Gadolinium oxysulfide nanoparticles as multimodal imaging agents for T2-weighted MR, X-ray tomography and photoluminescence†
Sèmiyou. A.
Osseni
a,
Sévérine
Lechevallier
a,
Marc
Verelst
a,
Pascal
Perriat
b,
Jeannette
Dexpert-Ghys
a,
David
Neumeyer
a,
Robin
Garcia
c,
Florian
Mayer
d,
Kristina
Djanashvili
d,
Joop A.
Peters
d,
Eddy
Magdeleine
e,
Hélène
Gros-Dagnac
f,
Pierre
Celsis
f and
Robert
Mauricot
*a
aCentre d'Elaboration de Matériaux et d'Etudes Structurales, Université de Toulouse - UPS, 29 rue Jeanne Marvig, BP 94347, 31055 Toulouse, Cedex 4, France. E-mail: Robert.Mauricot@cemes.fr; Fax: +33 5 62 25 79 99; Tel: +33 5 62 25 78 49
bUniversité Claude Bernard Lyon 1, UMR 5510, Materials: Engineering and Science, 7 avenue Jean Capelle 69622, Villeurbanne Cedex, France
cInstitut Sainte Catherine, Service de radiothérapie, 1750 chemin du lavandin, Avignon, France
dBiocatalysis and Organic Chemistry, Department of Biotechnology, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands
eICELLTIS, Parc Technologique Cap delta Sud, 09340 Verniolle, France
fPlateau Technique IRM, INSERM UPS/UMR 825, Pavillon Baudot, CHU Purpan, 31024 Toulouse Cedex3, France
First published on 24th October 2013
Abstract
We have synthesized gadolinium oxysulfide nanoparticles (NPs) doped with other lanthanides (Eu3+, Er3+, Yb3+) via a hydroxycarbonate precursor precipitation route followed by a sulfuration process under a H2S–Ar atmosphere at 750 °C in order to propose new multimodal nanoplatforms for Magnetic Resonance (MR), X-ray and photoluminescence imaging. Gd2O2S:Eu3+ NPs strongly absorb near UV (≈300–400 nm) and re-emit strong red light (624 nm). They can be easily internalized by cancer cells, and imaged by epifluorescence microscopy under excitation in the NUV (365 nm). They are not cytotoxic for living cells up to 100 μg mL−1. Consequently, they are well adapted for in vitro imaging on cell cultures. Gd2O2S:Eu3+ NPs also show strong transverse relaxivity and strong X-ray absorption allowing their use as contrast agents for T2-weighted MRI and X-ray tomography. Our study shows that Gd2O2S:Eu3+ NPs are considerably better than commercial Ferumoxtran-10 NPs as negative contrast agents for MRI. Upconversion emission of Gd2O2S:Er; Yb (1; 8%) NPs under infrared excitation (λex = 980 nm) shows mainly red emission (≈650–680 nm). Consequently, they are more specifically designed for in vivo deep fluorescence imaging, because both excitation and emission are located inside the “transparency window” of biological tissues (650–1200 nm). Magnetic relaxivity and X-ray absorption behaviors of Gd2O2S:Er; Yb NPs are almost similar to Gd2O2S:Eu3+ NPs.
1. Introduction
Nanoparticles (NPs) in which magnetic and optical properties coexist have been explored intensively for diverse biomedical applications, ranging from in vivo imaging and therapy1 to cellular imaging and manipulation.2 To date, approaches towards multimodal imaging agents have been focused on combining two different systems, such as magnetic materials and organic dyes,3 or on formulating magnetic NPs and fluorescent materials via polymers.4 Over the past decade, there have also been some developments in optical bio-labeling and bio-imaging involving upconversion nanoparticles, simultaneously bringing to the forefront the desirable characteristics, strengths and weaknesses of these luminescent nanomaterials.5–7There is an urgent need for the development of high-performance multimodal imaging agents with a simple and novel synthetic method.
We performed in our previous work, the synthesis and characterization of monodispersed Gd2O2S:Eu3+ NPs (called GADOX).8 This material is, in fact, a very well known scintillator which strongly absorbs X-rays and re-emits red light with a high quantum yield. Another advantage of gadolinium compounds, such as GADOX, is the high content of Gd element which is a contrast agent for MRI. Since MRI has an almost unlimited penetration depth, it allows to follow the NPs. In the same way, X-ray excitation may be used for in vivo tracking instead of NIR excitation used with up-converting probes.
To date, the majority of commercially available contrast agents (CAs) are Gd3+ chelates and many review papers on this topic have appeared in the literature over the past decades.9–11 Gd3+ chelates (also called positive contrast agents) are used as T1-weighted magnetic resonance (MR) imaging contrast agents, because they mainly shorten the longitudinal relaxation time of protons causing an increased signal intensity. More recently, ferumoxide, which is composed of dextran-coated superparamagnetic iron oxide (SPIO) NPs with a diameter around 150 nm, has been approved by the FDA for MRI contrast imaging on human beings, mainly for detection of liver metastases. Ferumoxide is a negative contrast agent because it causes predominant T2 (transverse relaxation time) shortening with a reduction in the signal intensity.12
In this work, we developed gadolinium oxysulfide NPs as multimodal imaging agents for T2-weighted magnetic resonance (MR) imaging, X-ray tomography and photoluminescence (PL) imaging via hydroxycarbonate precursor precipitation followed by sulfuration under a H2S–Ar atmosphere at 750 °C. It may be expected that these systems will gain in importance due to demand for mulitimodality probes and theragnostics.
2. Experimental section
2.a. Materials
Europium, erbium, ytterbium and gadolinium nitrates (99.99%) were stock solutions from Rhodia. Urea (98%) was purchased from Sigma Aldrich.2.b. Synthesis of the precursors
Gadolinium hydroxycarbonates were synthesized as previously described.8,13 Typically, gadolinium hydroxycarbonates were synthesized from nitrate precursors in water and ethanol following a procedure based on urea decomposition at temperatures above 80 °C. The optimum concentrations used in this work were [Gd3+] = 5.6 × 10−3 M and 0.5 M for urea. Europium doping concentration expressed in [Eu]/([Gd] + [Eu]) (mol%) was 5. For erbium/ytterbium co-doping, the molar composition was [Er]/([Gd] + [Er] + [Yb]) = 1 and [Yb]/([Gd] + [Er] + [Yb]) = 8. Solvent used was water–ethanol with 20 vol% of ethanol. Gd, Er, Yb and Eu nitrates and urea were dissolved in the solvent, and the solution was placed in a round-bottom flask at reflux for aging at 85 °C in an oil bath, during 100 min. The suspension was then centrifuged for 30 min at 4500 rpm. The supernatant solution was discarded, and the solid phase was resuspended in water and washed 3 times with water. The solid phase was then dried in an oven at 80 °C overnight.2.c. Gd2O2S NP synthesis
Gd2O2S NPs were synthesized in our laboratory and the details can be found in our earlier studies.8 The sulfuration of hydroxycarbonate was carried out by solid–gas reaction as previously described.8 Dried precipitates obtained were placed into a quartz tube and the sulfuration reaction was kept at 750 °C under an Ar–H2S atmosphere (83–17 vol%). After 90 min, the H2S gas flow was stopped. Only argon gas flow was supplied and samples were annealed at 850 °C for 4 h and after the samples were cooled to room temperature, Gd2O2S was obtained.2.d. Characterization techniques
Particle shape and size were examined via Transmission Electron Microscopy (TEM), using Philips Model CM20 and CM30 microscopes. The size was also analyzed by dynamic light scattering (DLS) using a Zetasizer Nano (Malvern instruments). Chemical bonding was analyzed by infrared spectroscopy, using a Perkin-Elmer 100 Series spectrometer. Samples were prepared by mixing the powders with potassium bromide (1/100 by weight) in a pellet.The luminescence of solid samples was studied with a Jobin-Yvon Model Fluorolog FL3-22 spectrometer that was equipped with a R928 Hammamatsu photomultiplier and a 450 W Xe excitation lamp. All intensities plotted on the spectra were corrected.
Water proton longitudinal relaxation times, T1, and transverse relaxation times, T2 were measured at low field (1.4 T) with a dedicated NMR relaxometer (Bruker Minispec 60 MHz, 25 °C). The Ln2O2S:Eu3+ suspensions for relaxometric studies at 1.4 T were prepared by mixing the particles with distilled water containing 1.5 wt% of agar gel as a surfactant. The τCP (half the time interval between successive 180° pulses in the CPMG pulse sequence) value was 1 ms. Relaxation rates (1/T1 and 1/T2) were plotted against Gd concentration values, and relaxivities (r1 and r2) were calculated from the slopes of the plots.
Water proton transverse relaxation times, T2, were also measured at 7 T with a Varian-INOVA 300 NMR spectrometer and at 9.4 T with a Bruker Avance 400 NMR spectrometer. The samples were prepared by mixing the solid particles with doubly distilled water containing 1 wt% of xanthan gum as a stabilizer followed by 5 min treatment with an ultrasonic probe (Cole Parmer CV 33, 1/2′′ tip, 40% amplitude, pulsed mode 10 s on/10 s off).
T 2 values were measured using the Carr–Purcell–Meiboom–Gill pulse sequence (CPMG). The values of T2* were evaluated from the line widths. The T1 measurements were performed with the inversion recovery pulse sequence. All experimental values of relaxation rates were corrected for diamagnetic contributions using a solution of 1 wt% of xanthan in water.
Magnetic Resonance Imaging (MRI) experiments were performed using a 3 T clinical imager (Philips Achieva, located in the Unit INSERM UMRS 825, hospital Purpan, Toulouse, France). We prepared the samples of Ln2O2S with various Ln3+ concentrations (0–15 mM) and they were dispersed in 1.5 wt% agar–agar gel. The acquired MR image was one slice (thickness 5 mm) with a field of view of 160 × 120 mm, and a voxel size of 1 × 1 × 5 mm. T2-weighted images were acquired using a spin-echo (T2-weighted) sequence with a repetition time/echo time (TR/TE) of 1500/11 ms.
X-ray tomography scan was done at Sainte Catherine Institute, Avignon (France). Computed tomography (CT) is a GE RT16 and set from 80 to 140 kV.
2.e. Cell culture, cytotoxicity test and fluorescence imaging
An indirect cytotoxicity test was performed using an elution method as described previously.14 The cells used are MDA-MB231 which are triple negative breast cancer cells.15 This type of cancer is one of the most aggressive. The cells were maintained in culture in RPMI 1640 medium complemented with 10% fetal bovine serum, 1% penicillin–streptomycin and incubated at 37 °C with 5% CO2. The cells were placed in 96 well plates at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well. The particles were added at different concentrations to the cell medium after sonication. The MTT (methylthiozoletetrazolium, Sigma) test was used to evaluate the viability of the MDA-MB231 cells in the presence of different concentrations of the NPs (Gd2O2S:Eu3+, 5%) one and three days after their addition to cell culture medium. The MTT test is a colorimetric assay for measuring the activity of enzymes that reduce MTT to formazan dye, giving a purple color. DMSO (dimethylsulfoxide) solution is added to dissolve the insoluble purple formazan product into a colored solution. The absorbance of this colored solution can be quantified spectrophotometrically by measuring at 570 nm.
000 cells per well. The particles were added at different concentrations to the cell medium after sonication. The MTT (methylthiozoletetrazolium, Sigma) test was used to evaluate the viability of the MDA-MB231 cells in the presence of different concentrations of the NPs (Gd2O2S:Eu3+, 5%) one and three days after their addition to cell culture medium. The MTT test is a colorimetric assay for measuring the activity of enzymes that reduce MTT to formazan dye, giving a purple color. DMSO (dimethylsulfoxide) solution is added to dissolve the insoluble purple formazan product into a colored solution. The absorbance of this colored solution can be quantified spectrophotometrically by measuring at 570 nm.
For in vitro labeling, cells were incubated with NPs (0.1 mg mL−1, 24 h) following identical conditions as for the cytotoxic test. Microscopic images were obtained using a “home made” Time Gated Luminescence Microscope (TGML) kindly built for us by Dr Dayong Jin from Macquarie University of Sydney. The main interest of a TGML is to be able to separate long lasting fluorescence coming from lanthanides from auto fluorescence coming from the biological media.16
3. Results and discussion
3.a. XRD and TEM characterization of the NPs
The XRD patterns (Fig. 1) of the final products exhibit well developed peaks, characteristic of gadolinium oxysulfide; all peaks are indexed following the ICDD data in the hexagonal phase, of Gd2O2S (File card no. 020-1422). No extra peak coming from any impurity can be detected. | ||
| Fig. 1 XRD patterns of: Gd2O2S (File card no. 020-1422) (a), Gd2O2S:Eu3+ (b), and Gd2O2S:Er; Yb (1; 8%) (c). | ||
TEM images corresponding to each NP are presented in Fig. 2. NPs are spherical and mono-dispersed in size with a mean diameter of 118 nm for Gd2O2S:Eu3+ and 100 nm for Gd2O2S:Er; Yb (1; 8%).
 | ||
| Fig. 2 TEM images of: Gd2O2S:Eu (a and b) and Gd2O2S:Er; Yb (1; 8%) (c and d). | ||
3.b. Photo luminescence characterization of the oxysulfide NPs
Fig. 3 shows excitation and emission spectra of Gd2O2S:Eu3+ NPs. The emission spectrum (Fig. 3a), recorded after excitation at 363 nm, exhibits the Eu3+ 5D0 → 7FJ (J = 0–4) transitions. The spectrum is dominated by a strong band centered at 624 nm, characteristic of the 5D0 → 7F2 electric dipole transition of Eu3+ ions in C3v symmetry. Some 5D1 → 7FJ transitions are also detected. The excitation spectrum (Fig. 3b), monitored at 624 nm, can be divided into three parts, each one corresponding to an excitation mode. The narrow lines observed between 390 and 500 nm correspond to the transitions between 4f levels of Eu3+. They correspond to the direct excitation of Eu3+ which is not very effective because the absorption coefficient of Eu3+ is weak. | ||
| Fig. 3 The PL spectra of the Gd2O2S:Eu3+ sample: (a) emission spectrum under a 363 nm excitation source; (b) excitation spectrum monitored at 624 nm. | ||
Between 390 and 320 nm, the excitation is due to the charge transfer state S2− → Eu3+, centered at 355 nm. This strong absorption band is very interesting because it corresponds to the main emission band of the Hg lamp available on most commercial epifluorescence microscopes.
Excitation below 330 nm causes poor transmission in most optics, needs bulky, expensive and limited light sources and finally most commercial microscopes do not work in these wavelengths.
The Eu3+ molar concentration for maximum red (5D0 → 7F2) emission has been established in a previous study at 5 mol%.8
Fig. 4 shows the upconversion emission of the Gd2O2S:Er; Yb (1; 8%) system under infrared excitation (λex = 980 nm). Red emission was mainly obtained under infrared excitation, with an additional slight green emission. These peaks are assigned to the following transitions: green emission in the region of 520–560 nm assigned to the {(2H11/2, 4S3/2) → 4I15/2} transition and red emission in the region of 650–680 nm assigned to the 4F9/2 → 4I15/2 transition for Er3+ ions. The best Yb and Er concentration for maximum red (670 nm) emission has been established to Yb3+ = 8 mol% and Er3+ = 1 mol% after a systematic study.
 | ||
| Fig. 4 Emission spectrum of Gd2O2S:Er; Yb (1; 8%) under a 980 nm excitation source with pump power. | ||
It is known that the intensity of the upconversion emission I, is proportional to some power n of the excitation intensity P; i.e.,
| I = Pn(n = 2, 3, …), | (1) |
 | ||
| Fig. 5 Dependence of upconversion emission intensities of Gd2O2S:Er; Yb (1; 8%) on pump power. | ||
The above results on Gd2O2S:Er; Yb (1; 8%) nano-phosphors should be very useful for deep (up to 1 cm) in vivo imaging using both excitation (980 nm) and emission (670 nm) radiation inside the “transparency window” of biological tissues (650–1200 nm).20,21
3.c. Cytotoxicity tests on nanoparticles
The optical density (OD) is directly proportional to the number of cells increasing the concentration of NPs. The comparison of the proliferation of MDA-MB231 cancer cells in the presence of growing concentrations of NPs 0.1; 0.5; 1; and 2 mg mL−1, emphasizes a significant decrease of cell viability with high doses of NPs (>0.5 mg mL−1) and shows an inhibition of cell growth whatever the time of culture (Fig. 6). However, for particle concentration around 100 μg mL−1 we consider that the cytotoxicity of particles is weak or negligible.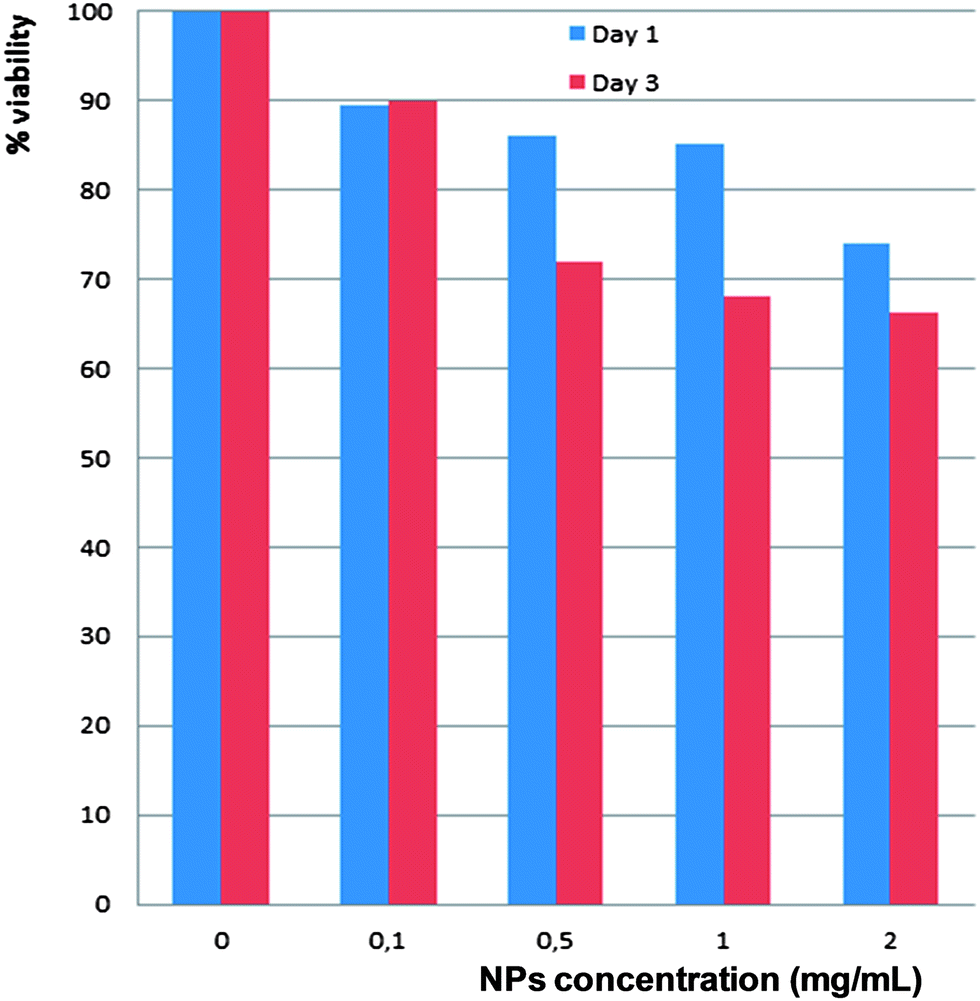 | ||
| Fig. 6 Cytotoxicity test of Gd2O2S:Eu3+ NPs. | ||
Compared to our previous work,8 where we performed the same experiments on NIH3T3 mouse cells using the same Gd2O2S:Eu3+, these NPs seemed to be more cytotoxic for MDA-MB231 cancer cells showing the variability of living cells.
3.d. Observation of particles fluorescence in living cells
The spherical Gd2O2S:Eu3+ NPs were incubated under controlled conditions, as described in Experimental section 2.e, with MDA-MB231 cancer cells. The results show (Fig. 7) that NPs have been internalized by the cells. Indeed, a strong red fluorescence is observed in their cytoplasm with a higher intensity in the perinuclear area. The nucleus, stained in blue with DAPI, appears to be totally free of NPs as shown by time gated detection. | ||
| Fig. 7 Internalization of Gd2O2S:Eu3+ NPs in MDA-MB231 cancer cells after exposure to Gd2O2S:Eu3+ NPs (overnight, 0.1 mg mL−1). (a) Representative bright field images + UV excitation (λex: 365 nm, the blue color comes from the nucleus colored with DAPI used to help cell detection). (b) UV excitation (λex: 365 nm) and time gated detection of Gd2O2S:Eu3+ NPs. | ||
3.e. Longitudinal and transverse relaxivity measurements; MR imaging of the oxysulfide NPs
The efficacy of the NPs as T1- or T2-weighted MR contrast agents was evaluated by measuring the longitudinal (r1) and the transverse (r2) nuclear magnetic relaxation rates of water protons in aqueous suspensions at low and high field.| r TEM (nm) | r 2 (s−1 mM−1) | r 1 (s−1 mM−1) | r 2/r1 |
|---|---|---|---|
| 25.5 | 15 | 0.46 | 32.6 |
| 59 | 20.1 | 0.33 | 60.9 |
| 110 | 24.9 | 0.38 | 65.6 |
MR contrast agents can be classified according to their T1 or T2 enhancement capability. When the r2/r1 value is close to 1.0, application as a positive contrast agent is favored. On the other hand, when this ratio is larger, the contrast agents are viewed as negative contrast agents.22
The longitudinal relaxivities r1 (see Table 1) were very small for Gd2O2S:Eu3+ NPs compared to Gd-DOTA – gadoterate meglumine (r1 = 3.4 s−1 mM−1 and r2 = 4.8 s−1 mM−1 at 1 Tesla; 37 °C),23 which suggests that the amount of water in close contact with the Ln3+ ion is limited, probably because of the relatively big size of these particles. Surely, these NPs will not show good performance as positive contrast agents. However, the quite good r2 value at low field is more promising. The ratio r2/r1 increased from 33 to 66 along with the NPs radius increase from 25.5 nm to 110 nm (see also Table 1). It indicates that Gd2O2S:Eu3+ NPs are more likely to be used as negative MR contrast agents (short T2 relaxation time).
| Material | r p (nm)b | r 1 (s−1 mM−1) | r 2 (s−1 mM−1)c | r 2* (s−1 mM−1) | Δω(rp) (106 s−1)d | Δω(rp)max (106 s−1)e | τ D(rdiff) (10−4 s)d | r p/rdiffd | r p (nm)f | r diff (nm)f |
|---|---|---|---|---|---|---|---|---|---|---|
| a 1 wt% xanthan. b From TEM. c At τCP = 5 ms. d From fitting of experimental data with eqn (2) and (3). e Calculated with magnetization of a single particle. f Calculated from the best-fit values of τD(rdiff), rp/rdiff, D0 = 1.9 × 10−9 m2 s−1, and R02 = 0. | ||||||||||
| Gd2O2S:Eu3+ | 25.5 | 0.24 | 55 | 483 | 7.763 ± 0.025 | 7.772 | 38 ± 49 | 0.022 ± 0.009 | 59.9 | 2673.4 |
| Gd2O2S:Eu3+ | 30.0 | 0.24 | 48 | 490 | 7.872 ± 0.005 | 7.772 | 211 ± 24 | 0.0114 ± 0.0004 | 72.2 | 6330.2 |
| Gd2O2S:Eu3+ | 59.0 | 0.23 | 114 | 639 | 10.263 ± 0.011 | 7.772 | 401 ± 33 | 0.0106 ± 0.0003 | 92.3 | 8731.3 |
The observed trends appeared to be similar to those reported previously for Ln2O3 NPs and this behavior is typical for the static dephasing regime (SDR),24 where the condition τD < Δω(rp)−1 holds (τD = r2p/D), where rp is the radius of the particle, D is the water diffusion coefficient and Δω(rp) is the difference in Larmor frequency of the water protons located at the surface of the particle and those at infinity. By contrast, dextran coated Dy2O3 NPs25 and Ho2O3 NPs stabilized by addition of 0.05% (w/w) of the cationic surfactant cetyltrimethylammonium bromide (CTAB)26 to the dispersion showed a behavior that is typical for the outer sphere regime (OS) with nearly equal values of R2 and R2* (in both cases no xanthan was needed to prevent precipitation). Therefore, the SDR behavior of the presently studied systems and of the previously studied Ln2O3 NPs can be ascribed to the adsorption of a thick layer of xanthan on the NPs.
The transverse relaxivities (r2 and r2*) were fitted to a model that has successfully been applied previously for the rationalization of the tranverse relaxivities of Ln2O3 NPs.25 Under SDR conditions, R2* can be ascribed to the dephasing of motionless magnetic moments in a non-uniform magnetic field created by the randomly distributed NPs. The value of R2* is then given by eqn (2), where f is the volume fraction occupied by the NPs and R02 is the contribution due to other relaxation mechanisms such as exchange. For the calculation of R2, a imaginary sphere around the particle is defined with radius rdiff for which τD(rdiff) = 1/Δω(rdiff) and which forms the border between contributing and non-contributing proton spins to R2 (e.g., of very slowly diffusing water molecules in the xanthan layer). Application of the theory of weak magnetic particles leads to eqn (3).27 The fitting was performed using Δω(rp), τD(rdiff), and rp/rdiff as adjustable parameters.
 | (2) |
 | (3) |
 and
and  .
.
The values of Δω(rdiff), τD(rdiff) and f(rdiff) can be expressed as follows:
 | (4) |
 | (5) |
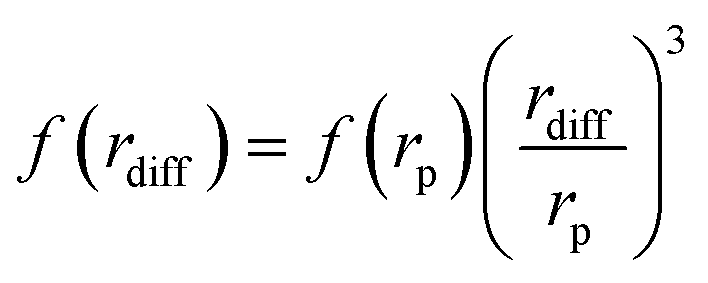 | (6) |
When R02 was adjustable, negative values for some of the other adjustable parameters or best-fit results with very large standard deviations were obtained. Therefore, R02 was fixed at 0. Very good agreement between the experimental and calculated values was achieved. The best-fit parameters are listed in Table 2. From the values of τD(rdiff) and D0 = 1.9 × 10−9 m2 s−1, the values for rdiff were calculated. From the latter and the best-fit parameters, rp was calculated. The values obtained were in reasonably good agreement with the values obtained from TEM, considering the crudeness of the model. Since, rdiff is the radius of the sphere, within which water protons are silent with regard to transverse relaxivity; it also reflects the thickness of the xanthan layer around the particle. From the obtained values it can be estimated to be 3–10 μm, which is an order of magnitude thicker than for the previously studied Ln2O3 nanoparticles.25 Another difference with the previously studied particles is that the best-fit values for Δω(rp) for the smallest NPs (25–35 nm) are in good agreement with the maximum calculated values for this parameter. In the previous study on Ln2O3 NPs, the evaluated values for Δω(rp) were significantly smaller than the calculated values for Δω(rp)max, which may be explained by the difference in their shapes. The previously studied material consisted, in the solid state, of fiber-like aggregates of plates with a size of 5–10 nm. In suspension, these aggregates disrupted into smaller aggregates with a radius of 50–100 nm. A non-spherical shape of those particles may explain the relatively low values for Δω(rp). In the previous study, in contrast to the present one, large values for R02 were needed to obtain good fits. Probably, R02 is mainly determined by exchange of water protons between the bulk and, for instance, intra-aggregate water protons which have a large chemical shift difference due to the neighboring paramagnetic Ln3+ ions.28
It should be noted that the evaluated values of Δω(rp) for the larger Gd2O2S particles (59–85 nm) are significantly higher than the calculated maximum values. Possibly magnetic interactions between the Gd3+ ions and the Eu3+ ions lead to μeff higher values and thus to higher relaxivities.
The relaxivities of the Gd2O2S NPs clearly demonstrate the effect of the radius. From the evaluated parameters, it can be concluded that the increase of the transverse relaxivities along with the particle size increase can be rationalized by the increase of τD(rdiff).
The transverse relaxivities are proportional to the effective magnetic moment of the system under study.
At B = 7.1 T, the relaxivities of the Gd2O2S NPs studied are in general better than those of commercial iron oxide based negative contrast agents. For example, the r2 value of Gd2O2S NPs of 59 nm in radius, is higher than that of Ferumoxtran-10 (AMI-227) which has r2 = 71 s−1 mM−1 at B = 7.1 T.29
The suspensions of the Gd2O2S NPs were also studied at a magnetic field strength of 9.4 T. The results show that both r2 and r2* are linearly proportional to the magnetic field strength B. Fig. 8 shows the case of Gd2O2S:Eu3+.
 | ||
| Fig. 8 Dependence of the transverse relaxivity of aqueous suspension of Gd2O2S:Eu3+ on the external magnetic field B. | ||
The value of r2 varies linearly with the field whatever the particle size. This linear dependence confirms that the SDR holds for these NPs under the conditions applied; for the OS mechanism a quadratic dependence of B would be expected.30
The longitudinal relaxivities of the Gd2O2S systems at 7.1 (see Table 2) are lower than those at 1.4 T, which is in agreement with the general trend predicted by the Solomon–Bloembergen–Morgan equations for slowly tumbling systems above about 1.5 T.10
At present, there is a tendency to perform MRI examinations at higher magnetic fields. Gadolinium oxysulfide has favorable T2-weighted relaxivity properties for high field MRI. It should be noted however, that various factors are important for application in molecular imaging, including the toxicity protection of the particle by the coating for leaching, and the effect of the particle size on the biodistribution. Further research to confirm this is required.
 | ||
| Fig. 9 T 2-weighted MR imaging of Gd2O2S:Eu3+ and Gd2O2S:Er; Yb (1; 8%) NPs with intensity profile measurements. | ||
This attenuation of the signal intensity is much larger when the NP radius increases. Thus the transverse relaxation time T2 becomes shorter when the NP size increases. These results confirm those obtained during the relaxometric studies and proved the usability of Gd2O2S:Eu3+ and Gd2O2S:Er; Yb (1; 8%) NPs as negative CAs in magnetic resonance imaging.
3.f. X-ray computed tomography
To investigate the in vitro Computed Tomography (CT) imaging of Gd2O2S:Eu3+ NPs, 7 phantom samples, containing an increasing amount of NPs, were embedded in a perforated plastic plate (Fig. 10). | ||
| Fig. 10 (a) Plate image “phantom” filled with holes containing Gd2O2S:Eu3+ NPs suspended in an agar gel, (b) plate image inside the CT, and (c) CT image (100 kV): 0: control, 1: 0.52 mM, 2: 1.04 mM, 3: 2.08 mM, 4: 4.15 mM, 5: 8.3 mM, and 6: 16.6 mM. | ||
The darkest spot represents the empty hole, which had no absorption. Sample 0 containing agar gel (1.5% wt), was used as a control. The X-rays were absorbed efficiently by the samples, and the intensity of the CT value increased as the concentration of the samples increased. Then, X-ray absorption increases with Gd concentration and thus with the electron density of the sample. From a concentration around 4 mM of Gd we can clearly detect the sample spot on the CT image. Thus, the Gd2O2S:Eu3+ NPs could be potentially applied as a CT imaging contrast agent.
When the sample is irradiated with X-rays, it produces radiation–matter interactions such as absorption and Compton scattering. The effect of X-ray attenuation on CT image was studied by measuring the mean Hounsfield Unit (HU) variation of the acquired image depending on Gd concentration at different voltages of used energy. Phantom images were treated using a standard image viewer application (scanner). Fig. 11 shows a linear relationship between NP concentration and the shift in HU from the background.
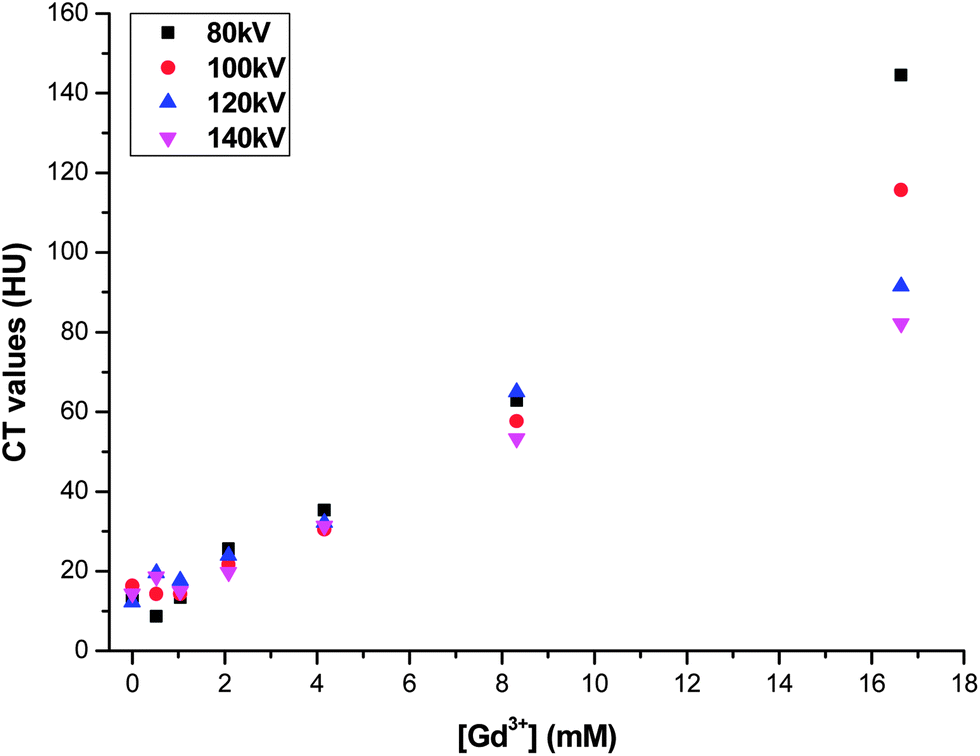 | ||
| Fig. 11 Plot of ΔHU versus Gd2O2S:Eu3+ concentration in agar–agar phantom. | ||
This figure also shows that the contrast effect is more effective for low energy (80 kV) than for higher energy (140 kV). Note that Gd2O2S:Eu3+ NP behavior is exactly opposite to gold NPs which give a better contrast effect at high energy (140 kV) than at lower (80 kV).31 This difference is clearly observed for [Gd3+] ≈ 16.6 mM. When [Gd3+] < 4 mM the number of Hounsfield units is less than 30 which we can assume is the limit to visualize the target against a background.
4. Conclusion, additional comments and perspectives
We have synthesized gadolinium oxysulfide NPs as multimodal imaging agents for T2-weighted MR imaging, X-ray tomography and photoluminescence imaging. Gd2O2S:Eu3+ NPs strongly absorb X-rays or near UV (363 nm) and re-emit red light with a high intensity. They are well adapted for in vitro fluorescence microscopy, MR imaging and X-ray tomography.Upconversion emission of the Gd2O2S:Er; Yb system under infrared excitation (λex = 980 nm) showed mainly red emission centered around 670 nm. Consequently, these NPs are more specially designed for deep in vivo fluorescence imaging, because both excitation and emission are located inside the “transparency window” of biological tissues (650–1200 nm).
The r2 value (114 s−1 mM−1 at B = 7 T) of Gd2O2S NPs for a radius of 59 nm, is higher than for Ferumoxtran-10 (Sinerem) (r2 = 71 s−1 mM−1 at B = 1.5 T), almost equal to Endorem (r2 = 120 s−1 mM−1 at B = 1.5 T) and slightly lower than for Resovist (r2 = rdiff 190 s−1 mM−1 at B = 1.5 T) which are very well known commercial iron oxide negative contrast agents. However comparison must be done with care because the T2 relaxivity of gadolinium oxysulfide NPs increases with the magnetic field whereas the transverse relaxivity of iron oxide particles gets saturated at magnetic field strengths >1 T. The particle size has also an influence here. Consequently, gadolinium oxysulfide NPs will be mainly interesting for high field and high resolution MRI (>3 T) where other contrast agents saturate.
X-rays are efficiently absorbed by Gd2O2S:Eu3+ NPs, and the contrast detected by Computed Tomography (CT) increased linearly as the concentration of the samples increased. Clear contrast can be detected on CT images beyond a concentration of 4 mM of Gd. Consequently, this work has clearly demonstrated the high potentiality of gadolinium oxysulfide NPs doped with other lanthanides (Eu3+, Er3+, Yb3+) as multimodal imaging agents.
These Gd2O2S NPs have been characterized without any functionalization because any surface change will modify both the luminescence and relaxivity properties. Consequently, data reported here constitute the reference before any surface modification for biological application. These nude Gd2O2S NPs are hydrophiles and a colloidal suspension in pure water is quite stable. Suspension in agar gel, leads to well dispersed non-precipitating particles allowing precise relaxivity measurements. However, the ionic strength of physiological serum causes an immediate flocculation preventing any biological use without surface modification.
Suspension of Gd2O2S NPs in water or physiological serum does not degrade the nano-objects even after several weeks and no release of toxic Gd3+ is detected. Moreover, as preliminary trials we have injected (in the tail vein) to five mice (C57 black mouse), 200 μL of a 3.3 mg mL−1 of Gd2O2S NPs dispersed in physiological serum using polyethylene glycol (mol wt 3000) as a dispersing agent with [Gd3+]/[PEG] = 5. Dynamic Light Scattering (DLS) measurements and photographs of the colloidal suspension were obtained and presented in the ESI.† As expected, Gd2O2S NPs move quickly (5 minutes) into the liver and kidneys where they give a strong negative contrast easily detected by MRI. The contrast is maximum after 50 minutes and remains almost constant for 15 days. After one month, the contrast begins to decrease little by little and disappears totally after 4 months showing that even if Gd2O2S NPs are very stable, they are finally eliminated by the body. During this preliminary trial, no mouse died and no mouse showed any pathology, weight loss or abnormal behavior. This preliminary toxicity test will be soon completed by further studies and will be the topic of the next publication.
Abbreviations and acronyms
| NPs | nanoparticles |
| CA | contrast agent |
| MR | magnetic resonance |
| MRI | magnetic resonance imaging |
| NMR | nuclear magnetic resonance |
| OS | outer sphere |
| SDR | static dephasing regime |
| D | relative diffusion constant |
| r diff | radius of a sphere around a particle inside which refocusing pulses are fully effective |
| R i (=1/Ti) | relaxation rate (i = 1, 2) |
| r i | relaxation rates expressed in s−1 mM−1 concentration of paramagnetic ions (i = 1, 2) |
| r p | radius of a particle |
| T 1 | longitudinal proton relaxation time |
| T 2 | transverse proton relaxation time |
| T 2* | observed time constant of the free induction NMR signal |
| τ CP | half the time interval between successive 180° pulses in the CPMG pulse sequence |
| τ D | diffusion correlation time, τD = r2/D (r, particle radius; D, diffusion coefficient) |
| CT | Computed Tomography. |
Acknowledgements
The “Agence National de la Recherche” (ANR) and the “Pôle de Compétitivité Cancer Bio Santé” from the Midi-Pyrénées region are thanked for their support of the PDTX program.References
- J. Kim, Y. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372–390 RSC.
- J. H. Lee, E. S. Kim, M. H. Cho, M. Son, S. I. Yeon, J. S. Shin and J. Cheon, Angew. Chem., Int Ed., 2010, 49, 5698–5702 CrossRef CAS PubMed.
- J. E. Lee, N. Lee, H. Kim, J. Kim, S. H. Choi, J. H. Kim, T. Kim, I. C. Song, S. P. Park, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2010, 132, 552–557 CrossRef CAS PubMed.
- K. J. Chen, S. M. Wolahan, H. Wang, C. H. Hsu, H. W. Chang and A. Durazo, Biomaterials, 2011, 32, 2160–2165 CrossRef CAS PubMed.
- F. Wang, D. Banerjee, Y. Liu, X. Chen and X. Liu, Analyst, 2010, 135, 1839–1854 RSC.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- W. Fan, B. Shen, W. Bu, F. Chen, K. Zhao, S. Zhang, L. Zhou, W. Peng, Q. Xiao, H. Xing, J. Liu, D. Ni, Q. He and J. Shi, J. Am. Chem. Soc., 2013, 135, 6494–6503 CrossRef CAS PubMed.
- S. A. Osseni, S. Lechevallier, M. Verelst, C. Dujardin, J. Dexpert-Ghys, D. Neumeyer, M. Leclercq, H. Baaziz, D. Cussac, V. Santran and R. Mauricot, J. Mater. Chem., 2011, 21, 18365 RSC.
- J. A. Peters, J. Huskens and D. J. Raber, Prog. Nucl. Magn. Reson. Spectrosc., 1996, 28, 283–350 CrossRef CAS.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- C. F. G. C. Geraldes and S. Laurent, Contrast Media Mol. Imaging, 2009, 4, 1–23 CrossRef CAS PubMed.
- E. Senéterre, P. Taourel, Y. Bouvier, J. Pradel, B. Van Beers, J. P. Daures, J. Pringot, D. Mathieu and J. M. Bruel, Radiology, 1996, 3, 785–792 Search PubMed.
- S. Lechevallier, P. Lecante, R. Mauricot, H. Dexpert, J. Dexpert-Ghys, H. Kong, H. K. Kong, G. L. Law and K. L. Wong, Chem. Mater., 2010, 22, 6153–6161 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- B. R. Brinkley, P. T. Beall, L. J. Wible, M. L. Mace, S. T. Donna and R. M. Cailleau, Cancer Res., 1980, 40, 3118–3129 CAS.
- J. Dayong and P. A. James, Anal. Chem., 2011, 83(6), 2294–2300 CrossRef PubMed.
- F. E. Auzel, Proc. IEEE, 1973, 61, 758–786 CrossRef CAS.
- M. Pollnau, D. R. Gamelin, S. R. Lüthi and H. U. Güdel, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337 CrossRef CAS.
- H. R. Page, K. I. Schaffers, P. A. Waide, J. B. Tassano, S. A. Payne and W. F. K. Krupke, J. Appl. Phys., 1972, 43, 595–600 CrossRef.
- Y. Yang, Q. Shao, R. Deng, C. Wang, X. Teng, K. Cheng, Z. Cheng, L. Huang, Z. Liu, X. Liu and B. Xing, Angew. Chem., Int. Ed., 2012, 51, 3125–3129 CrossRef CAS PubMed.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- M. A. Fortin, R. M. Petoral, J. F. Soderlind, A. Klasson, M. Engstrom, T. Veres, P. O. Kall and K. Uvdal, Nanotechnology, 2007, 18, 395501 CrossRef.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, E. L. Vander and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- M. Norek, G. A. Pereira, C. F. G. C. Geraldes, A. Denkova, W. Zhou and J. A. Peters, J. Phys. Chem. C, 2007, 111, 10240–10246 CAS.
- M. Norek, E. Kampert, U. Zeitler and J. A. Peters, J. Am. Chem. Soc., 2008, 130, 5335–5340 CrossRef CAS PubMed.
- F. Mayer, J. A. Peters and K. Djanashvili, Chem.–Eur. J., 2012, 18, 8004–8007 CrossRef CAS PubMed.
- J. H. Jensen and R. Chandra, Magn. Reson. Med., 2000, 44, 144–156 CrossRef CAS.
- Q. L. Vuong, P. Gillis and Y. Gossuin, J. Magn. Reson., 2011, 212, 139–148 CrossRef CAS PubMed.
- M. M. J. Modo, J. W. M. Bulte and E. E. Kim, J. Nucl. Med., 2007, 48, 2087 CrossRef.
- M. Norek and J. A. Peters, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 64–82 CrossRef CAS PubMed.
- E. Boote, G. Fent, V. Kattumur, S. Casteel, K. Katti, N. Chanda, R. Kannan, K. Katti and R. Churchill, Acad. Radiol., 2010, 17, 410–417 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nr03982j |
| This journal is © The Royal Society of Chemistry 2014 |