
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tautomerisation of thymine acts against the Hückel 4N + 2 rule. The effect of metal ions and H-bond complexations on the electronic structure of thymine†‡
Olga A.
Stasyuk
*a,
Halina
Szatylowicz
*a and
Tadeusz M.
Krygowski
b
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: ostasyuk@ch.pw.edu.pl; halina@ch.pw.edu.pl
bDepartment of Chemistry, Warsaw University, Pasteura 1, 02-093 Warsaw, Poland
First published on 30th June 2014
Abstract
The stability and aromaticity of thirteen known thymine tautomers were studied in the gas phase at the B3LYP/6-311++G(2d,2p) computational level. It was found that they do not follow the Hückel 4N + 2 rule when the energetic criterion is considered, but they follow it when aromaticity indices, such as NICS, HOMA and the sum of the Wiberg bond indices, are applied. It was shown that the stability of a given tautomer is strongly dependent on the number of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups attached to the ring. The most stable tautomer i.e. with two carbonyl groups exhibits low π-electron delocalization (HOMA = 0.490, NICS(0) = −1.5). Its stability results from specific interactions between Nδ−Hδ+ and Cδ+Oδ− bond dipoles. A qualitative rule, which implies an increase in stability and a loss of aromaticity with increasing number of CO groups, holds in the case of thymine tautomers. Effects of intermolecular interactions (H-bonding and metal ion complexation) on the geometry and π-electron structure were analyzed for the five most stable tautomers with the following partners: HF/F− and Li+/Na+/K+. The magnitude of these effects strongly depends on the site and type of intermolecular interaction. The electronic structure of the most aromatic tautomers is more weakly influenced by external perturbations such as H-bonding and is almost entirely resistant to metal complexation.
O groups attached to the ring. The most stable tautomer i.e. with two carbonyl groups exhibits low π-electron delocalization (HOMA = 0.490, NICS(0) = −1.5). Its stability results from specific interactions between Nδ−Hδ+ and Cδ+Oδ− bond dipoles. A qualitative rule, which implies an increase in stability and a loss of aromaticity with increasing number of CO groups, holds in the case of thymine tautomers. Effects of intermolecular interactions (H-bonding and metal ion complexation) on the geometry and π-electron structure were analyzed for the five most stable tautomers with the following partners: HF/F− and Li+/Na+/K+. The magnitude of these effects strongly depends on the site and type of intermolecular interaction. The electronic structure of the most aromatic tautomers is more weakly influenced by external perturbations such as H-bonding and is almost entirely resistant to metal complexation.
Introduction
The aromatic sextet concept has been widely applied in organic chemistry as a convenient tool for explaining distinctly different chemical behavior of cyclic molecules containing six electrons.1–6 Hückel molecular orbital7,8 and free electron molecular orbital9 theories clearly explain higher stability of the cyclic π-electron molecules containing six electrons rather than eight or four. The Hückel rule10–12 states that cyclic π-electron systems are more stable when they consist of 4N + 2 π-electrons than those with 4N π-electrons. A great advantage of its application in organic chemistry was presented in Streitwieser's monograph.13 Currently it is considered as the leading rule in the studies of cyclic π-electron systems. As has been recently shown this rule can be deduced on the basis of graph-topological considerations.14 Typical application of this rule is presented for annulenes,15,16 where the resonance energy of 4N + 2 systems is always greater than that for 4N ones. A similar picture is found when the reactivity-based aromaticity index KK17 is applied.18 In ref. 18 a classification of the cyclic π-electron systems was proposed, which showed that both 1,2- and 1,4-benzoquinones are much less aromatic than 4N annulenes: cyclobutadiene and cyclooctatetraene. Thus, double-bonded substituents attached to the ring dramatically decrease its aromatic character. Similar effects appear in some tautomers of thymine, deciding their stability and aromaticity.Thymine is a six-membered heterocyclic molecule which can exist in the form of 13 tautomers/rotamers. The 1H,3H-diketo tautomer, which contains two CO groups and two N–H groups constituting inherent parts of the ring, is the most stable in the gas phase, solution and the solid state19,20 which was confirmed by a series of theoretical21–23 and experimental studies.24–26 Generally, this diketo tautomer exists as the main form in the double helix,27 interacting with adenine via two H-bonds. A–T pairs with other tautomers of thymine are significantly less stable than the Watson–Crick pair comprising the canonical forms.28 If thymine forms pairs with other bases, the regular DNA structure is disrupted and inadvertent mutations arise. The most frequent damage is the formation of thymine dimers induced by UV radiation.29,30 They inhibit the enzymes carrying out DNA replications and transcription, and are the main source of carcinogenic mutation known as melanoma.31 Another factor which can cause a mismatch in the base pairing is the metalation of nucleobases.32 Metal cations contribute to the stabilization of rare nucleobase tautomers23,33 and increase the probability of gene mutations.34–36 Such interactions of thymine with alkali metal ions are well described by theoretical23,37–39 and experimental40–43 methods. It was found that metal cations in all types of complexes studied preferentially bind to the oxygen at the C4 atom of thymine as well as in uracil. However, the influence of metal binding on the aromaticity of the thymine ring has not yet been investigated. It is also interesting to compare the effect of metal binding and other important interactions of the nucleobases – hydrogen bonds, on their π-electron structure. Along with the studies of conventional H-bonds formed between nucleobase pairs, interest in interactions of DNA with anions has recently arisen.44–46 Based on an extensive survey of the Nucleic Acid Database (NDB), Auffinger et al.44 concluded that anions (mostly Cl− and SO4−) do intrude on the first hydration shell of nucleic acids and bind to electropositive amino, imino, and hydroxyl groups. Two types of such complexes formed by H-bonding of the most stable thymine tautomer with anions have recently been studied.46 It was found that the complexes with N1H centers are stronger and more stable than those with N3H ones. Additionally, a correlation between the interaction energy and the proton affinity for these complexes was established.
The purpose of this work was to undertake a systematic study of the electronic structure of the five most stable thymine tautomers and to evaluate the effect of two types of intermolecular interactions caused by different environments (HF/F− and metal cations) on geometric and π-electron structures of these tautomers in the gas phase. In particular, all these changes were elucidated by the use of the structural aromaticity index HOMA.47,48 It should be stressed that the gas phase computational results indicate stronger interactions than those in which solvent effect is taken into account.49,50 Therefore, in order to be able to observe a tendency in the changes of the thymine electronic structure the influence of these stronger intermolecular interactions on structural properties of thymine tautomers was investigated.
Methodology
All geometry optimizations of the studied systems, without any symmetry constraints, were performed using the Gaussian 09 series program51 at the B3LYP/6-311++G(2d,2p) computational level. To prove that the obtained structures are minima on the potential energy surface frequencies were calculated at the same level of theory. Justification of the theoretical method choice is given in our previous work.52Geometry-based index of aromaticity HOMA47,48 was used as a measure of π-electron delocalization in the ring. It is defined as:
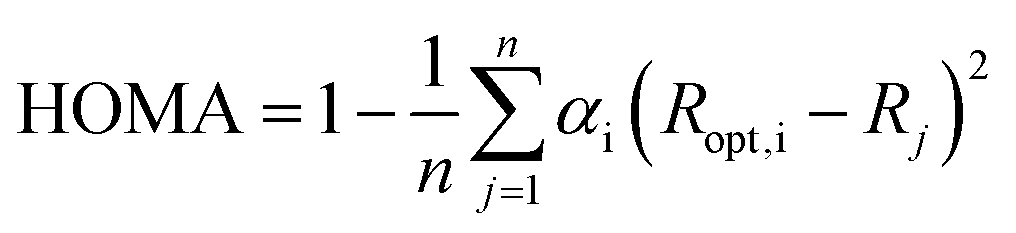 | (1) |
For free tautomers the following nucleus independent chemical shifts (NICS) were calculated: (i) in the center of the ring,53 NICS(0); (ii) 1 Å above the center,54 NICS(1); and (iii) the component of the tensor perpendicular to the molecular plane,55,56 NICS(1)zz. Calculations of the NICS's were performed with Gaussian 09 program51 at the HF/6-31+G(d) level of theory (following recommendation given in ref. 53) using the GIAO method.
As an additional tool for the electronic structure description, natural bond orbital (NBO) analysis57 was applied to study atomic charge distribution and carry out a population analysis. Additionally, the sum of the Wiberg bond indices58 of all bonds in the ring was taken as a numerical characteristic of aromaticity, WBIΣ.
Three types of complexes of thymine tautomers were studied: (i) neutral (with HF), (ii) anionic (with F−) and (iii) cationic (with M+, M = Li, Na, K). In order to estimate the energy of the intermolecular interaction, the difference between the energy of the complex A⋯B and the sum of the energies of its components (A and B) was used. The basis set superposition error (BSSE)59 was taken into account. In the next stage of the used approach, the total energy of interaction was decomposed into deformation (Edef) and interaction (Eint) components:
| Etot = Edef + Eint | (2) |
The first term of the above equation represents the amount of energy required to deform the equilibrium geometries of both fragments (E0A and E0B) into their geometries in the complex (EA and EB).
| Edef = (EA − E0A) + (EB − E0B) | (3) |
The second term of eqn (2) corresponds to the actual energy change when the two distorted fragments are combined in the final structure. The interaction energy was estimated using a calculation procedure described elsewhere.60
It should be stressed that the above methodology has already been used for a series of nucleobases.52,61,62 MarvinSketch editor was used for drawing of all chemical structures.63
Results and discussion
For clarity, the results are discussed in three sections dealing with (i) stability and aromaticity of free tautomers, (ii) their intermolecular interactions via H-bonding, and (iii) their complexation with metal ions.Stability and aromaticity of free thymine tautomers
To find the most stable tautomers of thymine the energies of all 13 tautomers were calculated. The resulting order of their stability is consistent with previous findings based on DFT calculations.22 It slightly differs from that obtained by MP2 one20,21 (Table S1‡). However, the set of five most stable tautomers, whose relative energies are within 15 kcal mol−1, is the same for all mentioned above calculations. In the subsequent part of the paper only these five tautomers will be subjected to deeper analysis. Their chemical structures are shown in Fig. 1, whereas their main characteristics (stability and aromaticity data) are presented in Table 1. Additionally, numbers of π-electrons in the ring, N, obtained from the formal approach and from NBO calculations57 are given in Table 1 (for details see Table S2‡). Some differences arise when comparing the formal number of π-electrons in the ring and the number obtained by NBO calculations. This can be explained by the fact that the formal approach counts electrons in atoms which are not interacting with each other, in contrast to the NBO method, which counts the occupation of interacting atoms in real molecules by taking into account their electronegativity and resonance/inductive/field interactions between particular parts of the molecule.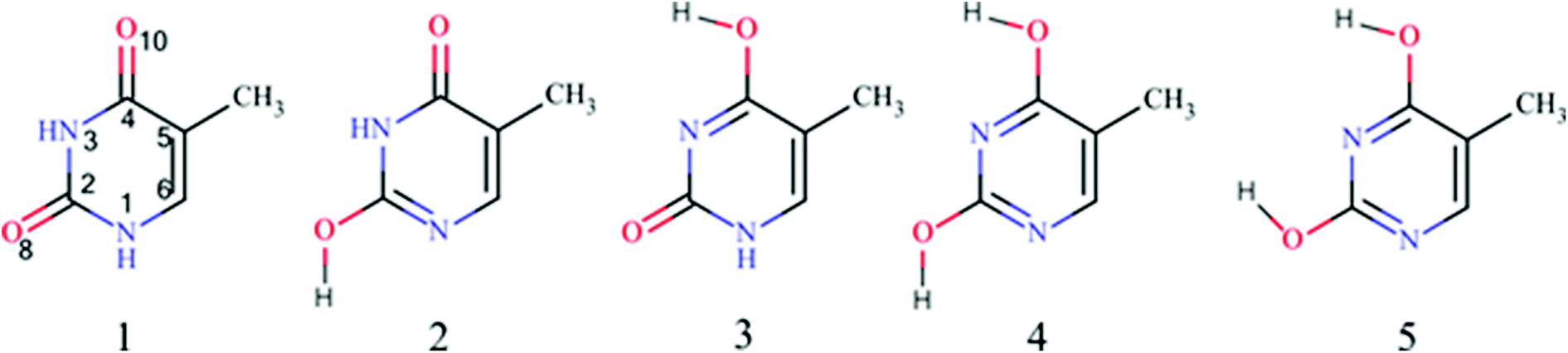 | ||
| Fig. 1 Structures of the most stable thymine tautomers. | ||
| Tautomer | N formal | N NBO | E rel/kcal mol−1 | HOMA | NICS(0) | NICS(1) | NICS(1)zz | WBIΣ |
|---|---|---|---|---|---|---|---|---|
| thy1 | 8 | 6.98 | 0.00 | 0.490 | −1.5 | −2.2 | −2.1 | 7.07 |
| thy2 | 7 | 6.63 | 10.83 | 0.629 | −2.8 | −4.3 | −8.2 | 7.64 |
| thy3 | 7 | 6.69 | 12.38 | 0.718 | −2.7 | −4.2 | −7.5 | 7.62 |
| thy4 | 6 | 6.24 | 13.02 | 0.984 | −7.2 | −8.8 | −20.9 | 8.28 |
| thy5 | 6 | 6.29 | 14.17 | 0.987 | −7.3 | −8.9 | −21.1 | 8.19 |
When relative energies, Erel, are considered (see Table 1), it appears that the most stable tautomer (thy1) contains formally 8 π-electrons, next two less stable ones – 7 π-electrons, and the least stable tautomers – 6 π-electrons. This is opposite to the Hückel rule. It is important to note that the Hückel rule was formulated for cyclic unsaturated hydrocarbons. In our case functional groups such as NH, CO and COH interfere and induce additional interactions, which may significantly affect the stability of thymine tautomers. However, when the aromaticity indices are concerned, particularly NICS(1)zz and HOMA, it can be noticed that π-electron delocalization follows the Hückel rule well. It is worth mentioning that HOMA and NICS(1)zz are well correlated (see Fig. S1,‡ cc = −0.982). The same kind of correlation was found between the sum of Wiberg bond indices, WBIΣ, and HOMA (cc = 0.976) as well as NICS(1)zz (cc = −0.976). The values of Wiberg bond indices for the most stable thymine tautomers are shown in Fig. S2.‡ Values of HOMA and NICS(1) were very close to those reported previously.64Fig. 2 shows the dependence of HOMA on Erel of the tautomers. Three groups of thymine tautomers can be distinguished: (i) very aromatic (HOMA is close to 1.0); (ii) the least aromatic (HOMA < 0.4) and (iii) with intermediate aromaticity (in this case an increase of aromaticity is connected with a decrease of stability). From the structural point of view, the most stable tautomer (thy1) may also be included into the second group for its quinoid-like structure. A very similar dependence was found by Raczynska et al. for uracil tautomers.65
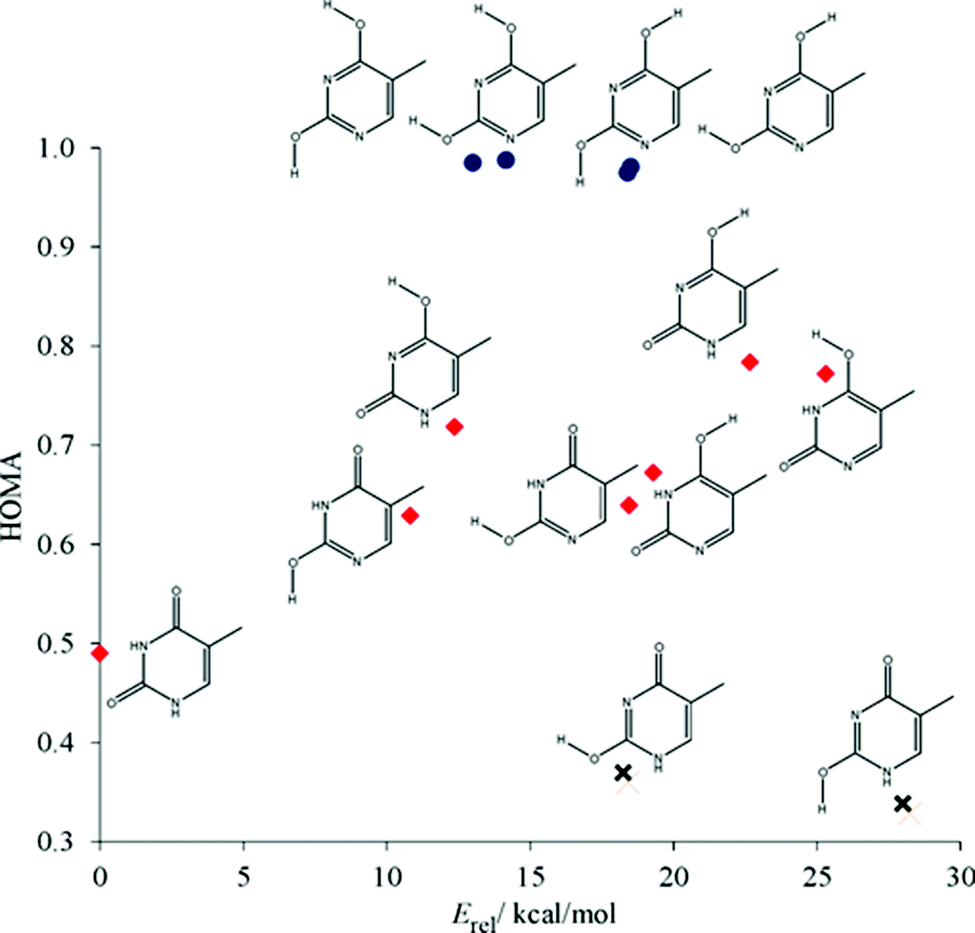 | ||
| Fig. 2 Dependence of HOMA on relative energy, Erel, for thymine tautomers. Group (i) blue circles, group (iii) crosses, and group (ii) red diamonds. | ||
Two potential explanations could be postulated:
(i) some kind of resonance effect between the electron-rich NH and the electron attracting CO groups;
(ii) stabilization/destabilization effects due to bond dipole interactions between Nδ−Hδ+ and Cδ+Oδ− as well as with the lone pair of the aza-type nitrogen atom.
The first reason implies a substantial change in the CO bond length. However, in thymine tautomers the CO bond lengths for carbonyl groups are between 1.213 Å and 1.219 Å, which are comparable to the values obtained for 1,4-benzoquinone (1.219 Å), acetone (1.211 Å) and urea (1.216 Å). Additionally, the obtained values of Wiberg bond indices for thymine tautomers (Fig. S2‡) confirm that in thy1 the resonance effect does not occur. Thus, this hypothesis should most likely be excluded. The next (ii) explanation assumes stabilizing interactions between charges at Hδ+ and Oδ− of NH and CO groups as well as destabilizing interactions between the nitrogen lone pairs and CO bond dipole in COH groups.
Such an assumption is confirmed by the NBO charge distribution in thymine tautomers (Fig. 3), which indicates that for thy1 stabilizing interactions between NH and CO groups are the strongest, for thy2 and thy3 they are weaker, for thy4 and thy5 they are essentially non-existent. A negative charge localized at aza-type nitrogen atoms is provided by the lone pairs located in the plane of the molecules. Therefore, interactions of these lone pairs with CO bond dipole of COH groups lead to the destabilization of such systems. To put these problems into a wider perspective a few demonstrative examples will be presented.
 | ||
| Fig. 3 NBO atomic charges for free tautomers of thymine. | ||
First, to estimate the magnitude of the repulsive energy between two identical bond dipoles CF in ortho- and para-positions, the energies of homodesmotic reactions for 1,2- and 1,4-difluorobenzene were calculated (see Fig. 4). The difference between their energies amounts to ca. 3 kcal mol−1 that indicates the destabilization of 1,2-difluorobenzene in comparison to the 1,4-isomer.
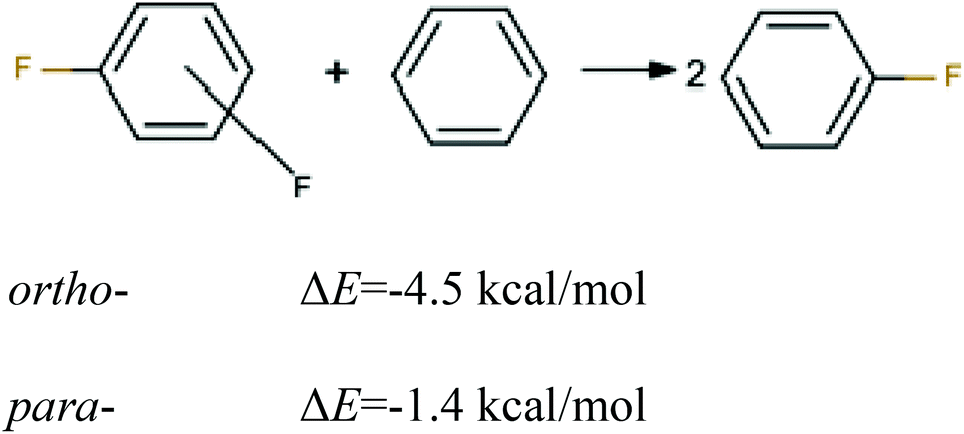 | ||
| Fig. 4 Homodesmotic reaction for 1,2- and 1,4-difluorobenzenes. | ||
2- and 3-Hydroxypyrroles can be considered as additional instructive examples. The results of their optimization show that 2-hydroxypyrrole is more stable than 3-hydroxypyrrole by 2.1 kcal mol−1. Total charge distribution in 2-hydroxypyrrole, presented in Fig. 5, clearly confirms the stabilizing interactions between the NH and CO(H) dipoles.
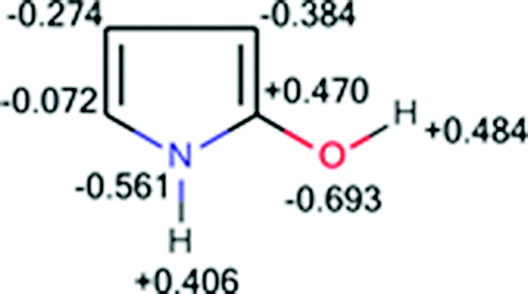 | ||
| Fig. 5 NBO charge distribution for 2-hydroxypyrrole. | ||
Finally, one more example, illustrating repulsive interactions between nitrogen lone pairs, comes from a comparison of energies of pyridazine and pyrimidine, where the intramolecular interactions are of 1,2- and 1,3-types, respectively. The difference in energy between them is 22.61 kcal mol−1 which indicates a substantial destabilization of the system in the case of 1,2-interactions as compared to 1,3-ones, see Table S3.‡
Taking into account these illustrative data, we can find good qualitative arguments for the earlier mentioned assumption (ii) that the reasons of anti-Hückel properties of thymine tautomers originate from specific intramolecular interactions between bond dipoles of NH, lone pairs of aza-nitrogen atoms and two forms of CO bonds. This interpretation differs from that applied to uracil. In that case it has been suggested that such interactions play a rather secondary role and the stability of the functional groups is the main factor responsible for the observed tautomeric preference.65 It is important to note that due to the conclusive remarks presented above H-bonding and metal complexation may play a crucial role in the stability and π-electron delocalization of thymine complexes.
H-bonded complexes of thymine tautomers
Energetic and structural characteristics of H-bonded complexes of the discussed thymine tautomers are shown in Table S4,‡ data for free tautomers are added for comparative reasons. Relationships between the strength of intermolecular interactions and the H-bond distance, shown in Fig. 6, resemble those obtained for cytosine complexes.62 Also in this case, three types of H-bonds can be distinguished: (i) charge-assisted, (ii) classical X⋯HF with sp2 hybridized X, X = O or N, and (iii) also X⋯HF with sp3 hybridized X, X = O of the hydroxyl group. It is found that their strength decreases approximately twice in a given sequence. Since the range of the H-bond lengths is not large and intermolecular interactions are less diversified than in the case of cytosine, the EHBvs. dHB dependences can be described by linear equations. The obtained slopes (Table S5‡) are almost three times greater for O⋯H interactions than for N⋯H ones (see Fig. 6) both for neutral and for charge-assisted H-bonds. This agrees with the expectation that for the O⋯H and N⋯H bonds of the same energy the latter should be longer.66,67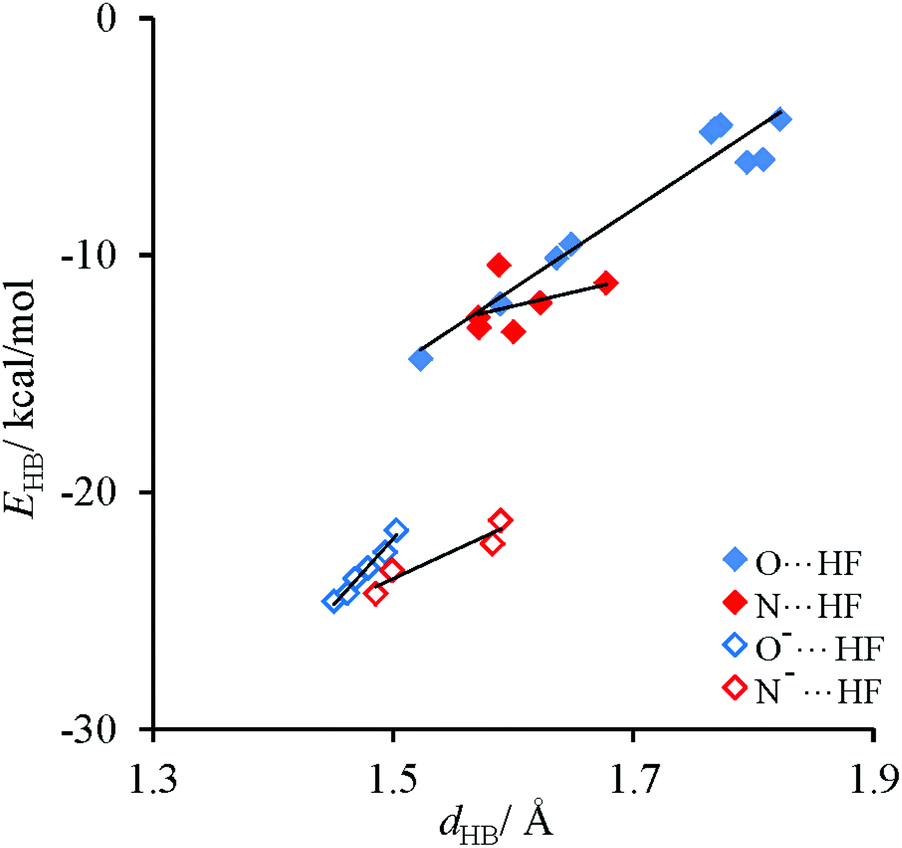 | ||
| Fig. 6 Dependencies of the H-bond energy on the H-bond length for H-bonded complexes of thymine tautomers. | ||
Almost all types of H-bondings induce significant structural changes in the region of interactions and in more distant parts of the complex (so-called long-distance consequences of the H-bond formation). The magnitude of these changes depends on the site and type of intermolecular interaction (see Table 2).
| Interaction | d C2O8 | d C4O10 | E HB/kcal mol−1 | Form | HOMA |
|---|---|---|---|---|---|
| N⋯HF | ↓ | ↓ | −10 to −13 | — | ↓ (Except N3 in thy3) |
| N−⋯HF | ↑ | ↑ | −23 to −25 | — | ↑ (Except N3 in thy1) |
| O8⋯HF | ↑ | ↓ | −4 to −6 | Enol | ↓ |
| −12 to −14 | Keto | ↑ | |||
| O8−⋯HF | ↓ | ↑ | −22 to −25 | — | ↓ (Except thy2) |
| O10⋯HF | ↓ | ↑ | −4 to −5 | Enol | ↑ |
| −10 | Keto | ↑ | |||
| O10−⋯HF | ↑ | ↓ | −21 to −25 | — | ↓ |
Due to the formation of the neutral H-bonds with O8 or O10 atoms the CO bond participating in this interaction is elongated, whereas the other CO bond is shortened. In the case of charge-assisted H-bonds involving the same oxygen atoms an opposite tendency is observed. Nevertheless, the long-distance effect of H-bonding is 3–5 times smaller than in the immediate region of the interaction. In addition, some long-distance consequences were also observed for interactions with the nitrogen atoms. The neutral H-bonds N⋯HF induce shortening of both CO bonds, whereas N−⋯HF interactions lead to their lengthening. The above-presented consequences of H-bonding result in a significant variability of the π-electron structure in the thymine rings. It is confirmed by changes in HOMA indices describing the aromaticity of studied systems (Table 2).
Interactions with metal cations (M+)
Two types of interactions between thymine and metal cation can be distinguished: (i) singular and (ii) bifurcated. Singular coordination, where the cation interacts only with one atom containing a lone pair (O or N), occurs in all tautomers. However, the case of bifurcated coordination is found only in thy3, thy4 and thy5, where the lone pair of nitrogen is located close to the lone pair of oxygen. Main characteristics of thymine interactions with M+ (M = Li, Na, K) are shown in Table S6.‡In agreement with previous studies,23,38 the most stable complexes of thymine tautomers with metal ions are found for thy1, where the cation interacts with the O10 atom of the carbonyl group (M+⋯O10). The decrease of thymine complexes stability up to Erel = 10 kcal mol−1 (Table S6‡) occurs in the following sequence: M+⋯N3,O8 of thy3, M+⋯O8 of thy1 and M+⋯O10 of thy2. However, their stability is not associated with the strength of interactions, which weaken in order: M+⋯N3,O8 of thy3, M+⋯O10 of thy2, M+⋯O10 of thy1 and M+⋯O8 of thy1. It should be noted that bifurcated interactions (M+⋯N3,O8 in thy3 complexes) are only slightly stronger (by ca. 8–12 kcal mol−1 for the above-mentioned complexes) than the singular ones. Only one exception to the above-presented stability order is found. The K+⋯N3,O8 of thy3 complex is less stable than K+⋯O8 of thy1 one, in contrast to the systems with Li+ and Na+ ions. It can be explained by a large ionic radius of K+, which tends to form singular coordination with O8 in thy1 (Table S6‡).
Structural consequences of the complexation by a metal cation are similar to those observed in H-bonded systems. However, since the first interactions are stronger they cause greater structural changes. The most representative and interesting consequences are observed in complexes in which the cation interacts with the oxygen atom of the carbonyl group. The CO bond length becomes longer and both adjacent CN (or CN and CC) bonds shorten. Opposite changes are observed for the second CO group as a consequence of the long distance interactions: the CO bond becomes slightly shorter and the adjacent CN (or CN and CC) bonds lengthen. A comparison of the structural parameters found for Li+⋯O8 and Li+⋯O10 of thy1 complexes (the strongest singular interaction) with the free tautomer is presented in Fig. S3.‡
The formation of thymine complexes with cations is associated with changes in the aromaticity of the rings (Table S6‡). Depending on the form of the oxygen atom, which participates in the interaction, π-electron delocalization of the ring, expressed by index HOMA, may increase or decrease as compared to the free tautomer. If the interaction takes place between the metal ion and the O atom of any carbonyl group, the HOMA index is higher for the complex than for the free tautomer. Rationalization of this observation is based on elongation of the double CO bond participating in the intermolecular interactions which leads to an increase of the ring aromaticity. The only exception is M+⋯O8 type of thy1 complex (see Fig. S3‡).
Aromaticity of a complex is, in turn, almost unchanged when the O atom of the hydroxyl group interacts with the cation. In such cases the intermolecular interaction cannot change the nature of the single CO bond to a large extent and thus does not significantly influence the ring system. A substantial decrease of aromaticity was observed only for complexes of thy2 (from 0.6 to 0.4 unit of HOMA). The reason for this decrease is non-planarity of such complexes, where M+ and the H atom of the OH group lie above and below the ring plane. This leads to a disturbance of conjugation between the lone pair at O8, which is almost coplanar with the ring plane and the π-electron system of the ring. In all other cases the lone pairs are almost perpendicular to the ring and can effectively interact with the ring π-electron system. Another important observation is that bifurcated coordination induces larger changes in the HOMA index than the singular one.
Additionally, correlations between ionic radii and HOMA values were found. The cation effect on the aromaticity changes is associated with the strength of the electrostatic interaction, which can play an important role in all discussed cases. The ionic radius increases in the following sequence of cations: Li+, Na+ and K+. Hence the electrostatic interaction between the ion and thymine diminishes. This leads to a slight increase of π-electron delocalization of the ring, following the above order, for complexes with O(hydroxyl)⋯M+ interactions. Furthermore, a general rule should be noted – a decrease of aromaticity in the above-given sequence of the ionic radius takes place in complexes where intermolecular interaction results in an increase of π-electron delocalization in comparison with the free tautomer. An opposite relation is observed when the complexation induces a decrease of the ring aromaticity relative to the non-interacting tautomer.
In complexes of highly aromatic thymine tautomers (HOMA > 0.98) intermolecular interactions with metal cations essentially do not change the π-electron delocalization of the ring, in contrast with H-bonding that may result in aromaticity reduction by ca. 0.2 HOMA units. In the case of less aromatic but more stable tautomers both cation complexation as well as H-bonding may significantly increase or decrease the ring aromaticity.
Both kinds of the studied intermolecular interactions can be also characterized by the extent of charge transfer from a thymine tautomer to a HF or metal cation. Good correlation is observed only between the total energy of H-bonds and the transferred charge (cc = 0.95), in the case of interactions with metal cations such correlation is significantly worse (cc = 0.64), see Fig. S4a.‡ However, the correlations between HOMA and CT (see Fig. S4b‡) do not exist.
Conclusions
(1) From the energetic viewpoint the tautomeric preference of thymine contradicts the Hückel rule. As a result, the most stable tautomer is not the most aromatic, as estimated by HOMA, NICS and the sum of the Wiberg bond indices (for the ring). Aromaticity strongly depends on the number of CO groups in the tautomer. The most aromatic systems are tautomers with two hydroxyl groups i.e. containing no carbonyl group. As is known, the Hückel rule was formulated for cyclic π-electron hydrocarbons, and an addition of any functional group to the cycle may affect the effectiveness of this rule, as earlier observed in benzoquinones.18
(2) The main factor responsible for the stability of tautomers is their stabilization due to the extra interactions between Nδ−Hδ+ and Cδ+Oδ− dipoles. Higher number of such interactions in the tautomer leads to its higher stability.
(3) Structural changes in the region of interactions and in its distant parts were found in all three types of the complexes of thymine tautomers studied: (i) neutral (with HF), (ii) anionic (with F−), and (iii) cationic (with M+, M = Li, Na, K). The magnitude of these geometrical and π-electron changes strongly depends on the site and type of intermolecular interaction.
(4) H-bonding and metal complexation affect aromaticity of the ring in thy1 to a greater extent, whereas the most aromatic tautomers are resistant to the interactions with cations. In contrast, H-bonding may decrease their aromaticity.
Acknowledgements
O. A. S., H. S. and T. M. K. gratefully acknowledge the Foundation for Polish Science for supporting this work under the MPD/2010/4 project “Towards Advanced Functional Materials and Novel Devices – Joint UW and WUT International PhD Programme” and the Interdisciplinary Center for Mathematical and Computational Modeling (Warsaw, Poland) for providing computer time and facilities.Notes and references
- E. Hückel, Z. Elektrochem., 1937, 43, 752–788 Search PubMed.
- M. S. J. Dewar, The electronic theory of organic chemistry, Clarendon Press, Oxford, 1949 Search PubMed.
- G. W. Wheland, Advanced Organic Chemistry, Wiley, New York, 1949 Search PubMed.
- C. K. Ingold, Structure and Mechanism in Organic Chemistry, Cornell University Press, Ithaca, New York, 1953 Search PubMed.
- W. Hückel, Theoretische Grundlagen der organischen Chemie, Band 1 and 2, Akademische Verlagsgesellschaft, Leipzig, 1956 Search PubMed.
- E. Müller, Neuere Anschauungen der Organischen Chemie, Springer-Verlag, Berlin, 1957 Search PubMed.
- E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef.
- E. Hückel, Z. Phys., 1932, 76, 628–648 CrossRef.
- J. R. Platt, J. Phys. Chem., 1949, 17, 484–495 CrossRef CAS PubMed.
- J. D. Roberts, A. Streitwieser Jr. and C. M. Regan, J. Am. Chem. Soc., 1952, 74, 4579–4582 CrossRef CAS.
- W. von Eggers Doering and F. L. Detert, J. Am. Chem. Soc., 1951, 73, 876–877 CrossRef.
- A. A. Frost and B. Musulin, J. Chem. Phys., 1953, 21, 572–573 CrossRef CAS PubMed.
- A. Streitwieser Jr., Molecular Orbital Theory for Organic Chemists, Wiley, New York, 1961 Search PubMed.
- A. Ciesielski, T. M. Krygowski, M. K. Cyranski and A. T. Balaban, Phys. Chem. Chem. Phys., 2011, 13, 3737–3747 RSC.
- M. S. J. Dewar and G. J. Gleicher, J. Am. Chem. Soc., 1965, 87, 685–692 CrossRef CAS.
- H. P. Figeys, Tetrahedron, 1970, 26, 5225–5234 CrossRef CAS.
- J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1970, 319–324 CrossRef CAS.
- T. M. Krygowski, Tetrahedron Lett., 1970, 1311–1312 CrossRef CAS.
- M. Piacenza and S. Grimme, J. Comput. Chem., 2004, 25, 83–98 CrossRef CAS PubMed.
- J. Rejnek, M. Hanus, M. Kabelac, F. Ryjacek and P. Hobza, Phys. Chem. Chem. Phys., 2005, 7, 2006–2017 RSC.
- T. K. Ha and H. H. Gunthard, J. Am. Chem. Soc., 1993, 115, 11939–11950 CrossRef CAS.
- J.-C. Fan, Z.-C. Shang, J. Liang, X.-H. Liu and H. Jin, J. Mol. Struct. (THEOCHEM), 2010, 939, 106–111 CrossRef CAS PubMed.
- J. Chen, H. Ai, Y. Zhao and J. Liu, J. Phys. Org. Chem., 2012, 25, 126–131 CrossRef CAS.
- M. A. Morsy, A. M. Al-Somali and A. Suwaiyan, J. Phys. Chem. B, 1999, 103, 11205–11210 CrossRef CAS.
- J. C. López, M. I. Peña, M. E. Sanz and J. L. Alonso, J. Chem. Phys., 2007, 126, 191103 CrossRef PubMed.
- M. Y. Choi and R. E. Miller, J. Phys. Chem. A, 2007, 111, 2475–2479 CrossRef CAS PubMed.
- J. D. Watson and F. H. Crick, Nature, 1953, 171, 737–738 CrossRef CAS.
- J. Rejnek and P. Hobza, J. Phys. Chem. B, 2007, 111, 641–645 CrossRef CAS PubMed.
- W. Harm, Biological Effects of Ultraviolet Radiation, Cambridge University Press, Cambridge, 1984 Search PubMed.
- I. Husain, J. Griffith and A. Sancar, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 2558–2562 CrossRef CAS.
- H. S. Black, F. R. de Gruijl, P. D. Forbes, J. E. Cleaver, H. N. Ananthaswany, E. C. de Fabo, S. E. Ullrich and R. M. Tyrrell, J. Photochem. Photobiol., B, 1997, 40, 29–47 CrossRef CAS.
- D. Galaris and A. Evangelou, Crit. Rev. Oncol. Hematol., 2002, 42, 93–103 CrossRef.
- P. Hobza and C. Sandorfy, J. Biomol. Struct. Dyn., 1985, 6, 1245–1252 Search PubMed.
- P. Strazewski and C. Tamm, Angew. Chem., Int. Ed. Engl., 1990, 29, 36–57 CrossRef.
- V. I. Danilov, V. M. Anisimov, N. Kurita and D. Hovorun, Chem. Phys. Lett., 2005, 412, 285–293 CrossRef CAS PubMed.
- Y. Podolyan, L. Gorb and J. Leszczynski, Int. J. Mol. Sci., 2003, 4, 410–421 CrossRef CAS PubMed.
- N. Russo, M. Toscano and A. Grand, J. Phys. Chem. B, 2001, 105, 4735–4741 CrossRef CAS.
- N. Russo, M. Toscano and A. Grand, J. Am. Chem. Soc., 2001, 123, 10272–10279 CrossRef CAS PubMed.
- M. Kabelac and P. Hobza, J. Phys. Chem. B, 2006, 110, 14515–14523 CrossRef CAS PubMed.
- B. A. Cerda and C. Wesdemiotis, J. Am. Chem. Soc., 1996, 118, 11884–11892 CrossRef CAS.
- M. T. Rodgers and P. B. Armentrout, J. Am. Chem. Soc., 2000, 122, 8548–8558 CrossRef CAS.
- E. A. L. Gillis, K. Rajabi and T. D. Fridgen, J. Phys. Chem. A, 2009, 113, 824–832 CrossRef CAS PubMed.
- S. A. Krasnokutski, G. S. Lee and D.-S. Yang, J. Chem. Phys., 2010, 132, 044304 CrossRef PubMed.
- P. Auffinger, L. Bielecki and E. Westhof, Structure, 2004, 12, 379–388 CrossRef CAS PubMed.
- A. Martinez, O. Dolgounitcheva, V. G. Zakrzewski and J. V. Ortiz, J. Phys. Chem. A, 2008, 112, 10399–10404 CrossRef CAS PubMed.
- M. Shakourian-Fard and A. Fattahi, Struct. Chem., 2012, 23, 17–28 CrossRef CAS.
- J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- T. M. Krygowski, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS.
- S. Grimme, Chem. – Eur J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- J. Poater, M. Swart, C. Fonseca Guerra and F. M. Bickelhaupt, Comput. Theor. Chem., 2012, 998, 57–63 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision B.01), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- O. A. Stasyuk, H. Szatylowicz and T. M. Krygowski, Org. Biomol. Chem., 2014, 12, 456–466 CAS.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- P. v. R. Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R. v. E. Hommes, Org. Lett., 2001, 3, 2465–2468 CrossRef CAS PubMed.
- C. Corminboeuf, T. Heine, G. Seifert, P. v. R. Schleyer and J. Weber, Phys. Chem. Chem. Phys., 2004, 6, 273–276 RSC.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.G, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2004 Search PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- H. Szatylowicz, J. Phys. Org. Chem., 2008, 21, 897–914 CrossRef CAS.
- O. A. Stasyuk, H. Szatylowicz and T. M. Krygowski, J. Org. Chem., 2012, 77, 4035–4045 CrossRef CAS PubMed.
- O. A. Stasyuk, H. Szatylowicz and T. M. Krygowski, Croat. Chem. Acta, 2014 Search PubMed , accepted.
- MarvinSketch 5.11.5, Copyright@ 1998–2013, ChemAxon Ltd. (http://www.chemaxon.com).
- M. K. Cyranski, M. Gilski, M. Jaskolski and T. M. Krygowski, J. Org. Chem., 2003, 68, 8607–8613 CrossRef CAS PubMed.
- E. D. Raczynska, K. Zientara, T. M. Stepniewski and K. Kolczynska, Collect. Czech. Chem. Commun., 2009, 74, 57–72 CrossRef CAS.
- T. Steiner, J. Phys. Chem. A, 1998, 102, 7041–7052 CrossRef CAS.
- P. Gilli, L. Pretto, V. Bertolasi and G. Gilli, Acc. Chem. Res., 2009, 42, 33–44 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to our friend, Professor Jacek Lipkowski of Guelph University, on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Tables: relative energies for thymine tautomers in comparison with previous studies (S1); natural atomic 2pz orbital occupancies for thymine tautomers (S2); energies of isomeric diazines, diazoles and triazoles (S3); the main characteristics for H-bonded complexes of thymine tautomers (S4); statistics of linear regressions between the H-bond energy and length for H-bonded complexes of thymine tautomers (S5); the main characteristics for metal complexes of thymine tautomers (S6); Cartesian coordinates of equilibrium geometries for free tautomers of the thymine (S7), their H-bonded complexes (S8), and complexes with metal ions (S9). Figures: correlations between aromaticity indices: NICS's, HOMA and WBIΣ (S1), Wiberg bond indices for the most stable thymine tautomers (S2), structural parameters of thy1 and its complexes with Li+ (S3), relationships, the total energy of interactions and aromaticity index HOMA against the amount of charge transfer for the studied complexes of thymine (S4). See DOI: 10.1039/c4ob00964a |
| This journal is © The Royal Society of Chemistry 2014 |