 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A “click” chemistry constructed affinity system for 2-oxoglutaric acid receptors and binding proteins†
Yang
Wang
ab,
Zeinab
Assaf
ac,
Xinjun
Liu
b,
Fabio
Ziarelli
d,
Amel
Latifi
c,
Otmane
Lamrabet
c,
Gilles
Quéléver
a,
Fanqi
Qu
b,
Cheng-Cai
Zhang
c and
Ling
Peng
*a
aAix-Marseille Université and CNRS, Centre Interdisciplinaire de Nanoscience de Marseille, UMR 7325, 13288, Marseille, France. E-mail: ling.peng@univ-amu.fr
bCollege of Chemistry and Molecular Sciences, Wuhan University, P. R. China
cAix-Marseille Université and CNRS, Laboratoire de Chimie Bactérienne, UMR 7283, Marseille, France
dAix-Marseille Université and CNRS, Spectropôle, FR 1739, Marseille, France
First published on 1st July 2014
Abstract
An ingenious and specific affinity resin designed to capture the 2-oxoglutaric acid (2-OG) binding proteins was constructed by appending a 2-OG tag to the solid resin via a Cu-catalyzed Huisgen “click” reaction. The so-obtained affinity resin was able to recognize, retain and separate the established 2-OG binding protein NtcA in both the pure form and crude cellular extract, thus constituting a valuable means of searching for novel 2-OG receptors with a view to exploring the signalling pathways of 2-OG, a key Krebs cycle intermediate with unprecedented signalling functions.
Introduction
2-Oxoglutaric acid (2-OG) (Scheme 1) is a strategically important intermediate of the Krebs cycle since it constitutes the precursor and the carbon skeleton for nitrogen assimilation, leading to the synthesis of various biomolecules and the production of cellular energy. In addition to its canonical roles, 2-OG has recently regained considerable attention as a key signalling molecule1–5 in different organisms as it plays important roles in various signalling pathways such as regulation of the balance in carbon and nitrogen metabolism,6–8 epigenetic regulation mediated by 2-OG dependent oxygenases,9 generation of the “onco-metabolite” 2-hydroxyglutarate via misregulated metabolism in various cancers, etc.10 The signalling pathways of 2-OG are rather complex and remain largely unexplored because of the difficulties faced in identifying the corresponding 2-OG receptors or 2-OG binding proteins. The prerequisite therefore in achieving the ultimate goal of better understanding the signalling role of 2-OG is finding a simple and effective way of searching and identifying potential 2-OG receptors. To this end, affinity chromatography appears to be the method of choice by virtue of its high specificity, easy manipulation and simple scale-up.11 In this method, a competent tag bearing 2-OG mimics shall be immobilized onto the solid support via an appropriate spacer arm, and the resulting affinity resin (Fig. 1) is expected to capture the 2-OG binding proteins via specific interactions between the 2-OG binding site of the proteins and the 2-OG mimic attached on the resin. An elution step using 2-OG containing eluent would then deliver the 2-OG binding proteins enabling their subsequent purification and identification.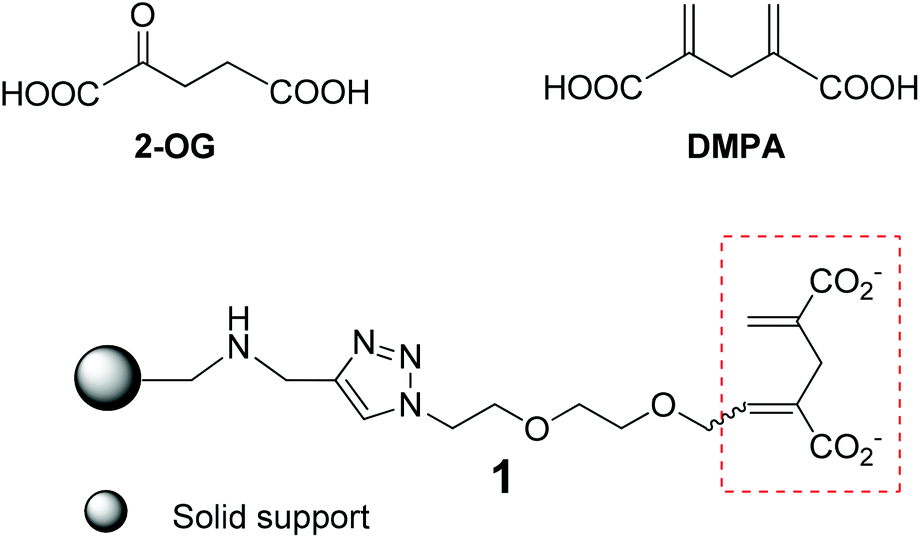 | ||
| Scheme 1 Structure of 2-OG, the 2-OG analogue DMPA and the affinity resin 1 with DMPA as a 2-OG tag on the solid support. | ||
 | ||
| Fig. 1 The principle of using 2-OG affinity chromatography to capture the potential 2-OG receptors. | ||
The foremost requirement to guarantee a successful affinity chromatographic study is establishing a competent affinity resin with high affinity and specificity, where the apt tag to sense the 2-OG binding proteins is of paramount importance. However, the native 2-OG does not appear to be the tag of choice as the α-keto carboxylic acid entity within 2-OG is rather labile, and ready to undergo different transformations in biological media containing various enzymes. This is certainly the biggest concern during our consideration to devise an appropriate affinity column to capture the 2-OG binding proteins in the crude cell extracts or biological media where many enzymes or proteins may react with the α-keto carboxylic acid entity. Furthermore, 2-OG is a small molecule, and any structural modification with relatively large functional moieties will inevitably modify binding characteristics towards 2-OG receptors.4,12–15 Taking all these into consideration, we opted for the nonmetabolisable 2-OG analogue, 2,4-dimethylene-pentanedioic acid (DMPA), as the 2-OG tag to construct the affinity resin 1 (Scheme 1) for its structural resemblance to, and resulting ability to mimic, 2-OG in signaling.14 In addition, it is practical for chemical modification and conjugation of DMPA onto the solid support. For linking the 2-OG tag onto the solid resin, we chose the hydrophilic ethyleneoxy spacer in order to provide a hydrophilic environment allowing the 2-OG tag to search for and bind to the 2-OG binding proteins. To anchor the 2-OG tag onto the resin, we wished to harness the Cu-catalyzed Huisgen reaction,16 a well acknowledged “click” reaction for affording clean products efficiently and rapidly in high yield under mild conditions, hence allowing a facile and economic solid support synthesis.
Based on this rationale, we report here the synthesis, characterization and biological validation of the so-devised affinity resin 1 that can selectively and specifically recognize, retain and separate the 2-OG binding protein NtcA, a known 2-OG receptor involved in regulating the carbon/nitrogen balance and heterocyst differentiation in diazotrophic cyanobacteria.4,12–15,17 To our knowledge, this is the first example of an affinity resin using a 2-OG mimic tag validated with a known 2-OG receptor.18 This affinity system hence holds great promise in searching for 2-OG receptors or 2-OG binding proteins involved in signal transduction and metabolic regulation.
Results and discussion
The construction of the affinity resin 1 was achieved according to the strategy presented in Scheme 2. The precursor of the 2-OG tag featuring the ethyleneoxy spacer (5 in Scheme 2) was obtained via an Horner–Wadsworth–Emmons (HWE) reaction20 by condensing the corresponding aldehyde 2 with the phosphonate 3 in the presence of sodium hydride, affording 4 with an excellent yield of 85% as a mixture of E/Z isomers (E/Z = 3/1).21 Subsequent treatment with NaN3 delivered the corresponding azides 5, which were further conjugated to the alkyne-bearing resin 6via a Cu-catalyzed Huisgen cycloaddition to yield the resin 7. Gratifyingly, the Cu-catalyzed “click” reaction proved to be very powerful and rewarding as it proceeded smoothly, and was accomplished rapidly within an hour (Fig. S1†). As shown in the IR spectra (Fig. 2A), the peak at 3289 cm−1 corresponding to the vibration of the C–H bond in the terminal alkynyl –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H moiety of 6 disappeared following the “click” reaction, and a very strong signal was generated around 1707 cm−1, relating to the carbonyl group of the ester functions of 7. The successful conversion of 6 to 7 was further affirmed by 13C cross polarization magic angle spinning (CPMAS) NMR, an excellent non-destructive method for the in situ analysis of the solid phase reaction.22–24 As illustrated in Fig. 2B, the disappearance of the signals associated with the alkynyl moiety at 70–80 ppm in 6 was accompanied by the emergence of the signals at 14 and 60 ppm corresponding to the ethyl groups within the ester functions of 7. Collectively, these results indicate a rapid and complete conversion of 6 to deliver 7via “click” chemistry. Subsequent alkaline hydrolysis of 7 furnished the final resin 1, which was successfully characterized by both IR and CPMAS NMR. As we can see in Fig. 2A, the hydrolysis of the ester groups in 7 to give the corresponding carboxylate functions was highlighted by the signal of the carbonyl group shifting from 1707 cm−1 (characteristic of the ester groups in 7) to 1658 cm−1 (characteristic of the carboxylate functions in 1). This was further confirmed with the results obtained by 13C CPMAS NMR. As clearly depicted in Fig. 2B, the signal concerning the ester carbonyl groups in 7 wore off at 166 ppm, and the new broad peak corresponding to the carboxylate carbonyl groups in 1 emerged at 175 ppm. Moreover, the signals relating to the ethyl moieties in the ester groups of 7 disappeared at 14 and 60 ppm, further stressing the successful hydrolysis of 7 to afford the resin 1.
C–H moiety of 6 disappeared following the “click” reaction, and a very strong signal was generated around 1707 cm−1, relating to the carbonyl group of the ester functions of 7. The successful conversion of 6 to 7 was further affirmed by 13C cross polarization magic angle spinning (CPMAS) NMR, an excellent non-destructive method for the in situ analysis of the solid phase reaction.22–24 As illustrated in Fig. 2B, the disappearance of the signals associated with the alkynyl moiety at 70–80 ppm in 6 was accompanied by the emergence of the signals at 14 and 60 ppm corresponding to the ethyl groups within the ester functions of 7. Collectively, these results indicate a rapid and complete conversion of 6 to deliver 7via “click” chemistry. Subsequent alkaline hydrolysis of 7 furnished the final resin 1, which was successfully characterized by both IR and CPMAS NMR. As we can see in Fig. 2A, the hydrolysis of the ester groups in 7 to give the corresponding carboxylate functions was highlighted by the signal of the carbonyl group shifting from 1707 cm−1 (characteristic of the ester groups in 7) to 1658 cm−1 (characteristic of the carboxylate functions in 1). This was further confirmed with the results obtained by 13C CPMAS NMR. As clearly depicted in Fig. 2B, the signal concerning the ester carbonyl groups in 7 wore off at 166 ppm, and the new broad peak corresponding to the carboxylate carbonyl groups in 1 emerged at 175 ppm. Moreover, the signals relating to the ethyl moieties in the ester groups of 7 disappeared at 14 and 60 ppm, further stressing the successful hydrolysis of 7 to afford the resin 1.
 | ||
| Scheme 2 Preparation of the affinity resin 1. | ||
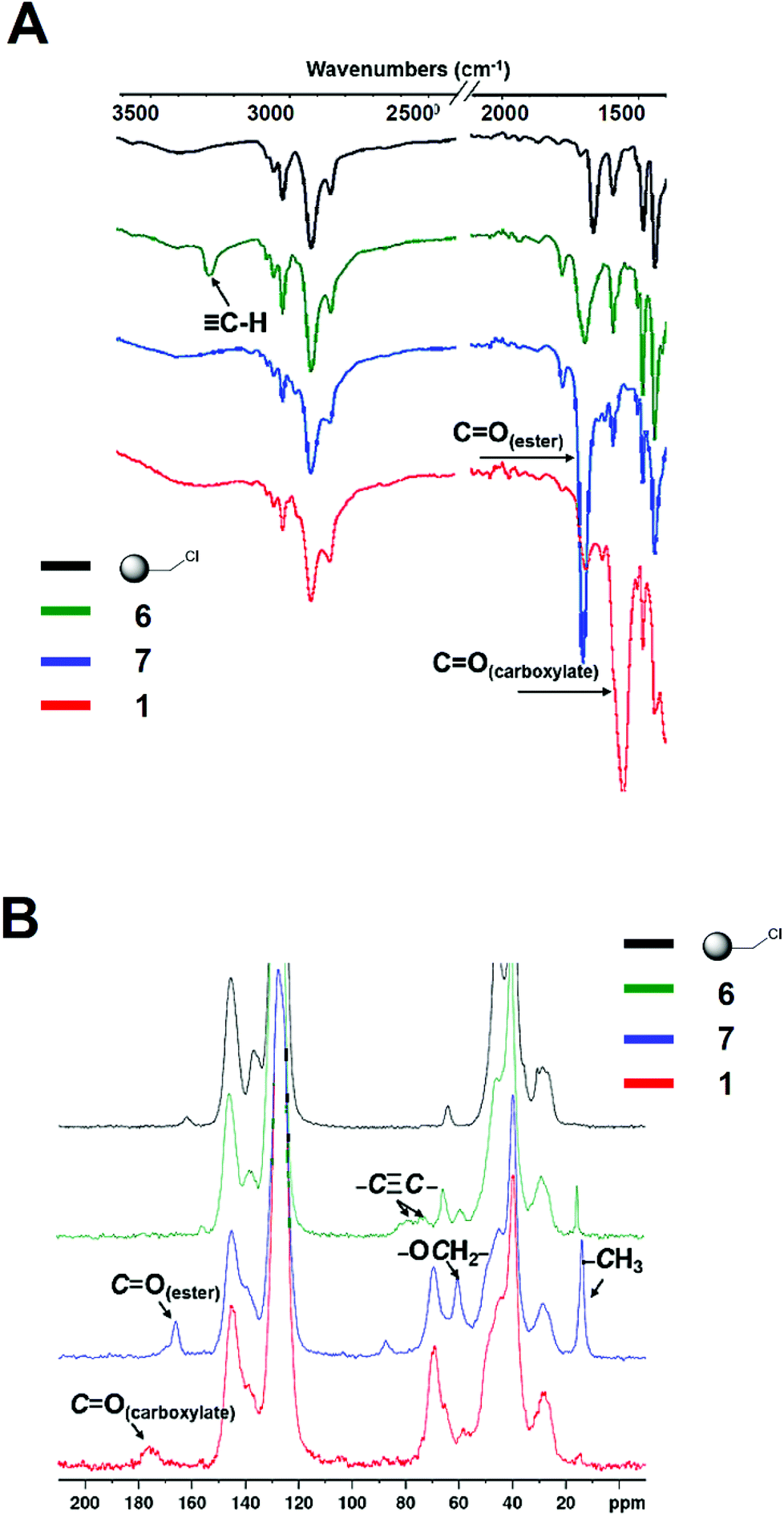 | ||
| Fig. 2 IR and 13C CPMAS NMR spectra of the intermediate products 6 and 7, and the final affinity resin 1. | ||
To verify that the above-obtained affinity resin 1 could indeed recognize and bind 2-OG binding proteins, we examined its interaction with NtcA, an established 2-OG receptor regulating nitrogen metabolism and responsible for heterocyst formation in cyanobacteria.4,17 Bovine serum albumin (BSA) was used as a negative control of non-2-OG binding protein. No retention of BSA on resin 1 was observed (data not shown), whereas 1 could effectively retain NtcA on the column, as indicated by the revelation of NtcA in the solution before and after passing through the affinity column composed of 1 (Fig. 3A, lanes 2, 3, 4 and 5). The retained NtcA on the affinity column was then effectively eluted out using a solution containing 2-OG (Fig. 3A, lane 6), authenticating that the retention of NtcA on the resin 1 is specific via the explicit interaction between NtcA and the 2-OG tag featured on 1.
 | ||
| Fig. 3 Protein revelation of resin 1 capturing and eluting NtcA from (A) a pure NtcA solution and (B) E. coli cell lysate. Lane 1: protein molecular weight marker; lane 2: NtcA or E. coli cell lysate as a reference; lane 3: the supernatant from the incubation buffer; lanes 4 and 5: elution with PBS; lane 6: elution with 2-OG solution (0.5 M) in PBS buffer; lane 7: elution with PBS; lane 8: elution with NaCl solution; lane 9: elution with PBS. The pH of PBS and 2-OG elution was controlled at pH = 7.5. | ||
We further tested the robustness of the affinity resin 1 in identifying and capturing the 2-OG receptor NtcA from cell lysate. To this end, we assessed the ability of resin 1 to fish out the NtcA protein from the total cellular extract of an E. coli strain which over-expresses NtcA. We found that 1 could indeed selectively and specifically anchor NtcA, and then release it when using 2-OG elution (Fig. 3B). Altogether, these results demonstrate the robust specificity of the affinity resin 1 and its potential use in detecting and searching for 2-OG receptors.
Conclusions
In summary, we have successfully established the affinity resin 1 for specific recognition, capture and release of the 2-OG receptor NtcA via affinity chromatography. This resin features an affinity tag bearing a 2-OG mimic, which was appended to the solid support resin via a hydrophilic linker. Cu-catalyzed “click” chemistry was implemented to conjugate the tag to the resin, and proved to be very efficient and rewarding. The so-obtained 2-OG resin could not only retain the 2-OG receptor NtcA in its pure form, but also identify and capture NtcA from the cell extract containing a mixture of various macromolecules, before allowing its release from the column with an eluent containing 2-OG. Altogether, these results illustrate the power and value of this resin in specifically recognizing, sufficiently binding and effectively releasing the 2-OG receptor NtcA. Consequently, this resin constitutes a novel useful means of searching for new 2-OG receptors with a view to investigate the signaling pathways of 2-OG and complete our understanding of its signaling roles. We are actively pursuing in this direction.Experimental section
General methods
All the reactions were carried out under argon. Monotosylated triethylene glycol was synthesized according to the literature.25 The detailed synthesis of compounds 2 and 3 and resin 6 is described in the ESI.† Anhydrous 1,2-dimethoxyethane (DME) was distilled in the presence of sodium-benzophenone. Anhydrous CH2Cl2 and DMSO were prepared by distillation in the presence of CaH2. 85% NaH was prepared by washing 60% NaH in mineral oil with petroleum ether and drying in a vacuum. The Merrifield resin (loading capacity: 3.1 mmol g−1) was purchased from Iris Biotech (Germany). All the other reagents were purchased from Sigma-Aldrich (China) or Acros Organics (China) without any further purification. Silica gel (200–300 mesh) used for flash chromatography was purchased from Qing Dao Hai Yang Chemical Industry Co. (China). 1H NMR and 13C NMR spectra were recorded at 300 MHz and 75 or 150 MHz respectively on Varian Mercury-VX300 and VX600 spectrometers. Chemical shifts are reported in parts per million (ppm) with TMS as an internal reference. The 31P NMR spectrum was recorded at 121 MHz on a Varian Mercury-VX300 spectrometer. Chemical shifts were reported relatively to 85% H3PO4 as the external standard. The solid-state 13C cross polarization magic angle spinning (CPMAS) NMR spectra were recorded on a Bruker Avance-400 MHz NMR spectrometer. HRMS were recorded using Waters Micromass GCT Premier and QStar Elite Mass spectrometers. IR spectra were recorded using a Bruker Alfa IR spectrometer. The microwave reaction was performed in a CEM Discover SP microwave reactor.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–4:1, v/v), yielding 4 as a colorless oil (2.22 g, 85%, E/Z ≈ 3/1). 1H NMR (300 MHz, CDCl3) δ 7.80 (d, J = 8.10 Hz, 2H, PhCH3), 7.35 (d, J = 8.40 Hz, 2H, –PhCH3), 6.98 (t, J = 5.70 Hz, 1H, E: C
:1–4:1, v/v), yielding 4 as a colorless oil (2.22 g, 85%, E/Z ≈ 3/1). 1H NMR (300 MHz, CDCl3) δ 7.80 (d, J = 8.10 Hz, 2H, PhCH3), 7.35 (d, J = 8.40 Hz, 2H, –PhCH3), 6.98 (t, J = 5.70 Hz, 1H, E: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2), 6.23 (s, 1H, Z: CCHH), 6.19–6.14 (m, 2H, E: CCHH, Z: CCHCH2), 5.55 (s, 1H, Z: CCHH), 5.49 (s, 1H, E: CCHH), 4.46 (d, J = 4.80 Hz, 1H, Z: CCHCH2), 4.23–4.14 (m, 8H, CCHCH2, CH2OTs, OCH2CH3), 3.70 (t, J = 4.80 Hz, CH2CH2OTs), 3.61–3.56 (m, 4H, OCH2), 3.31 (s, 2H, E: CH2), 3.29 (s, 2H, Z: CH2), 2.45 (s, 3H, –PhCH3), 1.32–1.25 (m, 6H, –CH2CH3); 13C NMR (150 MHz, CDCl3) δ 166.9, 166.8, 145.2, 144.0, 141.0, 138.6, 137.5, 133.2, 130.6, 130.2, 129.3, 128.2, 126.7, 125.6, 70.9–68.2, 61.2, 60.9, 35.3, 29.3, 21.9, 14.5; HRMS cacld for C23H32O9SNa+ 507.1659, found 507.1659.
:1–4:1, v/v), yielding pale yellow oil 5 as a mixture of E/Z isomers (863 mg, 54%, E/Z ≈ 3/1). 1H NMR (300 MHz, CDCl3) δ 7.00 (t, J = 5.90 Hz, 1H, E: CCHCH2), 6.23 (s, 1H, Z: CCHH), 6.19 (s, 2H, E: CCHH, Z: CCHCH2), 5.54 (s, 1H, Z: CCHH), 5.48 (s, 1H, E: CCHH), 4.49 (d, J = 4.80 Hz, 1H, Z: CCHCH2), 4.25–4.14 (m, 6H, CCHCH2, OCH2CH3), 3.70–3.63 (m, 6H, –OCH2), 3.41 (t, J = 4.95 Hz, CH2N3), 3.32 (s, 2H, E: CH2), 3.29 (s, 2H, Z: CH2), 1.32–1.24 (m, 6H, –CH2CH3); 13C NMR (150 MHz, CDCl3) δ 166.6, 166.5, 143.8, 140.8, 138.3, 137.2, 130.3, 129.0, 126.4, 125.2, 70.6–69.8, 68.0, 60.8, 60.6, 50.6, 35.0, 29.0, 14.2; IR (cm−1) ν 2106.1 (–N3); HRMS cacld for C16H26N3O6+ 356.1816, found 356.1815.
:1, v/v) were added 5 (196 mg, 0.552 mmol) and CuI (5.00 mg, 0.0263 mmol). The reaction mixture was stirred at 35 °C for 24 h, and then allowed to cool down to room temperature and filtered. The obtained resin was washed successively with THF, water, saturated EDTA solution, water, MeOH, CH2Cl2 and diethyl ether (30.0 mL for each) and dried in a vacuum, yielding 160 mg desired resin 7 as a pale green powder. IR (cm−1) ν 1707 (CO); 13C CPMAS NMR δ 166.0 (CO), 60.6 (OCH2CH3), 14.0 (OCH2CH3).
O); 13C CPMAS NMR δ 175.9 (CO).
CHCH2), 6.23 (s, 1H, Z: CCHH), 6.19–6.14 (m, 2H, E: CCHH, Z: CCHCH2), 5.55 (s, 1H, Z: CCHH), 5.49 (s, 1H, E: CCHH), 4.46 (d, J = 4.80 Hz, 1H, Z: CCHCH2), 4.23–4.14 (m, 8H, CCHCH2, CH2OTs, OCH2CH3), 3.70 (t, J = 4.80 Hz, CH2CH2OTs), 3.61–3.56 (m, 4H, OCH2), 3.31 (s, 2H, E: CH2), 3.29 (s, 2H, Z: CH2), 2.45 (s, 3H, –PhCH3), 1.32–1.25 (m, 6H, –CH2CH3); 13C NMR (150 MHz, CDCl3) δ 166.9, 166.8, 145.2, 144.0, 141.0, 138.6, 137.5, 133.2, 130.6, 130.2, 129.3, 128.2, 126.7, 125.6, 70.9–68.2, 61.2, 60.9, 35.3, 29.3, 21.9, 14.5; HRMS cacld for C23H32O9SNa+ 507.1659, found 507.1659.
:1–4:1, v/v), yielding pale yellow oil 5 as a mixture of E/Z isomers (863 mg, 54%, E/Z ≈ 3/1). 1H NMR (300 MHz, CDCl3) δ 7.00 (t, J = 5.90 Hz, 1H, E: CCHCH2), 6.23 (s, 1H, Z: CCHH), 6.19 (s, 2H, E: CCHH, Z: CCHCH2), 5.54 (s, 1H, Z: CCHH), 5.48 (s, 1H, E: CCHH), 4.49 (d, J = 4.80 Hz, 1H, Z: CCHCH2), 4.25–4.14 (m, 6H, CCHCH2, OCH2CH3), 3.70–3.63 (m, 6H, –OCH2), 3.41 (t, J = 4.95 Hz, CH2N3), 3.32 (s, 2H, E: CH2), 3.29 (s, 2H, Z: CH2), 1.32–1.24 (m, 6H, –CH2CH3); 13C NMR (150 MHz, CDCl3) δ 166.6, 166.5, 143.8, 140.8, 138.3, 137.2, 130.3, 129.0, 126.4, 125.2, 70.6–69.8, 68.0, 60.8, 60.6, 50.6, 35.0, 29.0, 14.2; IR (cm−1) ν 2106.1 (–N3); HRMS cacld for C16H26N3O6+ 356.1816, found 356.1815.
:1, v/v) were added 5 (196 mg, 0.552 mmol) and CuI (5.00 mg, 0.0263 mmol). The reaction mixture was stirred at 35 °C for 24 h, and then allowed to cool down to room temperature and filtered. The obtained resin was washed successively with THF, water, saturated EDTA solution, water, MeOH, CH2Cl2 and diethyl ether (30.0 mL for each) and dried in a vacuum, yielding 160 mg desired resin 7 as a pale green powder. IR (cm−1) ν 1707 (CO); 13C CPMAS NMR δ 166.0 (CO), 60.6 (OCH2CH3), 14.0 (OCH2CH3).
O); 13C CPMAS NMR δ 175.9 (CO).
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) -ATGATCGTGA CACAAGATAA3′ (NdeI site underlined) and the ntcA reverse primer 5′--AGTGAACTGT CTGCTGAGAG T3′ (SacI site underlined). The PCR product was cloned into the pET28 vector (Novagen, USA). A clone confirmed by DNA sequencing was transformed into the BLDE3 E. coli strain (Novagen, USA). The recombinant clones obtained were grown in LB rich medium supplemented with Kanamycin (50 μg mL−1) and glucose (0.2%) with an OD at 600 nm of 0.3–0.4. The culture was then washed twice with LB medium to eliminate the glucose. The expression of the recombinant protein was then induced by adding 1 mM isopropyl-β-D-thiogalactoside (IPTG) for 4 h. The recombinant proteins were purified using Hitrap columns as recommended by Pharmacia. Imidazole was removed from purified proteins using PD10 columns (Pharmacia, Sweden). Proteins were concentrated on Vivaspin columns and used for subsequent analyses. Proteins were fractionated by SDS-PAGE (12.5% gel) and stained using the SeeBand procedure (Euromedex, France).
300 rpm. The supernatant was loaded onto the resin 1 for affinity chromatography.
-ATGATCGTGA CACAAGATAA3′ (NdeI site underlined) and the ntcA reverse primer 5′--AGTGAACTGT CTGCTGAGAG T3′ (SacI site underlined). The PCR product was cloned into the pET28 vector (Novagen, USA). A clone confirmed by DNA sequencing was transformed into the BLDE3 E. coli strain (Novagen, USA). The recombinant clones obtained were grown in LB rich medium supplemented with Kanamycin (50 μg mL−1) and glucose (0.2%) with an OD at 600 nm of 0.3–0.4. The culture was then washed twice with LB medium to eliminate the glucose. The expression of the recombinant protein was then induced by adding 1 mM isopropyl-β-D-thiogalactoside (IPTG) for 4 h. The recombinant proteins were purified using Hitrap columns as recommended by Pharmacia. Imidazole was removed from purified proteins using PD10 columns (Pharmacia, Sweden). Proteins were concentrated on Vivaspin columns and used for subsequent analyses. Proteins were fractionated by SDS-PAGE (12.5% gel) and stained using the SeeBand procedure (Euromedex, France).
300 rpm. The supernatant was loaded onto the resin 1 for affinity chromatography.
Acknowledgements
We are grateful for the financial support from the “ProKrebs” project of the ANR program of “Physique, Chimie et du Vivant”, Région PACA, CNRS, Wuhan University and Aix-Marseille Université. Y.W. and X.L. are supported by the overseas Ph.D. fellowship from China Scholarship Council. We thank Dr Emily Witty for English correction.Notes and references
- S. Zhang and D. A. Bryant, Science, 2011, 334, 1551–1553 CrossRef CAS PubMed.
- W. He, F. J.-P. Miao, D. C.-H. Lin, R. T. Schwandner, Z. Wang, J. Gao, J.-L. Chen, H. Tian and L. Ling, Nature, 2004, 429, 188–193 CrossRef CAS PubMed.
- S. C. Hebert, Nature, 2004, 429, 143–145 CrossRef CAS PubMed.
- S. Laurent, H. Chen, S. Bédu, F. Ziarelli, L. Peng and C.-C. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9907–9912 CrossRef CAS PubMed.
- A. B. Feria Bourrellier, B. Valot, A. Guillot, F. Ambard-Bretteville, J. Vidal and M. Hodges, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 502–507 CrossRef PubMed.
- S. Gálvez, M. Lancien and M. Hodges, Trends Plant Sci., 1999, 4, 484–490 CrossRef.
- M. Hodges, J. Exp. Bot., 2002, 53, 905–916 CrossRef CAS.
- A. Ninfa and P. Jiang, Curr. Opin. Microbiol., 2005, 8, 168–173 CrossRef CAS PubMed.
- C. Loenarz and C. J. Schofield, Nat. Chem. Biol., 2008, 4, 152–156 CrossRef CAS PubMed.
- R. A. Cairns and T. W. Mak, Cancer Discovery, 2013, 3, 730–741 CrossRef CAS PubMed.
- S. Ziegler, V. Pries, C. Hedberg and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 2744–2792 CrossRef CAS PubMed.
- H. Chen, S. Laurent, S. Bédu, F. Ziarelli, H.-L. Chen, Y. Cheng, C.-C. Zhang and L. Peng, Chem. Biol., 2006, 13, 849–856 CrossRef CAS PubMed.
- X. Liu, H. Chen, E. Laurini, Y. Wang, V. Dal Col, P. Posocco, F. Ziarelli, M. Fermeglia, C.-C. Zhang, S. Pricl and L. Peng, Org. Lett., 2011, 13, 2924–2927 CrossRef CAS PubMed.
- X. Liu, Y. Wang, E. Laurini, P. Posocco, H. Chen, F. Ziarelli, A. Janicki, F. Qu, M. Fermeglia, S. Pricl, C.-C. Zhang and L. Peng, Org. Lett., 2013, 15, 4662–4665 CrossRef CAS PubMed.
- Y. Wang, X. Liu, E. Laurini, P. Posocco, F. Ziarelli, M. Fermeglia, F. Qu, S. Pricl, C.-C. Zhang and L. Peng, Org. Biomol. Chem., 2014, 12, 4723–4728 CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- M.-X. Zhao, Y.-L. Jiang, Y.-X. He, Y.-F. Chen, Y.-B. Teng, Y. Chen, C.-C. Zhang and C.-Z. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12487–12492 CrossRef CAS PubMed.
- Although Schofield et al. have established photoreactive affinity systems to probe 2-OG oxygenases,19 the effective tags that they employed to capture these proteins were based on 8-hydroxyquinoline, a known iron chelator presented in inhibitors of 2-OG oxygenases. Therefore, it is unlikely that the so-obtained affinity systems are generally applicable for all 2-OG binding proteins.
- D. Rotili, M. Altun, A. Kawamura, A. Wolf, R. Fischer, I. K. H. Leung, M. M. Mackeen, Y.-M. Tian, P. J. Ratcliffe, A. Mai, B. M. Kessler and C. J. Schofield, Chem. Biol., 2011, 18, 642–654 CrossRef CAS PubMed.
- W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733–1738 CrossRef CAS.
- The HWE reaction often favors the formation of E isomers. Since the E- and Z-isomers of 4 have similar polarity, it is very difficult for us to separate them by column chromatography. We therefore used the mixture of the E/Z isomers directly in the subsequent reaction.
- J. Schaefer and E. O. Stejskal, J. Am. Chem. Soc., 1976, 98, 1031–1032 CrossRef CAS.
- O. B. Peersen, X. Wu, I. Kustanovich and S. O. Smith, J. Magn. Reson., 1993, 104, 334–339 CrossRef CAS.
- R. L. Cook, C. H. Langford, R. Yamdagni and C. M. Preston, Anal. Chem., 1996, 68, 3979–3986 CrossRef CAS.
- H. Sashiwa, Y. Shigemasa and R. Roy, Carbohydr. Polym., 2002, 49, 195–205 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Schemes S1–S3, Fig. S1–S3, synthesis of 2, 3 and 6, 1H and 13C NMR spectra of compounds 2–5, IR and 13C HRMAS NMR spectra of resins 1, 6, 7. See DOI: 10.1039/c4ob01005a |
| This journal is © The Royal Society of Chemistry 2014 |