
A 2D–3D structure transition of gold clusters on CeO2−X(111) surfaces and its influence on CO and O2 adsorption: a comprehensive DFT + U investigation†
Zhong-Kang
Han
ab and
Yi
Gao
 *a
*a
aDivision of Interfacial Water and Key Laboratory of Interfacial Physics and Technology, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai, 201800, China. E-mail: gaoyi@sinap.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 3rd November 2014
Abstract
Detailed knowledge of the structures of gold nanoparticles on ceria surfaces is of fundamental importance to understand their extraordinary activities in catalysis. In this work, we employ density functional theory with the inclusion of the on-site Coulomb interaction (DFT + U) to investigate the structure evolution of small-sized gold (Au, Au4, Au8 and Au12) clusters on four types of reduced CeO2−X(111) surfaces: SSV (single surface oxygen vacancy), LSVT (linear surface oxygen vacancy trimer), dLSVC (double linear surface oxygen vacancy with a surface vacancy dimer and a subsurface vacancy), and TSVT (triangular surface oxygen vacancy trimer). Our results indicate that the relative stabilities of multilayer (3D) structures are strengthened gradually compared with the monolayer (2D) structures with increasing the number of gold atoms. In addition, the 2D–3D structure transition occurs on the size order of Au2D→3D@TSVT > Au2D→3D@dLSVC ∼ Au2D→3D@LSVT > Au2D→3D@SSV, which is determined by the charge transfer magnitude between the CeO2 surfaces and gold clusters. Meanwhile, two competitive nucleation patterns are observed, fcc-like nucleation and hcp-like nucleation, which highly affect the morphology of supported gold clusters. Further site-by-site investigations indicate that the coordination number and the charges of Au atoms are the dominant factors to influence the adsorption strength of CO and O2, and the interface plays a relatively minor role. These findings not only enrich our knowledge of the relationship between surface defects, gold cluster structures and small molecule adsorptions, but also provide a theoretical perspective to help design the optimal Au/CeO2 systems possessing high catalytic efficiency.
1. Introduction
Ceria can induce gold nanoparticles by strong metal–support bonding1 to exhibit extraordinary catalytic activities in many important reactions, such as CO oxidation reactions2–4 and water–gas-shift (WGS) reactions.5–8 Numerous efforts have been made to investigate the key factors that dominate their catalytic activities. These factors include the dimensionality and morphology of gold nanoparticles (NPs),9–12 the fluxionality and electronic state of gold clusters,13–19 the presence of gold NP–support interfaces,20–23etc. Nørskov and co-workers attributed the most important factor to the availability of low-coordinated gold atoms on supported Au NPs based on a set of experimental and theoretical studies.24 Kim et al. suggested that lowering the oxygen vacancy formation energy or increasing the concentration of reduced metal ions could improve the catalytic activity of ceria-supported gold clusters.3 Nevertheless, the experiments on the water–gas-shift (WGS) reaction suggested that nonmetallic Au particles were the catalytically active species on ceria-supported gold catalysts, while coexisting metallic Au particles did not take part in the reaction.5,26 Although some progress has been achieved in the past decade, the dominant factors for the high activity of ceria-supported gold catalysts have not been fully resolved due to the lack of their precise atomic structures.On the other hand, the structures of small-sized gold clusters were mostly characterized in free-standing style,25–36 while the structures and growth patterns of supported gold clusters have not been clarified yet.37–56 In particular, the structures of gold clusters on the CeO2−X(111) surface are very difficult to identify due to the high complexity of the substrate. Recently, the experiments proposed that Au preferentially nucleated at defects of the support exhibit a three-dimensional growth mode,6,52–54 which was partly confirmed by the theoretical studies.5,55 Zhang et al. examined clusters up to Au11 supported on SSV theoretically and found that 3D clusters were more stable than 2D clusters from Au4 (except for Au7), and notable electron transfer was observed between Au clusters and the ceria support.56 Kim et al. found that the most stable Au13 structure was a fcc-like 3D island when supported on TSVT.3 However, systematic studies of the structures of ceria-supported gold clusters are very scarce, which hinders fully understanding the properties and catalytic activities of this system.
In this work, we performed a systematic study of the structure evolution of gold nanoclusters on four types of reduced CeO2−X(111) surfaces. Our results provided a clear picture that different types of surface vacancies could not only adjust the relative stabilities of planar (2D) and compact (3D) structures of gold clusters, but also greatly influence their packing motifs and CO and O2 adsorption energies, which is crucial for their catalytic activities in CO oxidation.
2. Methodology
The low-lying structures of CeO2−X(111) supported gold clusters were explored using the basin-hopping (BH) global-minimum searching method combined with DFT geometry optimization. BH searching was carried out on the Au clusters with the fixed substrate. Different 2D and 3D structures were used as initial seeds for sampling. Due to the large size of the system and the limitation in computational capacity, we performed 50–100 Monte Carlo trial steps and more than 20 low-lying structures were considered, and we employed full geometry optimization for each system.Spin-polarized DFT calculations were performed using the Vienna ab initio simulation package (VASP, version 5.2.12) in which the valence electronic states were expanded in the basis of plane waves, with the core–valence interaction represented using the scalar relativistic projector augmented wave (PAW) approach57 and a cutoff of 400 eV. We treated the Ce (4f, 5s, 5p, 5d, 6s), O (2s, 2p) and Au (5d, 6s, 6p) electrons as valence states, whereas the remaining electrons were kept frozen as core states in the PAW method. The Perdew–Burke–Ernzerhof (PBE) GGA58 with the inclusion of the Hubbard-U term using the Dudarev approach59 was chosen to account for the exchange and correlation. In all calculations a value of U = 5 eV as suggested initially by Nolan et al.60 was used to obtain the localized electron distribution. The population was evaluated by Bader charge analysis. It should be noted that for free-standing Au clusters the structure transition between the planar (2D) and island (3D) might be sensitive to the choice of density functional.35,36,61 However, recent theoretical work by Zhang et al. has shown the same method used in this paper can give reasonable results for supported Au clusters on reduced ceria surfaces.56
The CeO2 support was modeled using a 4 × 4 supercell containing a slab of twelve atomic layers with a vacuum space of 15 Å. Six top atomic layers of the ceria support together with the gold clusters were allowed to relax while the other layers were kept fixed during geometry optimization. The force convergence criterion was set at 0.02 eV Å−1. The Brillouin zone sampling used a 2 × 2 × 1 Monkhorst-Pack grid, with the third vector perpendicular to the surface plane. The adsorption energy between the gold cluster and the ceria support was defined as ΔEad(Aun) = E(ceria–Aun) − E(Aun) − E(ceria), where E(ceria–Aun), E(Aun), and E(ceria) were the energies of the ceria–Aun system, the Aun cluster and the ceria substrate.
For CO and O2 adsorptions, all binding sites on ceria-supported gold systems were considered. The adsorption energies were evaluated using ΔEad(CO/O2) = E(ceria–Aun–CO/O2) − E(ceria–Aun) − E(CO/O2). In this work, CO binding energies on gold atoms are 1 eV, consistent with the experimental results.62
3. Results and discussion
The oxygen vacancies are abundant on ceria surfaces due to their low formation energies.63–67 Compared with the clean surface sites, these defect sites have stronger binding energies for Au atoms, which are generally considered as the Au nucleation sites.68 Previous studies showed that there are several electronic configurations for the CeO2−X(111) surfaces which differ in the positions of the reduced Ce3+ cations.69,70 However, these electronic configurations are almost energy degenerate and their energy differences are negligible when compared to the strong binding energies of Au atoms on these surfaces.The STM and non-contact AFM experiments suggested that the surface and subsurface vacancies were ordered into triangular clusters and chains.71–76 In this paper, four distinct types of CeO2−X(111) surfaces are considered, as shown in Fig. 1. Our results indicate that LSVT is most stable among the three configurations, with a vacancy formation energy of 7.06 eV, which is 0.21 eV lower in energy than TSVT (ΔE = 7.27 eV) and 1.05 eV lower than dLSVC (ΔE = 8.11 eV). It is consistent with previous calculations3 and some experiments,71,72 but has slightly deviated from the experiments by Namai et al.73 and Esch et al.74
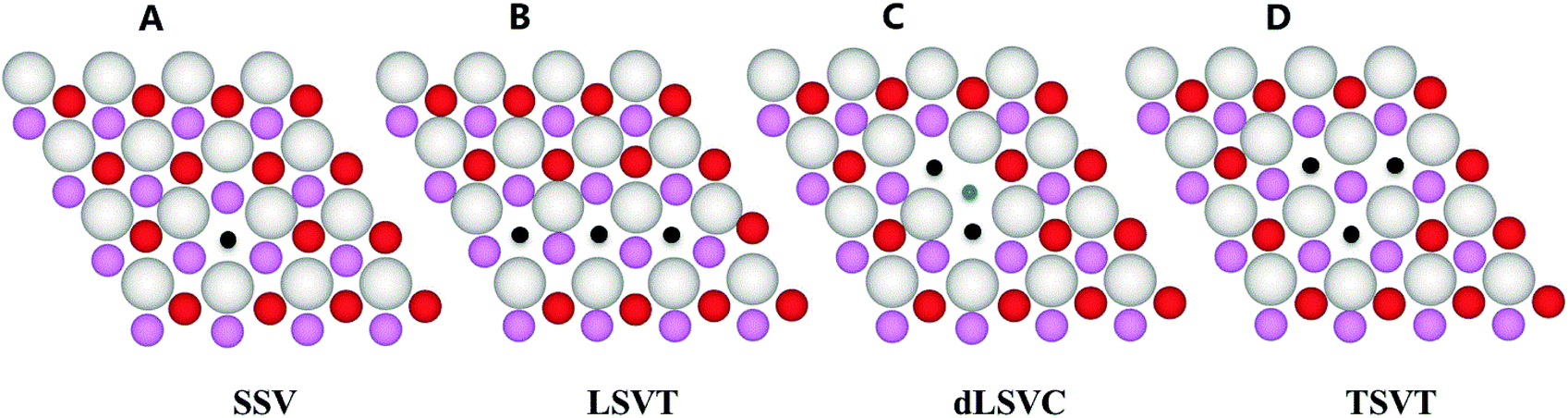 | ||
| Fig. 1 Structures of CeO2−X(111) surfaces. Surface oxygen, subsurface oxygen, and cerium atoms are in red, pink, and white respectively. The black dot represents the surface oxygen vacancy and the blue dot represents the subsurface oxygen vacancy. The remaining substrate is not shown for clarity. | ||
3.1. Structure evolution of gold clusters on CeO2−X(111) surfaces
Structural parameters for the most stable Au nanoparticles on CeO2−X(111) surfaces are collected in Table S1–S4,† and their corresponding structures are presented in Fig. 2. For the single Au atom, the most stable adsorption site is located at the oxygen vacancy of CeO2−X(111) surfaces. The adsorption energy of the Au atom on SSV is 2.57 eV, consistent with previous studies.77–79 For LSVT, we find an interesting phenomenon that a subsurface oxygen atom easily jumps up to the surface (energy barrier <0.01 eV) which is 0.34 eV more stable in the presence of the single gold atom. Furthermore, we calculated the energies of the pure LSVT surface before and after the jump of the subsurface oxygen atom and found that the pure former configuration is more stable by 0.24 eV and the jump barrier is 0.86 eV.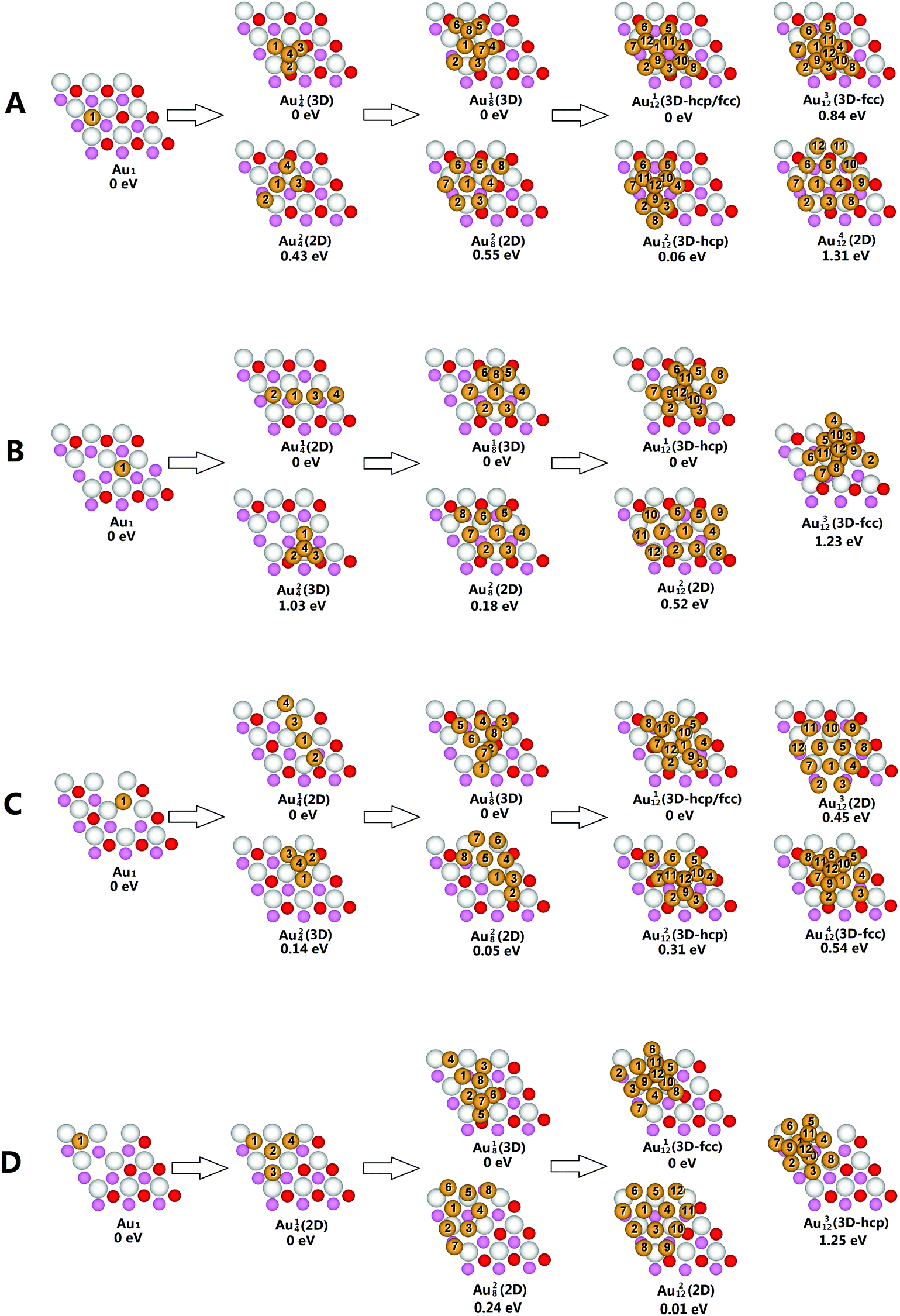 | ||
| Fig. 2 Structure evolution of small-sized gold clusters on CeO2−X(111). Gold, surface oxygen, subsurface oxygen, and cerium atoms are in gold, red, pink, and white respectively. Energy values are with respect to the most stable structure of each cluster size. The remaining substrate is not shown for clarity. For Aumn, n represents the number of gold atoms, m represents the label order of the gold clusters Aun. (A) Structure evolution of small-sized gold clusters on SSV; (B) structure evolution of small-sized gold clusters on LSVT; (C) structure evolution of small-sized gold clusters on dLSVC; and (D) structure evolution of small-sized gold clusters on TSVT. | ||
The most stable Au4 cluster on SSV is a pyramidal 3D structure with one Au atom occupying the vacancy site. As a result of strong Au–Au bonding, the vacancy-occupying Au atom shifts slightly toward the other Au atoms. The 3D structure is 0.43 eV lower in energy than the 2D structure, consistent with the previous results.56 The most stable Au4 clusters on LSVT and dLSVC are both linear structures with gold atoms adsorbed on the oxygen vacancies in a row. Both of them are more stable than their 3D counterparts by 1.03 eV and 0.14 eV, respectively. Their stabilities can be attributed to the stronger adsorption energies of Au atoms on the vacancy sites, which induce gold atoms to grow preferentially on the vacancies until all vacancies are occupied. A threefold symmetry planar structure is found to be the most stable structure for Au4 on TSVT and all initial 3D structures are transformed to 2D structures after geometry optimization.
The island (3D) structures are 0.55 eV and 0.18 eV lower in energy than the layered (2D) structures for Au8 on SSV and LSVT, respectively. It reflects the fact that the Au–Au bonding is stronger than the Au–O bonding. Thus, moving an Au atom from the ceria surface to the 3-fold hollow site of the first layer Au atoms brings one more Au–Au bond to offset the energy loss of an Au–O bond to form a more stable structure. For Au8/dLSVC, we find that the most stable 3D structure and 2D structure are almost energetic degenerate with the slight stability of the 3D structure by 0.05 eV. Planar Au18 is the most stable structure of the Au8 cluster on TSVT, and is 0.24 eV lower in energy than 3D Au28.
It is interesting to find two types of competing 3D nucleation patterns, namely the face-centered cubic like (fcc-like) nucleation and the hexagonal close packing like (hcp-like) nucleation for small-sized gold clusters. For Au12/SSV (Fig. 2A), Au112 is the precursor of the hcp/fcc mixture-like structure, while Au212 is an hcp-like structure (i.e., ABA stacking). Furthermore, the fcc-like cluster (i.e., ABC stacking) Au312 is less stable compared with the hcp-like structure with an energy difference of 0.78 eV. The most stable 2D cluster (Au412) is at least 1.31 eV higher in energy than the most stable 3D isomer. Au112 (Fig. 2B) is the most stable Au12 cluster on LSVT with an hcp-like structure. Similarly to Au12/SSV, the hcp-like structure is more energetic favorable than the fcc-like configuration (Au212) and the 2D structure (Au312) by 1.23 eV and 0.52 eV, respectively. For Au12/dLSVC, the most stable structure, Au112 (Fig. 2C), is the precursor of the hcp/fcc mixture-like 3D structure. It is more stable than the 2D and other 3D structures on the order of hcp/fcc > hcp > 2D > fcc. The most stable structure of Au112 on TSVT is very similar to the most stable Au13 structure on TSVT as proposed by Kim and co-workers,3 which is also a fcc-like 3D structure (Fig. 4e in ref. 3). Interestingly, the hcp-like structure is at least 1.25 eV higher in energy than the fcc-like structure for TSVT, but is more stable than fcc-like structure on other types of reduced ceria surfaces.
In general, the transition from the 2D structure to 3D structure occurs at a small cluster size of four gold atoms on SSV, eight gold atoms on LSVT and dLSVC and a relatively larger size of twelve Au atoms on TSVT.
3.2. The mechanism of structure evolution and electronic analysis
Charge strongly influences the dimensionality35,36,80 and catalytic activities81 of gold clusters. Here, the charges of each gold atom and gold cluster are shown in Table 1, and the results for the most stable gold structure of each cluster size are discussed below.| 1st | 2nd | 3rd | 4th | 5th | 6th | 7th | 8th | 9th | 10th | 11th | 12th | e t | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SSV | Au1 | −0.58 | −0.58 | |||||||||||
| Au4 | −0.61 | +0.19 | +0.20 | −0.08 | −0.30 | |||||||||
| Au8 | −0.11 | +0.04 | +0.15 | +0.18 | +0.22 | +0.16 | −0.09 | −0.05 | +0.50 | |||||
| Au12 | −0.27 | +0.13 | +0.21 | +0.21 | +0.14 | +0.11 | +0.14 | +0.10 | −0.10 | −0.05 | −0.08 | −0.08 | +0.48 | |
| LSVT | Au1 | −0.68 | −0.68 | |||||||||||
| Au4 | −0.23 | −0.44 | −0.25 | −0.44 | −1.36 | |||||||||
| Au8 | −0.04 | +0.03 | +0.06 | −0.47 | +0.17 | +0.16 | −0.48 | −0.03 | −0.60 | |||||
| Au12 | −0.21 | +0.13 | +0.08 | −0.10 | +0.12 | +0.13 | −0.54 | −0.09 | +0.01 | −0.03 | −0.02 | −0.09 | −0.62 | |
| dLSVC | Au1 | −0.70 | −0.70 | |||||||||||
| Au4 | −0.39 | −0.04 | −0.37 | −0.08 | −0.89 | |||||||||
| Au8 | −0.64 | +0.16 | +0.07 | +0.13 | −0.04 | −0.11 | −0.08 | −0.03 | −0.55 | |||||
| Au12 | −0.33 | +0.14 | +0.12 | −0.04 | +0.03 | −0.53 | +0.16 | +0.05 | −0.08 | −0.08 | −0.05 | −0.05 | −0.65 | |
| TSVT | Au1 | −0.62 | −0.62 | |||||||||||
| Au4 | −0.42 | −0.12 | −0.39 | −0.42 | −1.36 | |||||||||
| Au8 | −0.20 | −0.12 | −0.34 | −0.26 | −0.08 | +0.02 | −0.01 | −0.09 | −1.08 | |||||
| Au12 | −0.11 | +0.02 | 0.00 | −0.10 | −0.53 | +0.05 | +0.02 | +0.05 | +0.07 | −0.02 | −0.01 | −0.05 | −0.62 | |
For gold clusters on CeO2−X(111) surfaces, the trend of the charges of gold atoms can be summarized as follows: (1) the gold atoms filling the oxygen vacancies are all negatively charged. (2) Other first-layer gold atoms are positively charged when adsorbing on the bridge sites of Ce atoms and top or bridge sites of surface oxygen atoms, whereas they are negatively charged when adsorbing on the top sites of Ce atoms or top sites of subsurface oxygen atoms. These results are in very good agreement with the work of Branda et al.82 (3) The second-layer and third-layer gold atoms are always slightly negatively charged. Meanwhile, the oxidation and reduction of gold atoms are always associated with the reduction and oxidation of the surface cerium atoms (Au→Auδ+ and Ce4+→Ce3+; Au→Auδ− and Ce3+→Ce4+), while the oxidation states of the surface oxygen atoms are kept unchanged during these processes. It means that the charge transfer occurs only between Au clusters and their neighboring Ce atoms.
It is known that more negative charges favor the formation of planar structures for free-standing gold clusters.35,36,80 Our results indicate this is also applicable to gold clusters on CeO2−X(111) surfaces. For Au4, the most stable structure (3D structure) on SSV takes the charge of −0.30e, while the most stable structure (2D structure) on LSVT, dLSVC, and TSVT takes the charges of −1.36e, −0.89e, and −1.36e, respectively. Much less negative charge makes the Au4 cluster easier to transform from the 2D structure to 3D structure at the fewest gold atoms on SSV. A similar case could also be found in Au8. The charges of the most stable Au8 isomer on SSV, LSVT, dLSVC, and TSVT are +0.50e, −0.55e, −0.60e, and −1.08e, respectively, which makes Au8 prefer a planar structure on TSVT and a 3D structure on SSV and LSVT, but maybe co-existence of 2D and 3D motifs on dLSVC due to their competitive stabilities. For Au12 clusters, the most stable structures are all 3D structures except for TSVT, where 2D and 3D motifs may co-exist. The transition size from the 2D structure to 3D structure is Au2D→3D@TSVT > Au2D→3D@LSVT ∼ Au2D→3D@dLSVC > Au2D→3D@SSV. Thus, our results suggest that the charge transfer between Au clusters and the ceria surface determines the transition size from the 2D structure to 3D structure for gold clusters on CeO2−X(111) surfaces.
The DOS of Ce f orbitals of CeO2−X(111) surfaces are plotted in Fig. 3. The integral values of the occupied 4f (Ce) orbitals which lie just below the Fermi level (occupancy of the 4f (Ce) orbitals) are collected in Table 2, corresponding to the valence electrons of cerium atoms. It is clear that these values first decrease upon the adsorption of the vacancy-filling gold atoms and then increase with increasing the size of the gold clusters. It means that the electrons transfer from the oxygen vacancies to the gold clusters for smaller size initially, and gradually transfer reversely from the gold clusters to the surfaces for larger size. Furthermore, the charges and structures of the Au clusters are determined not only by their cluster sizes, but also by the size and type of oxygen vacancies. Thus, it is possible for experimentalists to control the morphologies of the Au clusters by adjusting the size and type of oxygen vacancies, and further to influence their catalytic capacities.
 | ||
| Fig. 3 Density of states projected onto f (Ce) orbitals of the CeO2−X(111) substrate before and after adsorption of gold atoms. The Fermi levels of each system are aligned and set to 0. For clarity we display only the occupancy states. (A) SSV; (B) LSVT; (C) dLSVC; and (D) TSVT. | ||
| Pure surface | Au1 | Au4 | Au8 | Au12 | |
|---|---|---|---|---|---|
| SSV | 1.79 | 0.94 | 1.96 | 3.68 | 3.91 |
| LSVT | 5.40 | 4.47 | 3.59 | 5.46 | 5.53 |
| dLSVC | 5.19 | 4.41 | 4.52 | 5.55 | 5.59 |
| TSVT | 6.20 | 4.48 | 3.48 | 4.59 | 5.42 |
3.3. CO and O2 adsorption
The adsorptions of CO and O2 are generally used as a probe to explore the catalytic activities of Au/CeO2 systems.24 In this paper, all distinctive sites of the most stable gold clusters and the CeO2−X(111) surfaces are thoroughly examined for the adsorption of CO and O2. The adsorption energies are schematically illustrated in Fig. 4. Different colors are used to describe the binding capability of every surface site. Dark green represents strong exothermic adsorption (adsorption energies <−0.9 eV); green represents moderate exothermic adsorption (adsorption energies are between −0.5 and −0.9 eV); light green represents weak exothermic adsorption (adsorption energies are between −0.2 and −0.5 eV); gold represents very weak exothermic adsorption (adsorption energies >−0.2 eV). Detailed CO and O2 adsorption configurations together with their corresponding adsorption energies are shown in Fig. S13–S26.† The charges and bond lengths are collected in Table S5–S9.† | ||
| Fig. 4 Schematic illustration of adsorption energies of CO and O2 on CeO2−X(111) supported gold clusters. Adsorption energy color code: dark green, <−0.9 eV; green, −0.5 to −0.9 eV; light green, −0.2 to −0.5 eV; gold, >−0.2 eV. Negative sign represents the exothermic process in adsorption. Surface oxygen, subsurface oxygen, and cerium atoms are in red, pink, and white respectively. The remaining substrate is not shown for clarity. (A) Gold clusters on SSV; (B) gold clusters on LSVT; (C) gold on dLSVC; and (D) gold clusters on TSVT. | ||
As shown in Fig. 4, O2 adsorption is much weaker than CO adsorption on all types of Au/CeO2−X systems. CO preferentially adsorbs on cationic Au atoms than anionic or metallic Au atoms, because the empty 6s orbitals of cationic Au atoms could easily accept electrons from the 5σ orbital of CO to strengthen the bonding.56 Both CO and O2 are preferentially adsorbed on low-coordinated gold atom sites. All details can be found in Table S5–S9.† Next we will discuss the CO and O2 adsorption capability of the four types of Au/CeO2−X systems, respectively.
The favorable CO and O2 adsorption sites and their strength on Au/SSV are shown in Fig. 4A. Au4 is a pyramid structure. CO strongly binds with the Au atom of the pyramid top, the lowest coordinate site in the gold cluster. O2 adsorptions are very weak for all positions. Au8 is a 3D structure with strong CO adsorption for all gold atoms except the inner fully-coordinated Au atom. O2 binds moderately on the Au8 with a binding energy of −0.49 eV and an elongated O–O bond length of 1.32 Å, known as superoxide adsorption. The calculated results showed that for O2 molecular adsorption the spin state was triplet, while for superoxide adsorption the spin state was singlet. Au12 is a 3D cluster with the hcp/fcc mixed structure. The positively-charged interfacial gold atoms become the strongest CO adsorption sites, but all sites are not favorable for O2 adsorptions.
For Au/LSVT, as shown in Fig. 4B, the strong CO adsorption sites are much fewer than those for Au/SSV. For linear Au4, CO more favorably adsorbs on the interfacial Ce3+ ion than on the gold atoms. Au8 is a quasi-2D structure with one gold atom shifting from the plane to the second layer, which induces the neighboring Au atoms to be under-coordinated. Thus, these Au atoms and the contacting Ce atom are favorable for CO adsorption than other sites. The only favorable site for O2 adsorption is the second-layer Au atom with a binding energy of −0.46 eV. For the Au12 hcp-like cluster, both CO and O2 prefer to adsorb on the top of the cluster with binding energies of −0.90 eV and −0.24 eV, respectively.
CO and O2 adsorptions are stronger for Au/dLSVC than for Au/LSVT, as shown in Fig. 4C. For linear Au4, CO adsorption energies are between −0.22 eV and −0.85 eV on gold atoms and −0.21 eV on the neighboring Ce3+ ion. O2 prefers to adsorb on gold atoms and the Ce3+ ion with binding energies of −0.84 eV and −0.29 eV, respectively. Au18 has three strong CO adsorption sites on gold with one at the interfacial triangle and two at the second layer. Besides gold atoms, a surface Ce3+ ion is also preferred for CO adsorption. On the other hand, a superoxide O2 adsorption could also be observed with a binding energy of −0.37 eV. Au12 is an hcp/fcc mixed 3D structure with multiple low-coordinate Au atoms. These gold atoms are metallic and more favorable for CO adsorptions. Besides, CO adsorption on surface Ce atoms is also observed. On the other hand, two favorable O2 adsorption sites are found on gold clusters with a binding energy of −0.27 eV.
The CeO2 surface plays a more important role in CO and O2 adsorption for Au/TSVT, as shown in Fig. 4D. For Y-shaped Au4, CO adsorptions are much weaker in gold atoms but are stronger in the neighboring Ce3+ ion. Besides, O2 could stably bind to the three corners of Au4 and the surface Ce3+ ion, much favorable than other reduced surfaces. Au8 is a planar structure. The low-coordinate metallic gold atoms are the most favorable CO binding sites, in addition to a surface Ce3+ ion. There are three O2 preferable binding modes with two on gold (superoxide: −0.58 eV; molecular: −0.58 eV) and one on the surface (−0.58 eV). Au112 is an fcc-like structure. Its top site is most favorable for CO and O2 binding. It should be noted that the gold cluster could help the neighboring surface O atom to bind strongly with CO to form a highly stable triangular CO2− species. This is consistent with the observation of Kim and co-workers that CO2− species were highly stable on Au13@TSVT.3
Besides, it should be noted that for Au12 clusters the bilayer structures (i.e., Au12@SSV and Au12@dLSVC) present more and stronger CO and O2 adsorption sites than the trilayer structures (i.e., Au12@LSVT and Au12@TSVT). Similarly, the bilayer structures of Au8 clusters also present more CO and O2 favorable adsorption sites. These results could be compared with the studies of Goodman and co-workers,9,83 which suggested that the bilayer gold structures exhibited a significantly higher performance for CO oxidation on reduced TiO2 surfaces. Thus, the thickness of the gold clusters may also be an important factor for their extraordinary catalytic activities.
4. Conclusion
Through a comprehensive DFT + U investigation, we found the types of oxygen surface defects strongly affect the electron transfer between the CeO2−X(111) surface and Au clusters, and further influence the 2D–3D transition size for gold clusters on CeO2−X(111) surfaces. The transition occurs at cluster sizes on the order of Au2D→3D@TSVT > Au2D→3D@dLSVC ∼ Au2D→3D@LSVT > Au2D→3D@SSV. Furthermore, we found two competitive nucleation patterns for supported gold clusters: the hcp-like nucleation and the fcc-like nucleation. In the case of Au@TSVT, gold atoms prefer the fcc-like nucleation pattern, while for other cases gold atoms prefer the hcp-like nucleation pattern in small size.Detailed site-by-site studies of the adsorption of CO and O2 show that CO preferentially adsorbs on cationic Au atoms and low coordinate metallic Au atoms, while O2 preferentially adsorbs on low coordinate gold atoms and Au–Ce3+ interfacial sites.
Different types of surface vacancies could adjust not only the thickness and structure motifs of gold clusters, but also the charges and oxidation states of gold atoms, which were all highly correlated with CO and O2 binding strength. Our results may provide a new method to control the catalytic activity of ceria-supported gold clusters by regulating the structures of gold clusters on the ceria substrate.
Acknowledgements
This work was supported by the startup funding from Shanghai Institute of Applied Physics, Chinese Academy of Sciences (Y290011011), National Natural Science Foundation of China (21273268), “Hundred People Project” from Chinese Academy of Sciences, and “Pu-jiang Rencai Project” from the Science and Technology Commission of Shanghai Municipality (13PJ1410400). The computational resources utilized in this research were provided by Shanghai Supercomputer Center, National Supercomputing Center in Tianjin and Supercomputing Center of Chinese Academy of Sciences in Beijing.References
- J. A. Farmer and C. T. Campbell, Science, 2010, 329, 933–936 CrossRef CAS PubMed
.
- M. F. Camellone and S. Fabris, J. Am. Chem. Soc., 2009, 131, 10473–10483 CrossRef PubMed
- H. Y. Kim, H. M. Lee and G. Henkelman, J. Am. Chem. Soc., 2012, 134, 1560–1570 CrossRef CAS PubMed
- M. Cargnello, V. V. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS PubMed
- Z.-P. Liu, S. J. Jenkins and D. A. King, Phys. Rev. Lett., 2005, 94, 196102 CrossRef
- J. A. Rodriguez, P. Liu, J. Hrbek, J. Evans and M. Pérez, Angew. Chem., Int. Ed., 2007, 46, 1329–1332 CrossRef CAS PubMed
- Y. Chen, J. Cheng, P. Hu and H. Wang, Surf. Sci., 2008, 602, 2828–2834 CrossRef CAS PubMed
- M. B. Boucher, S. Goergen, N. Yi and M. Flytzani-Stephanopoulos, Phys. Chem. Chem. Phys., 2011, 13, 2517–2527 RSC
- M. Chen and D. W. Goodman, Acc. Chem. Res., 2006, 39, 739–746 CrossRef CAS PubMed
- M. Chen and D. Goodman, Catal. Today, 2006, 111, 22–33 CrossRef CAS PubMed
- M. Chen and D. W. Goodman, Chem. Soc. Rev., 2008, 37, 1860–1870 RSC
- Y. Gao, N. Shao, Y. Pei and X. C. Zeng, Nano Lett., 2010, 10, 1055–1062 CrossRef CAS PubMed
- N. Lopez and J. K. Nørskov, J. Am. Chem. Soc., 2002, 124, 11262–11263 CrossRef CAS PubMed
- J. Guzman and B. C. Gates, J. Am. Chem. Soc., 2004, 126, 2672–2673 CrossRef CAS PubMed
- L. Molina, M. Rasmussen and B. Hammer, J. Chem. Phys., 2004, 120, 7673–7680 CrossRef CAS PubMed
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS PubMed
- G. J. Hutchings, M. S. Hall, A. F. Carley, P. Landon, B. E. Solsona, C. J. Kiely, A. Herzing, M. Makkee, J. A. Moulijn and A. Overweg, J. Catal., 2006, 242, 71–81 CrossRef CAS PubMed
- X. Lin, B. Yang, H.-M. Benia, P. Myrach, M. Yulikov, A. Aumer, M. A. Brown, M. Sterrer, O. Bondarchuk and E. Kieseritzky, J. Am. Chem. Soc., 2010, 132, 7745–7749 CrossRef CAS PubMed
- Y. Gao, N. Shao, Y. Pei, Z. Chen and X. C. Zeng, ACS Nano, 2011, 5, 7818–7829 CrossRef CAS PubMed
- M. M. Schubert, S. Hackenberg, A. C. Van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef CAS
- I. N. Remediakis, N. Lopez and J. K. Nørskov, Angew. Chem., Int. Ed., 2005, 117, 1858–1860 CrossRef
- J. A. Rodríguez, J. Evans, J. s. Graciani, J.-B. Park, P. Liu, J. Hrbek and J. F. Sanz, J. Phys. Chem. C, 2009, 113, 7364–7370 Search PubMed
- B.-T. Teng, J.-J. Lang, X.-D. Wen, C. Zhang, M. Fan and H. G. Harris, J. Phys. Chem. C, 2013, 117, 18986–18993 CAS
- N. Lopez, T. Janssens, B. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Nørskov, J. Catal., 2004, 223, 232–235 CrossRef CAS PubMed
- H. Häkkinen and U. Landman, Phys. Rev. B: Condens. Matter, 2000, 62, R2287 CrossRef
- J. Wang, G. Wang and J. Zhao, Phys. Rev. B: Condens. Matter, 2002, 66, 035418 CrossRef
- H. Häkkinen, B. Yoon, U. Landman, X. Li, H.-J. Zhai and L.-S. Wang, J. Phys. Chem. A, 2003, 107, 6168–6175 CrossRef
- J. Li, X. Li, H.-J. Zhai and L.-S. Wang, Science, 2003, 299, 864–867 CrossRef CAS PubMed
- R. M. Olson, S. Varganov, M. S. Gordon, H. Metiu, S. Chretien, P. Piecuch, K. Kowalski, S. A. Kucharski and M. Musial, J. Am. Chem. Soc., 2005, 127, 1049–1052 CrossRef CAS PubMed
- S. Bulusu, X. Li, L.-S. Wang and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8326–8330 CrossRef CAS PubMed
- R. M. Olson and M. S. Gordon, J. Chem. Phys., 2007, 126, 214310 CrossRef PubMed
- Y. Dong and M. Springborg, J. Phys. Chem. C, 2007, 111, 12528–12535 CAS
- P. Gruene, D. M. Rayner, B. Redlich, A. F. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Science, 2008, 321, 674–676 CrossRef CAS PubMed
- D. Tian and J. Zhao, J. Phys. Chem. A, 2008, 112, 3141–3144 CrossRef CAS PubMed
- M. P. Johansson, A. Lechtken, D. Schooss, M. M. Kappes and F. Furche, Phys. Rev. A, 2008, 77, 053202 CrossRef
- L. Ferrighi, B. Hammer and G. K. Madsen, J. Am. Chem. Soc., 2009, 131, 10605–10609 CrossRef CAS PubMed
- T. Risse, S. Shaikhutdinov, N. Nilius, M. Sterrer and H.-J. Freund, Acc. Chem. Res., 2008, 41, 949–956 CrossRef CAS PubMed
- D. Pillay and G. S. Hwang, Phys. Rev. B: Condens. Matter, 2005, 72, 205422 CrossRef
- D. Ricci, A. Bongiorno, G. Pacchioni and U. Landman, Phys. Rev. Lett., 2006, 97, 036106 CrossRef
- M. Sterrer, M. Yulikov, E. Fischbach, M. Heyde, H. P. Rust, G. Pacchioni, T. Risse and H. J. Freund, Angew. Chem., Int. Ed., 2006, 45, 2630–2632 CrossRef CAS PubMed
- M. Sterrer, T. Risse, M. Heyde, H.-P. Rust and H.-J. Freund, Phys. Rev. Lett., 2007, 98, 206103 CrossRef
- M. Walter, P. Frondelius, K. Honkala and H. Häkkinen, Phys. Rev. Lett., 2007, 99, 096102 CrossRef
- P. Frondelius, H. Häkkinen and K. Honkala, New J. Phys., 2007, 9, 339 CrossRef
- V. Simic-Milosevic, M. Heyde, X. Lin, T. König, H.-P. Rust, M. Sterrer, T. Risse, N. Nilius, H.-J. Freund and L. Giordano, Phys. Rev. B: Condens. Matter, 2008, 78, 235429 CrossRef
- C. Harding, V. Habibpour, S. Kunz, A. N.-S. Farnbacher, U. Heiz, B. Yoon and U. Landman, J. Am. Chem. Soc., 2008, 131, 538–548 CrossRef PubMed
- N. Shibata, A. Goto, K. Matsunaga, T. Mizoguchi, S. Findlay, T. Yamamoto and Y. Ikuhara, Phys. Rev. Lett., 2009, 102, 136105 CrossRef CAS
- N. Mammen, S. Narasimhan and S. d. Gironcoli, J. Am. Chem. Soc., 2011, 133, 2801–2803 CrossRef CAS PubMed
- D.-e. Jiang, S. H. Overbury and S. Dai, J. Phys. Chem. Lett., 2011, 2, 1211–1215 CrossRef CAS
- X. Shao, N. Nilius and H.-J. Freund, Phys. Rev. B: Condens. Matter, 2012, 85, 115444 CrossRef
- L. B. Vilhelmsen and B. Hammer, Phys. Rev. Lett., 2012, 108, 126101 CrossRef
- B.-T. Teng, F.-M. Wu, W.-X. Huang, X.-D. Wen, L.-H. Zhao and M.-F. Luo, ChemPhysChem, 2012, 13, 1261–1271 CrossRef CAS PubMed
- T. Akita, M. Okumura, K. Tanaka, M. Kohyama and M. Haruta, J. Mater. Sci., 2005, 40, 3101–3106 CrossRef CAS PubMed
- J.-L. Lu, H.-J. Gao, S. Shaikhutdinov and H.-J. Freund, Catal. Lett., 2007, 114, 8–16 CrossRef CAS
- C. Weststrate, A. Resta, R. Westerström, E. Lundgren, A. Mikkelsen and J. Andersen, J. Phys. Chem. C, 2008, 112, 6900–6906 CAS
- C. Zhang, A. Michaelides and S. J. Jenkins, Phys. Chem. Chem. Phys., 2011, 13, 22–33 RSC
- C. Zhang, A. Michaelides, D. A. King and S. J. Jenkins, J. Am. Chem. Soc., 2010, 132, 2175–2182 CrossRef CAS PubMed
- P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS
- S. Dudarev, G. Botton, S. Savrasov, C. Humphreys and A. Sutton, Phys. Rev. B: Condens. Matter, 1998, 57, 1505–1509 CrossRef CAS
- M. Nolan, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 595, 223–232 CrossRef CAS PubMed
- D. A. Götz, R. Schäfer and P. Schwerdtfeger, J. Comput. Chem., 2013, 34, 1975–1981 CrossRef PubMed
- M. Neumaier, F. Weigend, O. Hampe and M. M. Kappes, J. Chem. Phys., 2005, 122, 104702 CrossRef PubMed
- T. Sayle, S. Parker and C. Catlow, Surf. Sci., 1994, 316, 329–336 CrossRef CAS
- C. T. Campbell and C. H. Peden, Science, 2005, 309, 713–714 CrossRef CAS PubMed
- X. Wang, J. Rodriguez, J. Hanson, M. Perez and J. Evans, J. Chem. Phys., 2005, 123, 221101 CrossRef CAS PubMed
- M. Nolan, J. E. Fearon and G. W. Watson, Solid State Ionics, 2006, 177, 3069–3074 CrossRef CAS PubMed
- A. Migani, G. N. Vayssilov, S. T. Bromley, F. Illas and K. M. Neyman, J. Mater. Chem., 2010, 20, 10535–10546 RSC
- C. Zhang, A. Michaelides, D. A. King and S. J. Jenkins, J. Phys. Chem. C, 2009, 113, 6411–6417 CAS
- H.-Y. Li, H.-F. Wang, X.-Q. Gong, Y.-L. Guo, Y. Guo, G. Lu and P. Hu, Phys. Rev. B: Condens. Matter, 2009, 79, 193401 CrossRef
- M. V. Ganduglia-Pirovano, J. L. Da Silva and J. Sauer, Phys. Rev. Lett., 2009, 102, 026101 CrossRef
- H. Nörenberg and G. Briggs, Phys. Rev. Lett., 1997, 79, 4222 CrossRef
- H. Nörenberg and G. Briggs, Surf. Sci., 1999, 424, L352–L355 CrossRef
- Y. Namai, K.-i. Fukui and Y. Iwasawa, J. Phys. Chem. B, 2003, 107, 11666–11673 CrossRef CAS
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS PubMed
- S. Torbrügge, M. Reichling, A. Ishiyama, S. Morita and O. Custance, Phys. Rev. Lett., 2007, 99, 056101 CrossRef
- J. Kullgren, Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 896, 2012.
- Y. Chen, P. Hu, M.-H. Lee and H. Wang, Surf. Sci., 2008, 602, 1736–1741 CrossRef CAS PubMed
- C. Zhang, A. Michaelides, D. A. King and S. J. Jenkins, J. Chem. Phys., 2008, 129, 194708 CrossRef PubMed
- N. C. Hernández, R. Grau-Crespo, N. H. de Leeuw and J. F. Sanz, Phys. Chem. Chem. Phys., 2009, 11, 5246–5252 RSC
- M. Mantina, R. Valero and D. G. Truhlar, J. Chem. Phys., 2009, 131, 5 CrossRef PubMed
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed
- M. a. M. Branda, N. C. Hernández, J. F. Sanz and F. Illas, J. Phys. Chem. C, 2010, 114, 1934–1941 CAS
- M. Chen and D. Goodman, Science, 2004, 306, 252–255 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: All geometries of low-lying gold clusters on CeO2−X(111) surfaces, and all possible CO and O2 adsorption configurations are shown in the ESI. See DOI: 10.1039/c4nr03346a |
| This journal is © The Royal Society of Chemistry 2015 |