Chemical force microscopy of stimuli-responsive adhesive copolymers
Audrey
Beaussart
a,
T. Chinh
Ngo
b,
Sylvie
Derclaye
a,
Radostina
Kalinova
c,
Rosica
Mincheva
c,
Philippe
Dubois
c,
Philippe
Leclère
*b and
Yves F.
Dufrêne
*a
aUniversité catholique de Louvain, Institute of Life Sciences, Croix du Sud 1, bte L7.04.01., B-1348 Louvain-la-Neuve, Belgium. E-mail: Yves.Dufrene@uclouvain.be
bLaboratory for Chemistry of Novel Materials, Center of Innovation and Research in Materials and Polymers (CIRMAP), University of Mons (UMONS), 20 Place du Parc, 7000 Mons, Belgium. E-mail: Philippe.Leclere@umons.ac.be
cLaboratory of Polymeric and Composite Materials, Center of Innovation and Research in Materials and Polymers (CIRMAP), University of Mons (UMONS), 20 Place du Parc, 7000 Mons, Belgium
First published on 18th November 2013
Abstract
Atomic force microscopy with chemically sensitive tips was used to investigate the hydrophobic and electrostatic interaction forces of a stimuli-responsive adhesive polymer, and their dynamic changes in response to water immersion and salt concentration. Block copolymer-filled coatings were obtained by incorporating an amphiphilic block copolymer containing a polydimethylsiloxane (PDMS) block and a poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) block in a PDMS matrix. Topographic images of fresh samples revealed the presence of nanoscale domains associated with the presence of copolymers, covered by a thin layer of PDMS. Prolonged (30 days) immersion in aqueous solution led to the exposure of the hydrophilic PDMAEMA chains on the surface. Using adhesion force mapping with hydrophobic tips, we showed that fresh samples were uniformly hydrophobic, while aged samples exhibited lower surface hydrophobicity and featured nanoscale hydrophilic copolymer domains. Force mapping with negatively charged tips revealed remarkable salt-dependent force plateau signatures reflecting desorption of polyelectrolyte copolymer chains. These nanoscale experiments show how solvent-induced conformational changes of stimuli-responsive copolymers can be used to modulate surface adhesion.
Introduction
Stimuli-responsive materials offer a wide range of applications owing to their ability to change their conformation, properties and interactions in response to external triggers.1,2 Among these, adhesive polymers with switchable properties are being increasingly used to control adhesion.3–5 One of the most widely used material in this respect is polydimethylsiloxane (PDMS). Although PDMS shows remarkable material properties, its use as an underwater adhesive is strongly limited by its low surface free energy and chemical reactivity. This problem can be solved by introducing reactive functional groups onto the polymer surface, thus making it more hydrophilic.6–8 Knowledge of the nanoscale surface properties, interactions and dynamics of adhesive polymers is a key to gain a detailed understanding of their stimuli-responsive behaviour and to efficiently exploit them in industrial applications. Various tools are available to probe the adhesion of polymeric materials, such as the quartz crystal microbalance,9 the surface force apparatus,10–12 optical tweezers,13 and surface plasmon resonance.14 Currently, there is much interest in complementing these approaches with tools capable of probing the molecular interactions of adhesive polymers at high spatial resolution.During the past 20 years, atomic force microscopy (AFM) has been instrumental in characterizing the nanoscale properties of biological specimens and soft materials.15,16 In the field of macromolecules, single-polymer force spectroscopy has contributed in the elucidation of the mechanisms governing polyelectrolyte desorption.17–19 The principle consists of binding covalently a single polymer chain to an AFM tip and measuring the adhesion forces on different substrates, including polyelectrolyte multilayers,20 gold substrates,21,22 hydrophobic monolayers23 or glass.24 These single-molecule experiments have provided information on parameters such as the adsorption energy,21 the elasticity of the polymer backbone,24 the conformational state and the cohesion energy in polymer films.20 Another powerful method for polymer studies is chemical force microscopy (CFM),25 in which the AFM tip is functionalized with well-defined chemical groups, thereby enabling the quantification of the surface properties and interactions of thin organic films,25–27 block co-polymers28 and even living cells.29–31 Yet, the CFM method has never been applied to stimuli-responsive adhesive polymers.
In this report, CFM is used to quantify and map the nanoscale hydrophobic and electrostatic interactions of a wet adhesive block copolymer, and their dynamic changes in response to environmental conditions, i.e. aging in aqueous solution and increase of salt concentration. We focus on elastomeric polydimethylsiloxane (PDMS) coatings whose adhesive properties are improved by incorporation of an amphiphilic block copolymer made of a PDMS block and of a poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) block in a matrix of PDMS. The idea behind such mixed polymer coatings is to decrease the hydrophobic properties of the polymer surface to promote adhesion in aqueous media, and therefore making it more amenable for biological applications. Using CFM with hydrophobic tips, we show that the surface of fresh samples is uniformly hydrophobic, while prolonged immersion in water decreases the surface hydrophobicity due to the exposure of the hydrophilic polymer chains. As the PDMAEMA hydrophilic blocks exhibit positively charged groups, we then use CFM with negatively charged tips to unravel the desorption behavior of these polyelectrolytes, and its dynamic change with salt concentration. Overall, the results show that the use of AFM-based force mapping with chemically sensitive tips has great potential for probing the interaction forces of stimuli-responsive materials, and for tracking their surface dynamics in response to external cues.
Results and discussion
Nanoscale structure of block copolymers
The low reactivity, long-time durability and low surface energy of PDMS make this polymer very attractive for many biomedical applications. However, strong adhesion in aqueous solution requires increasing surface energy and reactivity of PDMS, which can be readily achieved by the incorporation of amphiphilic block copolymers. With this in mind, we prepared mixed coatings containing 90% PDMS and 10% of PDMS-b-PDMAEMA, a block-copolymer bearing pendent tertiary amino groups along the polyacrylate block (Fig. 1).32 The block copolymer (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) GPCn = 33
GPCn = 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1, Đ = 1.29, DPPDMS = 135, DPPDMAEMA = 116, TPDMSg = −129 °C; TPDMAEMAg = 21 °C) was obtained through PDMS initiated atom-transfer radical polymerization as described in Methods. Fig. 2a shows a contact mode AFM height image recorded in deionized water for a freshly prepared mixed copolymer sample. The surface clearly showed morphological heterogeneities in the form of nanoscale circular domains randomly distributed in a smooth, homogeneous matrix. As the surface coverage of these domains is ∼10%, it is reasonable to ascribe them to PDMS-b-PDMAEMA, while the surrounding matrix would be made of PDMS. However, X-ray photoelectron spectroscopy (XPS) and wettability analyses showed that fresh block copolymers are covered by a thin overlayer of PDMS.33 Supporting this view, the image contrast was often fuzzy, reflecting the presence of a thin layer on top of the domains.
300 g mol−1, Đ = 1.29, DPPDMS = 135, DPPDMAEMA = 116, TPDMSg = −129 °C; TPDMAEMAg = 21 °C) was obtained through PDMS initiated atom-transfer radical polymerization as described in Methods. Fig. 2a shows a contact mode AFM height image recorded in deionized water for a freshly prepared mixed copolymer sample. The surface clearly showed morphological heterogeneities in the form of nanoscale circular domains randomly distributed in a smooth, homogeneous matrix. As the surface coverage of these domains is ∼10%, it is reasonable to ascribe them to PDMS-b-PDMAEMA, while the surrounding matrix would be made of PDMS. However, X-ray photoelectron spectroscopy (XPS) and wettability analyses showed that fresh block copolymers are covered by a thin overlayer of PDMS.33 Supporting this view, the image contrast was often fuzzy, reflecting the presence of a thin layer on top of the domains.
 | ||
| Fig. 1 Chemical structure of PDMS-b-PDMAEMA. | ||
 | ||
| Fig. 2 Structural reorganisation of block copolymers in response to water immersion. (a and b) AFM height images (5 μm × 5 μm; z-scale = 100 nm) recorded in deionized water for mixed copolymer samples containing 90% PDMS and 10% PDMS-b-PDMAEMA, either freshly prepared (a) or after aging in aqueous solution for 30 days (b). The cartoons suggest the migration of the hydrophilic PDMAEMA chains towards the surface. As migration is hampered by the densely cross-linked PDMS, the data suggest that the resulting morphology is a ‘flower-like’ structure. | ||
Notably, we found that the morphology of the copolymer-filled coatings was clearly modified upon immersion in aqueous solution for 30 days. Fig. 2b shows that although nanoscale domains were still observed, they were more heterogeneous, with a flower-like structure. Analysis of multiple spots on three independent samples confirmed that these structures were representative and reproducibly observed. We hypothesize that this peculiar morphology is due to the reorganisation of the copolymer, the domains being composed of hydrophilic copolymer chains being exposed to the surface together with cross-linked PDMS.
Aging in aqueous solution decreases the nanoscale hydrophobicity
We used adhesion force mapping with hydrophobic tips (CH3-terminated alkanethiols) to probe the local hydrophobic properties of the copolymer surfaces. Fig. 3a and b shows the adhesion force maps, adhesion force histogram and representative force curves, recorded in deionized water with hydrophobic tips on fresh copolymer samples. Most force curves recorded across the surfaces showed large adhesion forces, with a mean magnitude of 5.5 ± 0.4 nN (n = 2048 force curves from two maps obtained using different tips and samples). Comparison with the data obtained on reference surfaces31 revealed that the polymer surface had a marked hydrophobic character, equivalent to that of a self-assembled monolayer composed of 80% CH3-terminated and 20% OH-terminated alkanethiols, and exhibiting a water contact angle of 105°. This value is very close to the contact angle reported for PDMS.34,35 Adhesion maps showed homogeneous contrast, meaning the surface was entirely hydrophobic. Taken together, these observations indicate that the surface of fresh copolymers is homogeneously covered with hydrophobic PDMS. | ||
| Fig. 3 CFM demonstrates that water immersion decreases surface hydrophobicity. (a and c) Adhesion force maps (5 μm × 5 μm, gray scale 10 nN; insets: second maps from independent experiments) and (b and d) corresponding adhesion force histograms (n = 2048 force curves from two independent experiments) recorded in deionized water with CH3-terminated tips on (a and b) fresh samples and (c and d) samples aged in aqueous solution. Black lines correspond to Gaussian fits. As emphasized in the cartoons, aging in aqueous solution leads to a major drop of hydrophobicity, reflecting the exposure (or migration) of hydrophilic PDMAEMA chains at the surface (red chains on the cartoons). | ||
We then probed the nanoscale hydrophobic properties of the samples after prolonged immersion in water. Fig. 3c and d shows that the adhesion forces were substantially lower in aged samples, indicating they were more hydrophilic. Interestingly, the adhesion maps were heterogeneous, hydrophilic nanoscale patches (0.9 ± 0.4 nN) being observed in a more hydrophobic matrix (3.1 ± 0.3 nN). As these patches correlate with the heterogeneous surface morphology (Fig. 2), we believe this heterogeneous hydrophobic contrast reflects the coexistence of the PDMAEMA hydrophilic and PDMS hydrophobic blocks on top of the immersed surface. The surface hydrophobicity of PDMS decreased upon contact with water is consistent with earlier measurements.32–34 Static contact angle measurements showed that both unfilled PDMS and PDMS-b-PDMAEMA-filled PDMS coatings exhibit a hydrophobic behavior, with contact angles exceeding 100° before immersion in water. After 4 weeks of immersion, the contact angle value measured for unfilled PDMS coatings decreased to 96°, an effect believed to be a consequence of a reversible reorganization of the siloxane backbone and the methyl groups at the coating surface. Interestingly, for the block copolymer-filled coatings, the average value of the contact angle shifted to even lower values (down to 84°), thus suggesting the occurrence of other processes, besides the rearrangement of the polysiloxane chains. Based on these results, one can imagine that the surface restructuring of PDMS chains allows penetration of water molecules into the near-surface layer. As a consequence, water molecules would provoke conformational changes in the PDMS-b-PDMAEMA-formed aggregates, bringing PDMAEMA segments to the surface. Thus, nitrogen atoms should be present at the surface of the PDMS-b-PDMAEMA-filled coatings after immersion in water, which was indeed confirmed by XPS analyses.33 Upon immersion in water, unfavourable interactions between methyl groups and water molecules are expected to induce molecular reorganisation of the polymer, tilting the polar oxygen groups toward the PDMS-water interface to minimise the surface energy.36 Accordingly, CFM is a valuable method for quantifying and mapping the nanoscale hydrophobic forces of stimuli-responsive surfaces, on a scale that was not accessible before.
Block copolymers show salt-dependent polyelectrolyte adsorption properties
As the PDMS-b-PDMAEMA chains carry permanent positive charges, we hypothesized that they should contribute to the overall adhesion properties of aged copolymer samples via polyelectrolyte adsorption interactions. To address this issue, aged samples were mapped using negatively charged tips (COO−-terminated alkanethiols; pH = 5.8). Fig. 4a–c shows the adhesion force maps, adhesion force histogram and representative force curves recorded in deionized water with negatively charged tips on aged copolymer samples. The adhesion maps were highly contrasted, a large fraction of the curves (59%) showing adhesion forces ranging from 100 to 1800 pN (n = 2048 force curves from two maps obtained using different tips and samples). The adhesive signatures exhibited two types of behaviours, that is, constant force plateaus with extended rupture lengths (14%) or single linear force peaks with short rupture length (45%). These two features were often sequential (Fig. 4c, top curves), force plateaus being frequently preceded with linear peaks. We attribute the initial force peaks to the rupture of cohesive electrostatic bonds between the charged tip and sample, and the force plateaus to the stretching of individual copolymer chains. Force plateaus exhibited variable rupture lengths (from 70 to 700 nm) but uniform rupture forces of 82 ± 7 pN magnitude. As this value is in the range of plateau forces reported in earlier single-polymer force spectroscopy studies,17,21 we believe that these forces correspond to the progressive stretching and desorption of single PDMAEMA polyelectrolyte chains from the tip surface (Fig. 4g). Multiple plateaus were sometimes observed, reflecting detachment of multiples chains (Fig. 4g).18,24 Note that adhesion maps did not show nanoscale domains and that the adhesion frequency (59%) was much larger than the fraction (10%) of block copolymer introduced into the PDMS matrix. As the polyelectrolyte chains are long and mobile, and our pixel size is very small (∼150 nm × 150 nm), the same polymer chains are probed several times, leading to an overestimation of the polyelectrolyte coverage. | ||
| Fig. 4 CFM unravels the polyelectrolyte behaviour of copolymer chains. (a and d) Adhesion force maps (5 μm × 5 μm, gray scale 1000 pN; insets: second maps from independent experiments), (b and e) corresponding adhesion force histograms (n = 2048), and (c and f) representative force curves recorded using COO−-terminated tips on aged samples in deionized water (a–c) or in 0.1 M NaCl solution (d–f). (g) Hydrophilic (protonated) PDMAEMA chains show remarkable force plateau signatures documenting polyelectrolyte desorption forces, which can be abolished by increasing the salt concentration. | ||
To assess whether these polyelectrolyte interactions can be controlled by environmental conditions, we probed aged copolymers at high ionic strength. Fig. 4d–f shows the force data obtained using the same tips and samples as in Fig. 4a–c following injection of 0.1 M NaCl solution. As can be seen, the presence of monovalent salts led to a major decrease of adhesion frequency, from 59% to 16%, and to the disappearance of most force plateaus (from 14% to 1%). Consistent with the literature data,24 this result indicates that increasing the ionic strength leads to a collapsed conformation of the PDMS-b-PDMAEMA chains, thus to a loss of polyelectrolyte properties (Fig. 4g, right).
We also investigated the dynamics of polymer desorption by recording force curves at various pulling speeds and contact times. As can be seen in Fig. 5a and b, plateau forces did not depend on the retraction speed, a behaviour clearly different from receptor–ligand unbinding forces37 and protein unfolding forces.38 This phenomenon, similar to that observed for amyloids39 and polymer chains,17,21 indicates that the measurements were made near thermodynamic equilibrium, thus the bonds involved in the desorption process dissociate and re-associate on a much faster timescale than the retraction speed of the tip. Fig. 5c shows that increasing the pulling speed from 100 nm s−1 to 10000 nm s−1 drastically decreased the plateau frequency, indicating that the occurrence of desorption events depends on the separation rate. To check whether this could be due to a time dependence in the desorption interaction, the contact time was varied while keeping the pulling rate constant (1000 nm s−1). Under these conditions, no variation in force plateau frequency was observed (Fig. 5d), suggesting that the electrostatic interaction bridging the polyelectrolyte to the tip is a fast process.
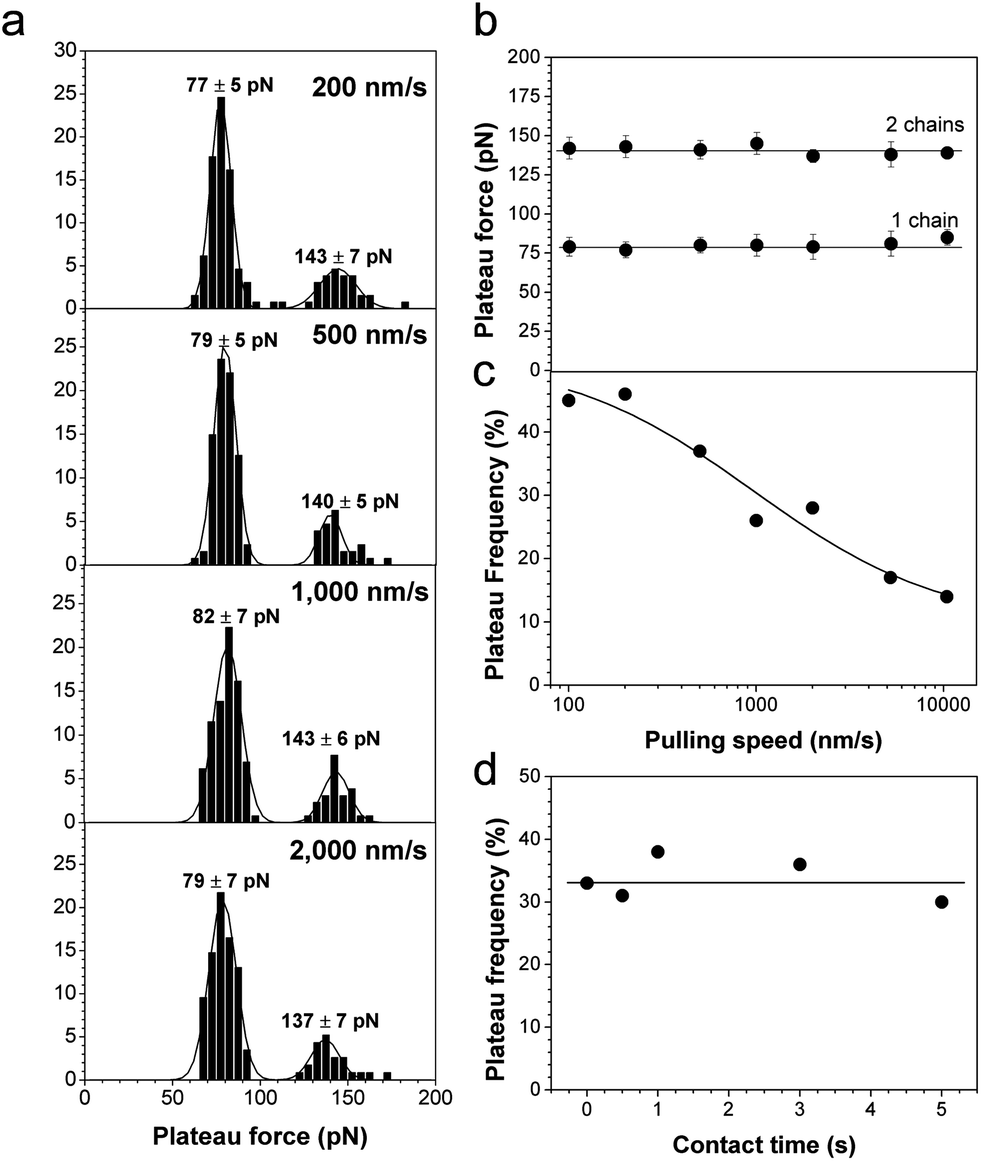 | ||
| Fig. 5 Dynamics of polyelectrolyte desorption. (a) Histograms showing the distribution of plateau forces at different pulling speeds. For each graph, the data correspond to more than 100 plateau curves from a total of 1280 force curves recorded using two different tips and surfaces. (b and c) Dependence of the mean plateau force (b) and force plateau frequency (c) on the pulling speed. Each data point in panel (b) has been extracted from a total of 256 force curves taken at different locations on the surface. Similar trends were observed in triplicate experiments using different tips and samples. (d) Dependence of the plateau force frequency on contact time measured at a constant pulling speed of 1000 nm s−1. Each data point corresponds to 256 force curves taken at different locations on the surface. Similar trends were observed in triplicate experiments using different tips and samples. | ||
Conclusions
Stimuli-responsive polymer materials respond to their environment by changing their physical properties, such as wettability and adhesion. Prominent examples are synthetic adhesive polymers which tune their adhesive properties on external triggers, like changes of aqueous media. Despite the tremendous potential of smart adhesive surfaces in nanotechnology, materials sciences and medicine, the strength and dynamics of their molecular interactions are poorly understood. We have shown that AFM force mapping with chemically sensitive tips (CFM) is a valuable approach to quantify and map the interaction forces of amphiphilic block copolymers on the nanoscale. Fresh PDMS-b-PDMAEMA block copolymer-filled coatings are highly structured on the nanoscale, yet show homogeneous hydrophobic properties due to the presence of a continuous layer of PDMS on the outermost surface. Aging in aqueous solution leads to major structural reorganization and to a major drop of hydrophobicity, reflecting exposure of hydrophilic block copolymer chains at the surface. In addition, PDMAEMA hydrophilic chains present on the surface show remarkable force plateau signatures documenting polyelectrolyte adsorption–desorption behaviors with negatively charged tips, and which can be readily abolished by increasing the salt concentration. As the lifetime of these desorption bonds is given by the plateau rupture length divided by the tip velocity, polyelectrolyte interactions are expected to substantially increase the lifetime – thus lowering the dissociation constant – of the polymer adhesive bonds. Our experiments demonstrate that CFM is a valuable method for quantifying and mapping the nanoscale adhesion forces of stimuli-responsive surfaces, on a scale that was not accessible before. They also show how solvent-induced conformational changes of stimuli-responsive copolymers can be used to control surface adhesion. In future applications, these stimuli-responsive interactions could be favoured (inhibited) to enhance (lower) the adhesive character of polymer coatings.Methods
Materials
2-(Dimethylamino)ethyl methacrylate (DMAEMA, Aldrich, 98%) was purified by passing through a basic alumina column, dried over CaH2 for 24 h and distilled under reduced pressure. Triethylamine (Sigma-Aldrich, ≥99%) was dried over CaH2 for 24 h and distilled under reduced pressure prior to use. Copper(I) bromide (CuBr, >99%, Aldrich) was purified first by stirring with acetic acid, then by washing with methanol, and finally by drying in a vacuum oven at 70 °C. Monocarbinol-terminated polydimethylsiloxane (α-hydroxy PDMS, ABCR, viscosity 250 cSt), 2-bromo-isobutyryl bromide (Sigma-Aldrich 99%), and 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, Aldrich, 97%) were used as received. Room temperature vulcanizing (RTV) silicone was purchased from ABCR, Germany. It consists of two parts: namely A and B, which have to be mixed together in a 10:1 weight ratio in order to obtain crosslinked materials. Part A initially contains a polymer base (silanol-terminated PDMS) and a crosslinking agent (poly(diethoxysiloxane)). Part B contains a catalyst (DBTL (dibutyl tin dilaurate, Aldrich, 95%)) and trimethylsiloxy-terminated PDMS (PDMS–CH3). A filler (silicon dioxide (silica), hexamethylsilazane surface-treated, ABCR Germany) was further added in order to improve the mechanical stability of the cured silicone. Toluene and THF were dried on a MB SPS-800 solvent purification system from MBRAUN. All other chemicals were of analytical grade of purity and used as received.
Syntheses
The PDMS-b-PDMAEMA block copolymer was synthesized by a two-step synthetic route involving (1) synthesis of an α-Br-PDMS macroinitiator followed by (2) atom-transfer radical polymerization as described before.32 Briefly: (1) 15 g of carbinol-terminated PDMS (1.5 mmol, 1 eq.), 30 mL toluene and 0.42 mL of Et3N (0.3 g, 3 mmol, 2 eq.) were introduced into a two-necked round bottom flask equipped with dropping funnel and condenser. 2-Bromo-isobutyryl bromide (0.689 g, 3 mmol) was then added dropwise to the PDMS solution. The reaction was carried out at 80 °C for 48 h. The resulting mixture was filtered to eliminate the insoluble salts and the solvent was evaporated under reduced pressure. The obtained oil was dissolved in CH2Cl2 and washed with aqueous saturated NaHCO3 solution and water. The organic layer was then isolated and dried over MgSO4. Finally, the solvent was evaporated and the obtained product was dried overnight at 80 °C under reduced pressure. Yield: 82%. 1H NMR (300 MHz, CDCl3, δ ppm): 0.04 –(Si(CH3)2–O)135–, 0.51 CH3–(CH2)2–CH2–(Si(CH3)2–O)135–Si(CH3)2–CH2–(CH2)2–O–, 0.86 CH3–(CH2)3–(Si(CH3)2–O)135–, 1.28 CH3–CH2–CH2–CH2–(Si(CH3)2–O)135–, 1.56 CH3–(CH2)3–(Si(CH3)2–O)135–Si(CH3)2–CH2–CH2–CH2–O–, 1.92 CH3–(CH2)3–(Si(CH3)2–O)135–Si(CH3)2–(CH2)3–O–(CH2)2–O–CO–C(CH3)2Br, 3.42 CH3–(CH2)3–(Si(CH3)2–O)135–Si(CH3)2–CH2–CH2–CH2–O–, 3.65 CH3–(CH2)3–(Si(CH3)2–O)135–Si(CH3)2–(CH2)3–O–CH2–CH2–O–, 4.30 CH3–(CH2)3–(Si(CH3)2–O)135–Si(CH3)2–(CH2)3–O–CH2–CH2–O–. (2) 3 g (0.3 mmol, 1 eq.) of the as obtained, purified and characterized α-Br-PDMS macroinitiator, 6.13 g DMAEMA (6.57 mL, 39 mmol, 130 eq.) and 6 mL dry toluene were charged in a round bottom flask A. The solution was degassed by three freezing/thawing cycles before transferring it into flask B, containing 0.056 g CuBr (0.39 mmol, 1 eq.) and 0.138 g HMTETA (0.6 mmol, 2 eq.). The polymerization was carried out at 80 °C for 3 h under N2. The polymerization was terminated by quenching in liquid N2. The reaction mixture was then diluted with THF and quickly filtered through a basic alumina column to remove the copper catalyst. The solvent was then evaporated and the resulting material dried overnight in a vacuum oven at 80 °C to yield the pure polymer. Yield: (85%). 1H NMR (300 MHz, CDCl3, δ ppm): 0.04 [m, –(Si(CH3)2–O)135–], 0.70–1.10 [m, –(CH2–C(CH3))n–], 1.70–2.10 [m, –(CH2–C(CH3))n–], 2.30 [m, –N(CH3)2], 2.60 [m, –CH2–N–(CH3)2], 4.07 [m, –OCH2–]. DP (PDMAEMA-block) = 116. Size exclusion chromatography (SEC, Agilent Technologies, 1200 chromatograph operating in THF 35 °C, flow rate 1 mL min−1, sample concentration 1 mg mL−1, poly(methyl methacrylate) standards): monomodal, Mn = 33300, Đ = 1.29. Differential scanning calorimetry (DSC, T.A. Instruments, Q2000 apparatus, N2 flow, heating rate 10 °C min−1): TPDMSg = −129 °C; TPDMAEMAg = 21 °C.
Sample preparation
Block copolymer-filled coatings were prepared as follows. First, PDMS–OH and 2 wt% silica were mixed with a mechanical stirrer for 30 min. Then the block copolymer (10 wt%) in the powder form was roll-milled within part A of the condensation-curing silicone as previously described.32 The obtained mixture was degassed in a vacuum oven, followed by addition of the crosslinker under stirring (silanol-terminated PDMS: crosslinker = 91/9 mass ratio). Then part B (PDMS–CH3 and the catalyst – DBTL, 1 wt%) (10:1 mass ratio) was added and the mixture stirred at 500 rpm for 1 min. The final mixture was spread on glass substrates (microscopy glass slides) by means of the doctor-blade method, which resulted in a coating thickness of about 300 μm. After that, the samples were cured at room temperature for 24 h.
Atomic force microscopy
AFM measurements were performed using a Nanoscope VIII (Bruker corporation, Santa Barbara) at room temperature (20 °C) and in ultrapure MilliQ water (Elga LabWater), unless otherwise stated. Imaging was performed in contact mode with a minimal applied force using oxide sharpened microfabricated Si3N4 cantilevers (Bruker corporation, Santa Barbara). For chemical force microscopy, gold coated cantilevers (OMCL-TR4, Olympus Ltd., Tokyo, Japan) were rinsed with ethanol, dried with N2, placed in a UV-ozone cleaner for 10 min, rinsed with ethanol and immersed for 12 h in 1 mM solutions of HS(CH2)11CH3 or HS(CH2)15COOH (Sigma), for hydrophobic and acid carboxylic tips, respectively, rinsed with ethanol, dried with N2 and immediately mounted on the AFM set-up. The spring constants of the cantilever were determined using the thermal noise method (Bruker corporation, Santa Barbara). Unless otherwise specified, all force measurements were recorded with an approach and retraction speed of 1000 nm s−1 and a contact time of <50 ms. Adhesion maps were obtained by recording 32 × 32 force distance curves on areas of given size, calculating the maximum adhesion peak and displaying the value as a gray pixel. For high ionic strength experiments, 0.1 M NaCl solution was injected into the liquid cell 15 min prior to force measurements.Acknowledgements
Work at the Université catholique de Louvain was supported by the Belgian Science Policy Office (Belspo, IAP-PAI P7/05: Functional Supramolecular Systems), the National Foundation for Scientific Research (FNRS), the Université catholique de Louvain, and the Research Department of the Communauté Française de Belgique. Work at University of Mons was supported by the ARC BIOMIME project (ARC AUWB-2008-08/12-UMH15), the Science Policy Office of the Belgian Federal Government (PAI 7/05) and FNRS-FRFC. Y.F.D. and P.L. are Research Director and Research Associate of the FNRS.References
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- T. P. Russell, Science, 2002, 297, 964 CrossRef CAS PubMed.
- M. Kamperman and A. Synytska, J. Mater. Chem., 2012, 22, 19390 RSC.
- A. K. Geim, S. V. Dubonos, I. V. Grigorieva, K. S. Novoselov, A. A. Zhukov and S. Y. Shapoval, Nat. Mater., 2003, 2, 461 CrossRef CAS PubMed.
- E. G. Kelley, J. N. L. Albert, M. O. Sullivan and I. I. I. T. H. Epps, Chem. Soc. Rev., 2013, 42, 7057 RSC.
- W. Mussard, N. Kebir, I. Kriegel, M. Estève and V. Semetey, Angew. Chem., Int. Ed., 2011, 50, 10871 CrossRef CAS PubMed.
- K. Yu and Y. Han, Soft Matter, 2006, 2, 705 RSC.
- F. Abbasi, H. Mirzadeh and A. A. Katbab, Polym. Int., 2001, 50, 1279 CrossRef CAS.
- R. R. Costa, C. A. Custõdio, F. J. Arias, J. C. Rodríguez-Cabello and J. F. Mano, Small, 2011, 7, 2640 CrossRef CAS PubMed.
- N. Maeda, N. Chen, M. Tirrell and J. N. Israelachvili, Science, 2002, 297, 379 CrossRef CAS PubMed.
- M. Valtiner, S. H. Donaldson, M. A. Gebbie and J. N. Israelachvili, J. Am. Chem. Soc., 2012, 134, 1746 CrossRef CAS PubMed.
- H. Zeng, Y. Tian, B. Zhao, M. Tirrell and J. Israelachvili, Langmuir, 2009, 25, 4954 CrossRef CAS PubMed.
- G. Knöner, B. E. Rolfe, J. H. Campbell, S. J. Parkin, N. R. Heckenberg and H. Rubinsztein-Dunlop, Biophys. J., 2006, 91, 3085 CrossRef PubMed.
- C. Zhao, J. Zhao, X. Li, J. Wu, S. Chen, Q. Chen, Q. Wang, X. Gong, L. Li and J. Zheng, Biomaterials, 2013, 34, 4714 CrossRef CAS PubMed.
- D. J. Müller, J. Helenius, D. Alsteens and Y. F. Dufrêne, Nat. Chem. Biol., 2009, 5, 383 CrossRef PubMed.
- R. García, R. Magerle and R. Perez, Nat. Mater., 2007, 6, 405 CrossRef PubMed.
- C. Friedsam, M. Seitz and H. E. Gaub, J. Phys.: Condens. Matter, 2004, 16, S2369 CrossRef CAS.
- C. Friedsam, H. E. Gaub and R. R. Netz, Biointerphases, 2006, 1, MR1 CrossRef CAS PubMed.
- M. I. Giannotti and G. J. Vancso, ChemPhysChem, 2007, 8, 2290 CrossRef CAS PubMed.
- B. N. Balzer, S. Micciulla, S. Dodoo, M. Zerball, M. Gallei, M. Rehahn, R. V. Klitzing and T. Hugel, ACS Appl. Mater. Interfaces, 2013, 5, 6300 CAS.
- M. A. Nash and H. E. Gaub, ACS Nano, 2012, 6, 10735 CrossRef CAS PubMed.
- L. Garnier, B. Gauthier-Manuel, E. W. Van Der Vegte, J. Snijders and G. Hadziioannou, J. Chem. Phys., 2000, 113, 2497 CrossRef CAS.
- T. Pirzer, M. Geisler, T. Scheibel and T. Hugel, Phys. Biol., 2009, 6, 025004 CrossRef CAS PubMed.
- T. Hugel, M. Grosholz, H. Clausen-Schaumann, A. Pfau, H. Gaub and M. Seitz, Macromolecules, 2001, 34, 1039 CrossRef CAS.
- C. D. Frisbie, L. F. Rozsnyai, A. Noy, M. S. Wrighton and C. M. Lieber, Science, 1994, 265, 2071 CAS.
- E. W. Van Der Vegte and G. Hadziioannou, Langmuir, 1997, 13, 4357 CrossRef CAS.
- D. V. Vezenov, A. Noy, L. F. Rozsnyai and C. M. Lieber, J. Am. Chem. Soc., 1997, 119, 2006 CrossRef.
- S. K. Sinniah, A. B. Steel, C. J. Miller and J. E. Reutt-Robey, J. Am. Chem. Soc., 1996, 118, 8925 CrossRef CAS.
- Y. F. Dufrêne, Biophys. J., 2000, 78, 3286 CrossRef.
- E. Dague, D. Alsteens, J. P. Latgé, C. Verbelen, D. Raze, A. R. Baulard and Y. F. Dufrêne, Nano Lett., 2007, 7, 3026 CrossRef CAS PubMed.
- D. Alsteens, E. Dague, P. G. Rouxhet, A. R. Baulard and Y. F. Dufrêne, Langmuir, 2007, 23, 11977 CrossRef CAS PubMed.
- E. Duquesne, J. Habimana, P. Degée and P. Dubois, Macromol. Chem. Phys., 2006, 207, 1116 CrossRef CAS.
- T. C. Ngo, R. Kalinova, D. Cossement, E. Hennebert, R. Mincheva, R. Snyders, P. Flammang, R. Dubois, R. Lazzaroni and P. Leclère, 2013, submitted.
- A. Beigbeder, M. Jeusette, R. Mincheva, M. Claes, P. Brocorens, R. Lazzaroni and P. Dubois, Journal of Nanostructured Polymers and Nanocomposites, 2009, 5, 37 Search PubMed.
- H. Schmolke, S. Demming, A. Edlich, V. Magdanz, S. Buttgenbach, E. Franco-Lara, R. Krull and C. P. Klages, Biomicrofluidics, 2010, 4, 3523059 CrossRef PubMed.
- C. Chen, J. Wang and Z. Chen, Langmuir, 2004, 20, 10186 CrossRef CAS PubMed.
- R. Merkel, P. Nassoy, A. Leung, K. Ritchie and E. Evans, Nature, 1999, 397, 50 CrossRef CAS PubMed.
- M. Rief, M. Gautel, F. Oesterhelt, J. M. Fernandez and H. E. Gaub, Science, 1997, 276, 1109 CrossRef CAS.
- D. Alsteens, C. B. Ramsook, P. N. Lipke and Y. F. Dufrêne, ACS Nano, 2012, 6, 7703 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |