatment, mainly in the cell's membranes, as evidenced by increased permeability, membrane rupture and cell lysis.10–14 More specifically, PDT's lethal action and the injury which is caused leads to damage of transport mechanisms, depolarization of electric potential and leakage across the plasma and intracellular membranes.12,15–22
The chain of cellular events that evolve and follow an initial oxidation by singlet oxygen is not clear, but probably depends on the cell, the sensitizer and other circumstantial conditions. We have given indications in previous studies that the electric depolarization of a membrane is not caused by a simple pore that is formed in it by lipid oxidation.23–25 The assumption is that damage to membrane-bound proteins is responsible for the depolarization. The fundamental understanding of subcellular sites of damage provoked by photodynamic treatment has two central aspects: one concerns the importance of damage to mitochondria versus plasma membrane in inducing cell death, while the other concerns the nature of cellular damage induced by hydrophilic vs. hydrophobic sensitizers, or endogenous vs. exogenous protoporphyrin IX in inducing membrane damage. Future developments in PDT sensitizers have to consider targeting these two major sites in the cells in order to increase PDT efficacy.
In this paper we report the relative rates of damage to the plasma membrane and the mitochondrial membranes, when different photosensitizers are employed. We followed the damage to the mitochondrial membrane by employing the potentiometric indicator JC-1, which monitors the electric cross-membrane potential. The integrity of the plasma membrane was monitored by the fluorometric indicator Draq7. The dyes were employed in multi-color experiments by FACS, after the cells were incubated with the photosensitizers and were illuminated for increased periods of time. We show that the depolarization of the mitochondrial membrane and the damage to the plasma membrane progress with different kinetics, which, in turn, depend on the hydrophobicity of the photosensitizers, which determine their intracellular locations.
Materials and methods
Cell culture
Human glioblastoma U251 cells were grown on tissue culture plates (Greiner, Stroudwater, UK) in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, US), supplemented with 10% fetal calf serum, and containing the antibiotics penicillin–streptomycin–nystatin and L-glutamine (Biological Industries, Beit-Haemek, Israel). The cells were incubated at 37 °C in a humidified atmosphere with 5% CO2 and 95% air, and were sub-cultured twice a week.Photosensitizers
5-Aminolevulinic-acid (ALA) was obtained from Sigma-Aldrich (St. Louis, MO, US). The exogenous photosensitizers protoporphyrin IX (PpIX) and hematoporphyrin IX (HP) were also from Sigma, and hypericin tetrasulfonic acid (HypS4) was a gift from the late Dr Mazur, Weizmann Institute of Science, Israel. A stock solution of ALA was 100 mM in a medium without serum, the stock of PpIX was 2 mM in DMSO, the stock of HypS4 was 2 mM in water, and the stock of HP was 0.5 mM in water. The sensitizers were added to the suspensions of cells at a final concentration of 0.7 mM, 75 nM, 2 μM and 75 nM respectively. With HypS4 we used two incubation protocols. In the first, we incubated it for 5 hours in medium without serum, in order to expose the cells directly to the sensitizer. The second protocol was carried out in a serum-enriched medium for 20 hours, during which the proteins may serve as transfer vehicles for the sensitizer. With HP we used two different incubation times: two hours, to allow the sensitizer to reach equilibrium of uptake, and 40 minutes, to test a shorter exposure time to examine the uptake of the sensitizer in the plasma membrane.Photodynamic action on cells U251
Cells were seeded and incubated with ALA or with the exogenous sensitizers in serum-free medium or with serum, as explained above. The cells were irradiated for 1–25 minutes, using a Vilber Lourmat light source VL-206BL, which has a Gaussian-shaped emission spectrum, peaking at 353 nm, with a spike at 365 nm and FWHM of 40 nm.Cellular ALA and exogenous photosensitizers’ analysis by flow cytometry
U251 cells (1 × 105) were seeded in 24-well plates and were incubated for one to four hours with ALA or exogenous photosensitizers. The cells were harvested in the dark, centrifuged at 900 rpm, washed twice with filtered PBS without magnesium and calcium and resuspended in 0.5 mL of the same PBS buffer. The fluorescence of photosensitizers was measured in 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per sample using a FACS (Gallios, Beckman Coulter, Nyon, Switzerland) with an excitation wavelength of 488 nm and an emission wavelength of 610 nm long pass filter.
000 cells per sample using a FACS (Gallios, Beckman Coulter, Nyon, Switzerland) with an excitation wavelength of 488 nm and an emission wavelength of 610 nm long pass filter.
Assessment of plasma membrane damage
Draq7 is a far-red fluorescent DNA-staining dye that stains only the nuclei in dead and permeabilized cells.26 U251 cells (2 × 105) were seeded in 2 mL plates (Greiner Bio-One, Germany), treated with our sensitizers for different times, and exposed to light illumination. Immediately after illumination, the plasma membrane's integrity of the cells was assessed using FACS, with an excitation wavelength of 605 nm and observing the fluorescence through the following filter: 695/30 nm (central wavelength/FWHM).Assessment of mitochondrial membrane potential (ΔΨm)
The mitochondrial membrane potential was quantified using the potentiometric dye indicator 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1), which undergoes a potential-dependent accumulation in the mitochondria. A mitochondrion accumulates the positively-charged dye, according to the Nernst equation. One expects that a typical ΔΨm of −180 mV would account for an internal concentration of a membrane-permeable, positively charged molecule that is 3 orders of magnitude higher than its external concentration. At a high concentration, JC-1 tends to form aggregates, which fluoresce in the red (590 nm). Upon depolarization of the mitochondrial membrane, JC-1 concentration is strongly depleted and a high concentration of monomers is formed, and these monomers fluoresce in the green (530 nm). The ratio of red/green fluorescence intensities indicates the physiological state of the mitochondrion, where a high red/green ratio indicates a healthy mitochondrion, and a low red/green ratio implies a damaged one. This ratio can be quantified spectroscopically and it was used to monitor the state of the mitochondrial population.U251 cells (2 × 105) were seeded in 2 mL plates (Greiner Bio-One, Germany), treated with our sensitizers for different times, and exposed to light irradiation. Immediately after irradiation, the mitochondrial membrane potential of the cells was measured by monitoring the ratio between the fluorescence intensities of the monomeric and dimeric forms of JC-1. This was done by a FACS (Gallios, Beckman-Coulter, Switzerland), with excitation with the following laser lines: 405 nm and 488 nm and observing the fluorescence through the following filters (central wavelength/FWHM): 525/40, 575/40 nm.
MTT cell viability assay
U251 cells (1 × 105) were seeded in 24-well plates and incubated with ALA or exogenous photosensitizers. The samples were irradiated as described above and 24 hours later, the medium was decanted and 5 mg mL−1 of MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, Sigma-Aldrich, St. Louis, MO) was added to each well for 2 hours. Afterwards, 200 μL of a water–dimethylformamide solution (1:1 in volume), containing 0.2 gram of sodium dodecyl sulphate per 1 mL, were added. Absorbance was then measured at 570 nm using a Tecan spectrophotometer (NeoTec, Melbourne, Australia).
Lactate dehydrogenase (LDH) activity
LDH activity was measured using a LDH-L reagent (Thermo Scientific, Waltham, US). Immediately after irradiation the cells were centrifuged and the supernatant was collected and diluted in the LDH-L reagent, using a sample:reagent ratio of 1:2 (v:v). The absorbance was measured after 1 hour at 340 nm, using a Tecan spectrophotometer (NeoTec, Melbourne, Australia).
Scanning electron microscopy (SEM)
Cells were treated with ALA or exogenous photosensitizers and were irradiated, as described above. 24 hours later, the cells were harvested and washed with calcium- and magnesium-free PBS. The cells were attached to 10 mm cover slips coated with poly-L-lysine 0.1% (Sigma-Aldrich, St. Louis, MO, US) for 1 hour. Samples were fixed according to a modification of the triple fixation GTGO method for scanning electron microscopy (SEM). Briefly, the samples were fixed with 2.5% glutaraldehyde in phosphate buffer (pH 7.2), followed by post-fixation in 4% OsO4. The third fixative was 2% tannic acid–guanidine hydrochloride. After fixation, the cells were dehydrated in graded ethanol solutions, and then the ethanol was exchanged for Freon 113, again using graded solutions. The samples were then air-dried, gold-coated, and examined using an FEI Quanta 200FEG scanning electron microscope (FEI, Hillsboro OR, US).Results
The effect of ALA on PpIX accumulation
ALA-PDT efficacy is mostly dependent on the PpIX synthesis capacity of the treated cells or the tumor. Supplementation of exogenous ALA to the cells bypasses the rate-limiting enzyme ALA synthase (ALAS) and therefore induces the biosynthesis of PpIX. PpIX that is synthesized endogenously is localized in and around mitochondria27–29 and eventually it equilibrates with other intracellular hydrophobic parts. However, due to PpIX's very low water solubility this equilibration is slow.To estimate the immediate effect of ALA on PpIX accumulation in U251 cells we treated them with ALA and detected the PpIX fluorescence by FACS analysis. The kinetics of PpIX accumulation in the cells showed significant positive correlation with the cells’ incubation time with ALA (Fig. 1). Up until 8 hours incubation, PpIX levels increased gradually, and reached a plateau and even slightly diminished after this time. These results indicate that incubation with ALA augments PpIX synthesis.30,31
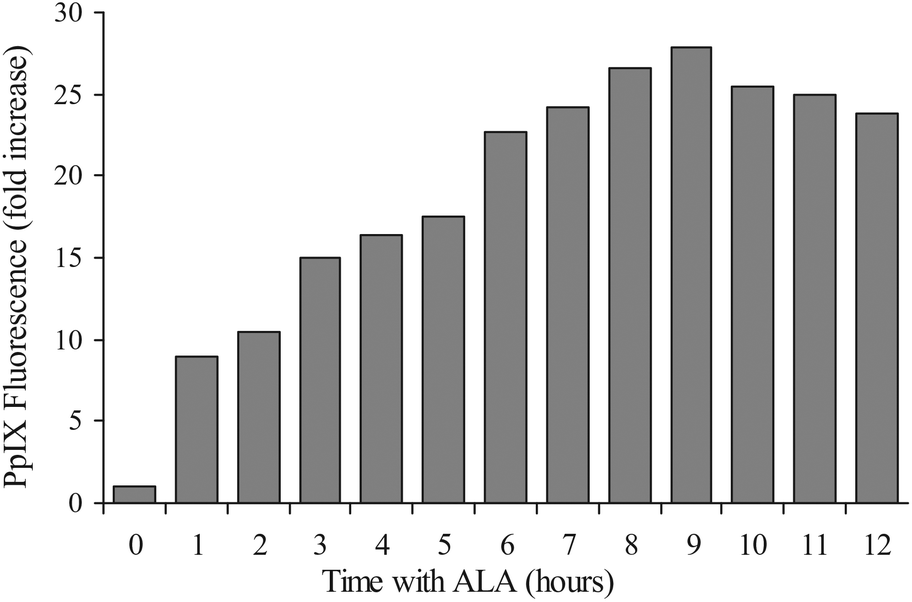 | ||
| Fig. 1 Time dependence of PpIX accumulation in U251 cells treated by ALA. U251 cells were exposed to 0.7 mM ALA for various durations. PpIX fluorescence was measured through a 610 nm long-pass filter of the FACS. The results represent average values of PpIX fluorescence (fold increase) obtained from 3 independent experiments. | ||
Comparing the rate of damage to mitochondrial and plasma membranes by ALA-PDT
In order to examine the effect of ALA-PDT on the mitochondrial membrane potential (ΔΨm) after light illumination, the following experiments were performed.Measuring the JC-1 ratio after ALA-PDT showed that the mitochondrial potential decreased very rapidly with the length of illumination (Fig. 2, dotted line); about 1 minute of illumination was sufficient to reduce the sample-average ΔΨm by ∼50%, in comparison to untreated cells. After 7 minutes of illumination, ΔΨm decreased to almost zero and remained unchanged after this illumination time.
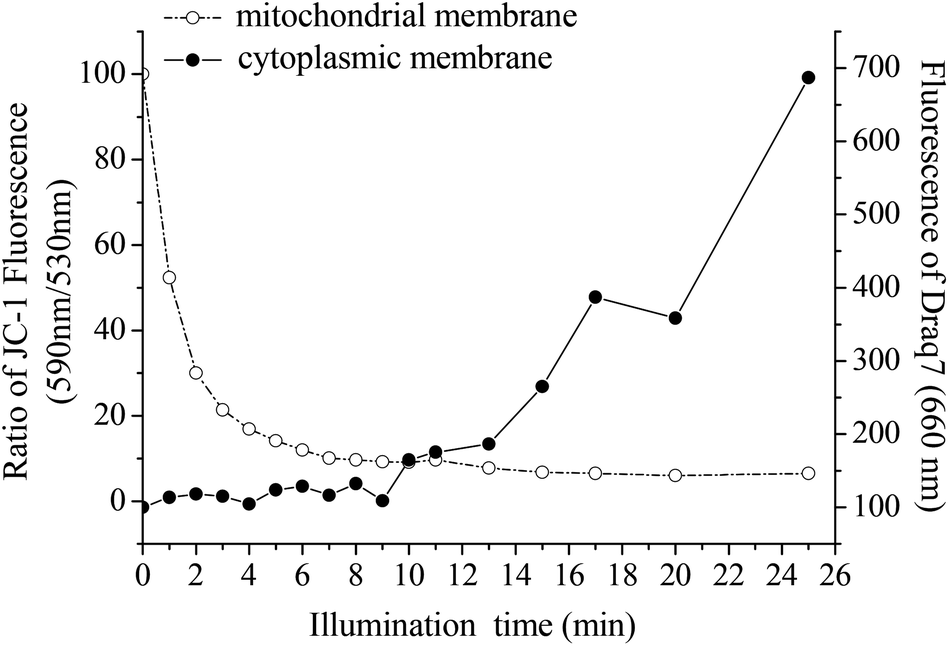 | ||
| Fig. 2 Illumination time-dependent effect of ALA-PDT on plasma membrane vs. mitochondrial membrane potential. U251 cells were treated with ALA for 4 hours, followed by addition of Draq7 and JC-1, and illuminated for different times (1–25 min). The damage to the mitochondrial membrane is represented by JC-1 red/green ratio (dotted curve), while the plasma membrane damage is shown by Draq7 (full curve). | ||
In addition to the above experiments, the cells were also assessed for the effect of the illumination on the level of damage that was caused to the plasma membrane by ALA-PDT treatment. The progress of this damage was tracked by using the new indicator dye Draq7, which is a far-red fluorescent DNA dye that specifically stains the nuclei of dead and permeabilized cells. Based on FACS results, no fluorescence of Draq7 was detected after up to 9 minutes of illumination, indicating that even this duration of illumination did not lead to membrane damage that would lead to penetration of the indicator dye into the cells’ nuclei (Fig. 2, full line). An increase in the fluorescence, indicating plasma membrane damage, was observed to increase rapidly when the samples were illuminated for times longer than 9 minutes.
Combining the two sets of data in Fig. 2, it can be seen that the mitochondrial membrane is more sensitive to ALA-PDT than the plasma membrane. While the mitochondrial membrane potential almost fully decreases within 7 minutes of illumination, the plasma membrane lags very much behind it and it starts to show signs of damage only after 9 minutes. This study does not point to the mechanism by which the plasma membrane is being damaged, nor did it intend to do so. It can be caused by singlet oxygen molecules that diffuse to the plasma membrane and cause damage to, most probably, membrane-localized protein systems. It can also be a result of a more indirect chain of events. Our aim was to show the big difference in the two types of mechanisms, which arises from the proximity of the origin of singlet oxygen generation, namely the photosensitizer, to one of the membranes, the mitochondrial membrane in the case of endogenous PpIX.
Comparing the rate of damage to mitochondrial and plasma membranes using exogenous photosensitizers
In addition to the above studies, we also examined the effect of different exogenous photosensitizers on the damage to mitochondrial and plasma membranes. The photosensitizers we chose were the hydrophobic protoporphyrin IX (PpIX), hydrophilic hypericin tetrasulfonic acid (HypS4) and hematoporphyrin (HP), an amphiphilic molecule. The examination of the effect of these exogenous photosensitizers was conducted in the same way as above, using the JC-1 and Draq7 probes. The concentrations of the sensitizers in the incubation medium and the incubation times were chosen so that the intracellular concentrations would be similar, based on their cellular fluorescence intensities and fluorescence–concentration calibration curves. At the concentrations that we used, none of the sensitizers affected toxicity to the cells in the dark.The results shown in Fig. 3 indicate that each photosensitizer affects the destruction of the mitochondrial and plasma membranes in a different way, as evidenced by the kinetics of the indicators JC-1 and Draq7. Table 1 summarizes the characteristic time that describes an exponential decrease in the mitochondrial electric potential as the illumination time increases. The table also gives the onset time of the damage to the plasma membrane, namely the length of illumination at which the fluorescence of Draq7 shows an inflection in its upward change of intensity.
 | ||
| Fig. 3 Illumination time-dependent effect of photosensitizers on mitochondrial membrane potential and on the plasma membrane. U251 cells were treated with different concentrations of photosensitizers, followed by addition of Draq7 and JC-1. The cells were irradiated for different time intervals (1–25 min), and the destruction kinetics of both plasma and mitochondrial membranes were examined. The damage of the mitochondrial membrane is represented by JC-1 fluorescence ratio (dotted curve), while the plasma membrane damage is shown by Draq7 (full curve). The cells were treated with (A) 75 nM exogenous PpIX for 2 hours, (B) 2 μM HypS4 for 5 hours in a medium without serum, (C) 2 μM HypS4 for 20 hours in a serum-enriched medium, (D) 75 nM HP for 40 minutes and (E) 75 nM HP for 2 hours. The descriptions of x and y axes in panels B to E are as in panel A. | ||
| Sensitizer and mode of incubation | Time to 1/e of the initial mitochondrial potential (minutes) | Onset point of damage to plasma membrane (minutes) |
|---|---|---|
| ALA | 1.79 ± 0.13 | 9 ± 1 |
| PpIX | 4.15 ± 0.60 | 1 ± 1 |
| HypS4 5 hours, without serum | 2.55 ± 0.30 | 6 ± 1 |
| HypS4 20 hours, with serum | 4.19 ± 0.45 | 1 ± 1 |
| HP (40 minutes) | 7.38 ± 1.49 | 6 ± 1 |
| HP (2 hours) | 2.52 ± 0.13 | 1 ± 1 |
Exogenous PpIX coupled with photodynamic treatment caused a decrease in the mitochondrial electric potential, ΔΨm, upon illumination, with a characteristic illumination time of about 4 minutes. The fluorescence of Draq7 with the same cells exhibited an instant increase, and the onset of the destruction of the plasma membranes was observed already after 1 minute of illumination (Table 1 and Fig. 3A). As explained above, PpIX is very hydrophobic. Therefore, it is expected that after diffusional partitioning into the plasma membrane, it will equilibrate into intracellular organelles quite slowly, much slower than the time scale of minutes that are relevant here. This is the basis for our understanding that the plasma membrane is damaged faster than the mitochondrial membrane when PpIX is given exogenously.
Two different incubation conditions were chosen for HypS4. The same concentration of 2 μM was applied for either 5 hours in a medium without serum, or for 20 hours in a serum-containing medium. The presence or absence of serum was tested since serum proteins can act as carriers for HypS4, and therefore they could affect the sensitizer's rate of penetration and its intracellular location. We employed a 20 hour incubation period to allow better infiltration of serum proteins into the cells, as it gives enough time for the protein–HypS4 complexes to reach equilibrium in the cells by endocytosis.32 The HypS4 penetrates the cell by endocytosis and the endosome fuses with a lysosome. The source of the damage is thus the photosensitizer in the endolysosome, but the effects of the damage extend to the mitochondrial and cytoplasmic membranes. HypS4 in serum-enriched medium coupled with photodynamic treatment caused ΔΨm to decrease with a time-constant of more than 4 minutes, and to reach saturation after just 8 minutes (Fig. 3C), while without serum the effect on the mitochondria was faster (Fig. 3B). On the other hand, the increase of the fluorescence of Draq7 was observed instantly when incubated with serum (Fig. 3C), while without serum the fluorescence of Draq7 began to rise significantly only after 6 minutes of illumination, indicating delayed damage of the plasma membrane under these conditions (Fig. 3B and Table 1).
Since hematoporphyrin is an amphiphilic photosensitizer, we wanted to assess whether subjection to different incubation times will affect its subcellular location, and thus the damage it causes to the various membranes. In order to measure this we selected a long incubation time (2 hours) and a short incubation time (40 minutes). The same concentration of HP (75 nM) was used with both incubations. The striking difference between these two conditions was that when 2 hours incubation was used the evolution of mitochondrial damage could be observed even after the shortest illumination time, it progressed rapidly with a time-constant of 2.52 minutes and was practically complete after 7 minutes of illumination (Fig. 3E). On the other hand, when HP was incubated with the cells for 40 minutes the illumination caused a slower effect on mitochondrial depolarization, having a time-constant of 7.38 minutes and about 16 minutes of illumination was required to reach maximal depolarization of the mitochondria (Fig. 3D). A similar difference was observed in the evolution of the damage to the plasma membrane. After the shorter incubation time, longer illumination was required (∼6 minutes) before the onset of membrane damage could be observed, whereas it started immediately, after only 1 minute of illumination, following the longer incubation.
Fig. 4 shows fluorescence microscopy pictures of the cells after the two protocols of incubation with HP. As can be seen, the sensitizer distributes itself into the cytoplasm rapidly and quite homogeneously, and one does not see any distinct staining of the plasma membrane at short time vs. staining of the cytosol after long incubation. What is evident, though, is stronger overall uptake of HP after longer incubation. This is reflected in the more rapid and efficient damage to both the mitochondrial and plasma membranes, as discussed above and shown in Table 1.
 | ||
| Fig. 4 Intracellular location of hematoporphyrin in U251 cells, after 40 minutes (left) and 2 hours (right) of incubation. | ||
Our results strongly suggest that the damage resulting from various photosensitizers affects the mitochondrial and plasma membranes differentially, as observed from both the onset points and the rates of the observed damage to the plasma and mitochondrial membranes. In addition, the photodynamic damage by the exogenous photosensitizers differs from that seen with the ALA-PDT alone. A correlation between the two damage pathways and the areas where the sensitizer's concentration is more pronounced is observed. As was mentioned earlier, this observation is independent of the mechanism that can be attributed to the real mechanism by which the damage is inflicted.
MTT and LDH tests of mitochondrial activity and plasma membrane integrity
In order to evaluate the activity of the mitochondria under the above conditions, the MTT assay was applied. The MTT assay33 is a rapid colorimetric assay, based on the tetrazolium salt MTT, which is cleaved in active mitochondria. The optical absorption of the cleaved form of MTT was read at 570 nm.The cells were treated with either ALA (Fig. 5A) or with the exogenous photosensitizers (Fig. 5B) and were illuminated for different time intervals (0–20 minutes). The results shown in Fig. 4 clearly indicate that the mitochondrial activity decreases with longer illumination time. When the cells were incubated with ALA, 3 minutes of illumination were sufficient to decrease the mitochondrial activity to 50% of the initial value. In contrast, the exogenous photosensitizers were able to reduce the mitochondrial activity by only 20% after the same illumination time. In addition, it can be seen that a good correlation is observed in the rate of mitochondrial membrane destruction caused by the various photosensitizers between the MTT assay results and the FACS results described earlier.
 | ||
| Fig. 5 The photodynamic effect by different sensitizers on mitochondrial activity. U251 cells were treated with either ALA (A) or various exogenous photosensitizers (B) and illuminated. The mitochondrial activity was evaluated by the MTT assay 24 hours after illumination, by reading the absorption at 570 nm. | ||
To assess the integrity of the plasma membrane, the lactate dehydrogenase (LDH) assay was employed. LDH is a cytosolic enzyme, and it leaks out of the cell in case of plasma membrane damage. The cells were incubated for 4 hours with ALA and illuminated for different times. Immediately after the illumination the release of the LDH protein into the medium was measured by monitoring the absorption band of LDH-L at 340 nm.
Fig. 6 shows the results of LDH activity outside the cells after photodynamic action with ALA or exogenous sensitizers. It can be seen that the longer the illumination time, the larger the extent of LDH leakage into the extracellular medium, which means larger destruction of the plasma membrane. Both the start point and the destruction rate of the plasma membrane differ for the examined conditions. For ALA-PDT, only after 8 minutes of illumination LDH begins to leak into the medium (Fig. 6A), similar to the result obtained by FACS with Draq7. However, with the exogenous photosensitizers the leakage was detected already in the first minutes of illumination, with a strong increase afterwards, as observed in the FACS experiments.
 | ||
| Fig. 6 LDH assay of plasma membrane integrity for different photosensitizers. U251 cells were treated with either ALA (A) or various exogenous photosensitizers (B) and illuminated for various times. The activity of LDH in the medium outside the cells was measured immediately after the illumination, by monitoring the absorption band of LDH-L at 340 nm. | ||
The location of the photosensitizers within the cell
As we have mentioned above, the difference between the observed effects of the various photosensitizers might be connected with their location inside the cells. In order to check this possibility, we examined the location of the photosensitizer HypS4 (hydrophilic molecule) incubated for 5 hours as compared with the photosensitizer PpIX produced endogenously by the mitochondria. The location was determined by fluorescence microscopy.Fig. 7 shows the difference between the fluorescence patterns of HypS4 (Fig. 7A) and PpIX (Fig. 7B). PpIX is seen to be uniformly distributed through the cell while HypS4 is observed to be present in vacuoles. Because PpIX is formed only in the mitochondria, and the mitochondria in nerve cells are scattered throughout the cell, we assumed a diffuse distribution of the fluorescence near mitochondria, which was confirmed by this fluorescence microscopy. However, HypS4 is a hydrophilic molecule, which we would expect to find in the cytoplasm within the cell, and also here, with fluorescence microscopy, we can see that the fluorescence comes from this area. We have shown in a previous publication that HypS4 exhibited mainly endolysosomal internalization into murine colon carcinoma CT26 cells.34
 | ||
| Fig. 7 Intracellular location of photosensitizers. (A) Fluorescence of exogenous HypS4 after 5 hours incubation in medium without serum. (B) Fluorescence of endogenous PpIX produced in the mitochondria. | ||
Morphological analysis of cells after photodynamic treatment
To further evaluate the difference between plasma membrane damage caused by externally provided versus endogenously synthesized PpIX, scanning electron microscopy (SEM) was employed. The U251 cells were incubated with ALA or PpIX for 4 hours or 2 hours respectively, followed by illumination of 5 or 10 minutes, and were then examined for the extent of plasma membrane damage, as compared to the control cells (Fig. 8). After 5 minutes of illumination, the plasma membrane starts to get damaged in cells treated with exogenous PpIX (Fig. 8B), as compared to the intact membrane of cells that were treated with endogenous PpIX from ALA (Fig. 8C). However, after 10 minutes of illumination, approximately the same damage is seen for both types of PpIX treatments (Fig. 8D and E). Thus, 10 minutes of illumination are long enough to destroy and perforate the plasma membrane with treatment by both types of sensitizers. The different kinetics of external and internal damage to the outer membrane and to mitochondria indicate the initiation of necrotic and apoptotic mechanisms by these two sensitization procedures. Indeed, the FACS results also showed, more quantitatively, that the damage to the plasma membrane with externally provided PpIX progressed more rapidly than when it was produced endogenously with ALA (Table 1).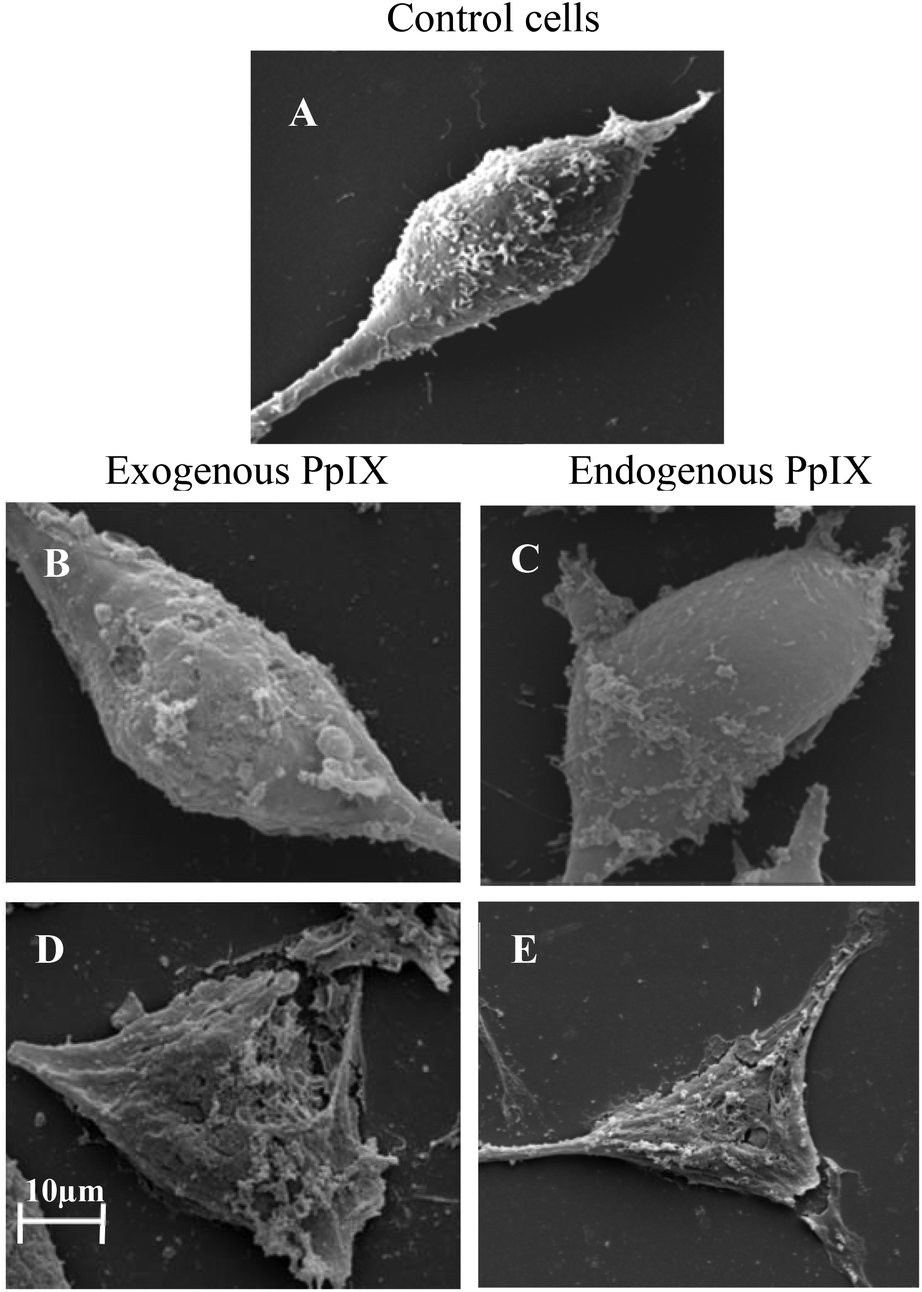 | ||
| Fig. 8 The damage induced to the plasma membrane by endogenous vs. exogenous PpIX, after different illumination times. (A) U251 control cells. (B) Cells incubated with exogenous PpIX, after 5 minutes of illumination. (C) Cells incubated with ALA (endogenous PpIX), after 5 minutes of illumination. (D) Cells incubated with exogenous PpIX, after 10 minutes of illumination. (E) Cells incubated with ALA, after 10 minutes of illumination. The scale bar for SEM images was 10 μm. | ||
Discussion
Our aim in this work was to study the rate at which the plasma membrane is being damaged by photodynamic action, and compare it with the rate of photosensitized damage to the mitochondrial inner membrane, in whichever mechanisms these reactions occur. Damage to either of them may lead to movement of ions across these membranes, thus depolarizing the cross-membrane electrochemical resting potential. This may lead to an immediate halt in the activity of ion pumps that reside in the plasma membrane, or decouple the flow of electrons across the mitochondrial membrane from the formation of a proton-gradient, as required by Mitchell's Chemiosmotic Theory.It is a reasonable assumption that the intracellular residence of a photosensitizer might determine the region in the cell where the damage would manifest itself first, because of closer proximity for singlet oxygen to diffuse. A previous study has shown that Rose Bengal and merocyanine 540, which are sensitizers that localize in the plasma membrane, caused damage to five membrane functions that were assessed.19 Another study used two structural isomers of an analog of tetraphenylporphyrin.22 This study found that the isomer that localized in the mitochondria of murine cells caused rapid loss of the mitochondrial electric potential, while the other isomer, which was internalized into lysosomes, caused extensive photodamage to the lysosomes.
The results of the present study directly address the old debate on the differences between PpIX that was synthesized endogenously from ALA versus PpIX or other sensitizers that were added externally, in inducing intracellular damage of major targets in the cell and of the plasma and mitochondrial membranes. The sensitizers differ in their hydrophobicity/hydrophilicity, which leads to their different localizations inside the cells. We also used different protocols of incubation of the cells with some of the sensitizers. We have observed that photosensitizers that are localized close to the mitochondria, such as protoporphyrin IX that is synthesized from δ-aminolevulinic acid, cause rapid and immediate damage to the mitochondrial membrane, while a long illumination is required before one can observe the onset of damage to the plasma membrane. In a sharp contrast, the same protoporphyrin IX, if supplied externally, is taken up also by a diffusive mechanism into the plasma membrane, which, as a consequence, exhibits an immediate onset of damage while the depolarization of the mitochondrial membrane is slower in this case than with endogenously-synthesized protoporphyrin IX. The other cases fall between these extremes. For example, hematoporphyrin that was incubated with the cells for a long, 2-hour, period and is distributed throughout the cell and its membranes causes fast depolarization of the mitochondrial membrane and also a fast onset of the damage to the plasma membrane.
In conclusion, the onset of membrane damage induced by PpIX that was provided externally or synthesized within the cells, as well as the damage that is caused by other sensitizers of different characteristics of polarity and that are taken up by various protocols of incubation, reflect differences in the kinetics of damage mechanisms. These may also involve differences between apoptotic and necrotic pathways, depicted by the decay in membrane potentials and collapse of membrane integrity.
Acknowledgements
This research was supported by a grant from the US–Israel Binational Science Foundation and by the Michael David Falk Chair in Laser Phototherapy.References
- T. J. Dougherty, J. E. Kaufman, A. Goldfarb, K. R. Weishaupt, D. Boyle and A. Mittleman, Photoradiation therapy for the treatment of malignant tumors, Cancer Res., 1978, 38, 2628–2635 CAS.
- T. J. Dougherty, Photosensitizers: therapy and detection of malignant tumors, Photochem. Photobiol., 1987, 45, 879–889 CrossRef CAS.
- B. W. Henderson and T. J. Dougherty, How does photodynamic therapy work?, Photochem. Photobiol., 1992, 55, 145–157 CrossRef CAS.
- G. Jori and S. B. Brown, Photosensitized inactivation of microorganisms, Photochem. Photobiol. Sci., 2004, 3, 403–405 CAS.
- M. Wainwright, Photoinactivation of viruses, Photochem. Photobiol. Sci., 2004, 3, 406–411 CAS.
- A. Juzeniene, Q. Peng and J. Moan, Milestones in the development of photodynamic therapy and fluorescence diagnosis, Photochem. Photobiol. Sci., 2004, 6, 1234–1245 Search PubMed.
- S. Verma, G. M. Watt, Z. Mal and T. Hasan, Strategies for enhanced photodynamic therapy effects, Photochem. Photobiol., 2007, 83, 996–1005 CrossRef CAS PubMed.
- J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Activatable photosensitizers for imaging and therapy, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS PubMed.
- S. K. Sharma, P. Mroz, T. H. Dai, Y. Y. Huang, T. G. St. Denis and M. R. Hamblin, Photodynamic Therapy for Cancer and for Infections: What Is the Difference?, Isr. J. Chem., 2012, 52, 691–705 CrossRef CAS PubMed.
- Z. Malik and M. Djaldetti, Destruction of erythroleukemia myelocytic leukemia and Burkitt lymphoma cells by photoactivated protoporphyrin, Int. J. Cancer, 1980, 26, 495–500 CrossRef CAS.
- J. P. J. Boegheim, J. W. M. Lagerberg, T. M. A. R. Dubbelman, K. Tijssen, H. J. Tanke, J. Van der Meulen and J. Van Steveninck, Photodynamic effects of HPD on the uptake of rhodamine 123 by mitochondria of intact murine L929 fibroblasts and Chinese hamster ovary K1 cells, Photochem. Photobiol., 1988, 48, 613–620 CrossRef CAS.
- M. Paardekooper, P. J. A. Van den Broek, A. W. De Bruijne, J. G. R. Elferink, T. M. A. R. Dubbelman and J. Van Steveninck, Photodynamic treatment of yeast cells with the dye toluidine blue: all-or-none loss of plasma membrane barrier properties, Biochim. Biophys. Acta, 1992, 1108, 86–90 CrossRef CAS.
- K. Berg and J. Moan, Lysosomes and microtubules as targets for photochemotherapy of cancer, Photochem. Photobiol., 1997, 65, 403–409 CrossRef CAS.
- H. Mojzisova, S. Bonneau and D. Brault, Structural and physico-chemical determinants of the interactions of macrocyclic photosensitizers with cells, Eur. Biophys. J., 2007, 36, 943–953 CrossRef CAS PubMed.
- D. A. Bellnier and T. J. Dougherty, Membrane lysis in Chinese hamster ovary cells treated with hematoporphyrin derivative plus light, Photochem. Photobiol., 1982, 36, 43–47 CrossRef CAS.
- B. W. Henderson and J. M. Donovan, Release of prostaglandin E2 from cells by photodynamic treatment in vitro, Cancer Res., 1989, 49, 6896–6900 CAS.
- K. G. Specht and M. A. J. Rodgers, Plasma membrane depolarization and calcium influx during cell injury by photodynamic action, Biochim. Biophys. Acta, 1991, 1070, 60–68 CrossRef CAS.
- B. Krammer-Reubel, Transmembrane potential measurements of normal and transformed human fibroblasts following photodynamic laser therapy, Bioelectrochem. Bioenerg., 1992, 27, 19–22 CrossRef.
- I. E. Kochevar, J. Bouvier, M. Lynch and C. W. Lin, Influence of dye and protein location on photosensitization of the plasma membrane, Biochim. Biophys. Acta-Biomembranes, 1994, 1196, 172–180 CrossRef.
- L. Kunz and G. Stark, Photofrin II sensitized modifications of ion transport across the plasma membrane of an epithelial cell line: I. Electrical measurements at the whole-cell level, J. Membr. Biol., 1998, 166, 179–185 CrossRef CAS.
- R. Chaloupka, T. Obsil, J. Plasek and F. Sureau, The effect of hypericin and hypocrellin-A on lipid membranes and membrane potential of 3T3 fibroblasts, Biochim. Biophys. Acta-Biomembranes, 1999, 1418, 39–47 CrossRef CAS.
- D. Kessel, R. Luguya and M. G. H. Vicente, Localization and photodynamic efficacy of two cationic porphyrins varying in charge distribution, Photochem. Photobiol., 2003, 78, 431–435 CrossRef CAS.
- B. Ehrenberg, E. Gross, Y. Nitzan and Z. Malik, Electric depolarization of photosensitized cells: lipid vs. protein alterations, Biochim. Biophys. Acta, 1993, 1151, 257–264 CrossRef CAS.
- S. Ytzhak, J. P. Wuskell, L. M. Loew and B. Ehrenberg, Lipid composition affects the rate of photosensitized dissipation of cross-membrane diffusion potential on liposomes, J. Phys. Chem. B, 2010, 114, 10097–10104 CrossRef CAS PubMed.
- S. Ytzhak, H. Weitman and B. Ehrenberg, The effect of lipid composition on the permeability of fluorescent markers from photosensitized membranes, Photochem. Photobiol., 2013, 89, 619–624 CrossRef CAS PubMed.
- http://www.biostatus.com/DRAQ7_s/1836.htm .
- S. Iinuma, S. S. Farshi, B. Ortel and T. Hasan, A mechanistic study of cellular photodestruction with 5-aminolaevulinic acid-induced porphyrin, Br. J. Cancer, 1994, 70, 21–28 CrossRef CAS.
- X. Wang, P. Wang, W. Tong and Q. Liu, Comparison of pharmacokinetics, intracellular localizations and sonodynamic efficacy of endogenous and exogenous protoporphyrin IX in sarcoma 180 cells, Ultrasonics, 2010, 50, 803–810 CrossRef CAS PubMed.
- H. Kobuchi, K. Moriya, T. Ogino, H. Fujita, K. Inoue, T. Shuin, T. Yasuda, K. Utsumi and T. Utsumi, Mitochondrial localization of ABC transporter ABCG2 and its function in 5-aminolevulinic acid-mediated protoporphyrin IX accumulation, PLoS One, 2012, 7, e50082 CAS.
- Z. Malik and M. Djaldetti, 5-Aminolevulinic acid stimulation of porphyrin and hemoglobin synthesis by uninduced Friend erythroleukemic cells, Cell Differ., 1979, 8, 223–233 CrossRef CAS.
- Z. Malik and H. Lugaci, Selective destruction of erythroleukemic cells by photoactivation of endogenous porphyrins, Br. J. Cancer, 1987, 56, 389–395 CrossRef.
- G. Siboni, H. Weitman, D. Freeman, Y. Mazur, Z. Malik and B. Ehrenberg, Photochem. Photobiol. Sci., 2002, 1, 483–491 CAS.
- G. Denis and T. Nicole, Use of MTT colorimetric assay to measure cell activation, J. Immunol. Methods, 1986, 94, 57–63 CrossRef.
- G. Siboni, H. Weitman, D. Freeman, Y. Mazur, Z. Malik and B. Ehrenberg, The correlation between hydrophilicity of hypericins and helianthrone: internalization mechanisms, subcellular distribution and photodynamic action in colon carcinoma cells, Photochem. Photobiol. Sci., 2002, 1, 483–491 CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2014 |