Supporting palladium metal on gold nanoparticles improves its catalysis for nitrite reduction†
Huifeng
Qian
a,
Zhun
Zhao
a,
Juan C.
Velazquez
a,
Lori A.
Pretzer
b,
Kimberly N.
Heck
a and
Michael S.
Wong
*abc
aDepartment of Chemical and Biomolecular Engineering, Rice University, 6100 Main Street, Houston, TX 77005-1892, USA. E-mail: mswong@rice.edu
bDepartment of Chemistry, Rice University, 6100 Main Street, Houston, TX 77005-1892, USA
cDepartment of Civil and Environmental Engineering, Rice University, 6100 Main Street, Houston, TX 77005-1892, USA
First published on 30th October 2013
Abstract
Nitrate (NO3−) and nitrite (NO2−) anions are often found in groundwater and surface water as contaminants globally, especially in agricultural areas due to nitrate-rich fertilizer use. One popular approach to studying the removal of nitrite/nitrate from water has been their degradation to dinitrogen via Pd-based reduction catalysis. However, little progress has been made towards understanding how the catalyst structure can improve activity. Focusing on the catalytic reduction of nitrite in this study, we report that Au NPs supporting Pd metal ("Pd-on-Au NPs") show catalytic activity that varies with volcano-shape dependence on Pd surface coverage. At room temperature, in CO2-buffered water, and under H2 headspace, the NPs were maximally active at a Pd surface coverage of 80%, with a first-order rate constant (kcat = 576 L gPd−1 min−1) that was 15x and 7.5x higher than monometallic Pd NPs (∼4 nm; 40 L gPd−1 min−1) and Pd/Al2O3 (1 wt% Pd; 76 L gPd−1 min−1), respectively. Accounting only for surface Pd atoms, these NPs (576 L gsurface-Pd−1 min−1) were 3.6x and 1.6x higher than monometallic Pd NPs (160 L gsurface-Pd−1 min−1) and Pd/Al2O3 (361 L gsurface-Pd−1 min−1). These NPs retained ∼98% of catalytic activity at a chloride concentration of 1 mM, whereas Pd/Al2O3 lost ∼50%. The Pd-on-Au nanostructure is a promising approach to improve the catalytic reduction process for nitrite and, with further development, also for nitrate anions.
1. Introduction
Nitrate (NO3−) is found in the groundwater and surface water globally, especially in agricultural areas due to the use of nitrate rich fertilizers.1–4 This anion, and its metabolic conversion to the nitrite anion (NO2−), cause adverse human health effects, including methemoglobinemia or the ‘blue baby’ syndrome.5 Nitrite can be also converted to carcinogenic N-nitroso compounds in the body,6 which may result in cancer and hypertension.7 The maximum contaminant levels (MCL) of NO3− and NO2− have been set at 10 mg N per L and 1.0 mg N per L (calculated by nitrogen weight, denoted as mg N per L), respectively, by the United States Environmental Protection Agency (US EPA).8 For the European Union countries, the European Drinking Water Directive set concentration limits for NO3− and NO2− at 11.3 mg N per L and 0.15 mg N per L, respectively.9 The World Health Organization guideline values of NO3− and NO2− are 11.3 mg N per L and 0.91 mg N per L.10Current methods to treat nitrate/nitrite contamination include ion exchange, reverse osmosis, biological denitrification, and catalytic reduction.11–21 The EPA-approved treatment technologies for removing nitrates/nitrites in waters destined for drinking purposes are ion exchange and reverse osmosis, with the former being the most widely used.22 The ion exchange approach removes nitrate/nitrite from water by replacing them with equivalent amount of anions such as chloride or bicarbonate ions.11 The main disadvantage is that, after the ion exchange material is regenerated for further use by contacting with a brine solution, the resulting brine which contains the nitrate/nitrite anions must undergo further treatment prior to disposal. Reverse osmosis also results in a nitrate/nitrite-containing secondary stream that needs further treatment. The biological denitrification and catalytic reduction routes have been studied at the laboratory and pilot scales but they are not yet commercially practiced. Biological denitrification can selectively remove nitrate/nitrite from water, but it is affected by pH and the presence of other salts in water; it can generate undesired by-products;15 and bacterial contamination of the treated water is a concern.11
The catalytic reduction of nitrate/nitrite has received intense research interest ever since Pd-based catalysts (e.g., Pd–Cu, Pd–In and Pd–Sn) were shown capable of converting nitrate/nitrite to harmless nitrogen (N2) using hydrogen.23 This chemistry was first reported by Vorlop and Tacke in 1989 using supported Pd–Cu/Al2O3 catalysts.14 Vorlop and co-workers reported improved reduction activity using Pd–Sn/Al2O3 and Pd–In/Al2O3 catalysts.24
The catalytic reduction of nitrate is generally accepted to occur in two stages: the reduction of NO3− to NO2−, and then reduction of NO2− to N2 or NH3 (Scheme 1).15,25–28 In greater detail, the second metal (i.e., Cu, In, Sn) reduces the chemisorbed NO3− species in the first step and the Pd then regenerates the oxidized metal via H2 reduction (reaction (1) of Scheme 1). In the second stage, Pd by itself reduces chemisorbed NO2− species in several surface reaction steps, eventually to form N2 (reaction (2)) or NH3 (reaction (3)). Ammonia is an undesirable reaction product because it is a groundwater pollutant with a US EPA MCL of 0.66 mg N per L.8 The Pd–Cu composition provides N2 selectivity values as high as 80–95%, depending on the reaction testing conditions used.19,29 Ammonia formation is favored at high pH values, and so N2 selectivity is improved by operating the reaction at non-alkaline conditions, for example.29
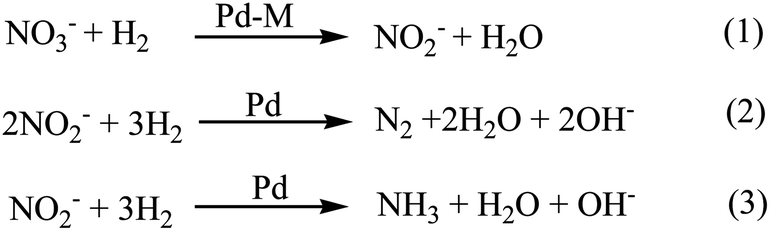 | ||
| Scheme 1 General reaction pathway for the reduction of nitrate or nitrite anions using a Pd-based catalyst. M is a promoter metal such as Cu, In, and Sn.27 | ||
While the compositional effects on catalytic nitrate/nitrite reduction have been amply studied, the effect of nanostructure is not well addressed yet. We have previously demonstrated that Pd catalytic activity for hydrodechlorination (a type of reduction reaction) can be varied with great control by depositing Pd metal on Au nanoparticles (to form Pd-on-Au NPs) at a pre-determined Pd surface coverage.30–33 Motivating this work, we hypothesized that Pd-on-Au NPs catalyze nitrite reduction (the second stage of nitrate reduction) and that catalytic activity is correlated to Pd surface coverage. We synthesized ∼4.3 nm Pd-on-Au NPs of varying Pd coverages, and quantified their reaction rate constants for nitrite reduction at room temperature, under H2 headspace, in water and at pH 5–7 (buffered using CO2 gas). We quantified the corresponding N2 selectivities, and studied the effect of chloride anions, a common groundwater constituent.
2. Materials and methods
2.1. Materials
Tetrachloroauric(III) acid (HAuCl4·3H2O, 99.99%), tannic acid (C76H52O46, >99.5%), potassium carbonate (K2CO3, >99.5%), palladium(II) chloride (PdCl2, 99.99%), 1 wt% Pd/Al2O3, modified Griess's reagent, and Nessler's reagent (K2HgI4) were purchased from Sigma-Aldrich. Trisodium citrate (Na3C6H5O7, >99.5%, Fisher), sodium nitrite (NaNO2, 99.7%) and sodium chloride (NaCl, 99.99%) were obtained from Fisher. Hydrogen gas (99.99%) was purchased from Matheson. 0.9 wt% Au/Al2O3 was obtained from Mintek Autek, South Africa. All experiments were conducted using Nanopure water (>18 MΩ cm, Barnstead NANOpure Diamond).2.2. NP synthesis
4 nm Au NPs, Pd NPs and Pd-on-Au bimetallic NPs were synthesized as previously reported.30 To synthesize Au NPs, 0.05 g tannic acid, 0.018 g K2CO3 and 0.04 g trisodium citrate were dissolved in 20 mL of water. In a second flask, 200 μL of HAuCl4 solution (0.127 mol L−1) was dissolved in 79.8 mL of water. Both solutions were heated to 60 °C, and the first solution was added to the second under vigorous stirring. The color of the resultant sol immediately changed from pale yellow to reddish-brown, indicative of the formation of Au NPs. The solution was heated to boiling, allowed to boil for 2 min and then cooled to room temperature (Scheme 2). The Au NP concentration was 1.26 × 1014 NP per mL.30 | ||
| Scheme 2 Two-stage synthesis of Pd-on-Au NPs with a specific Pd surface coverage. | ||
The Pd NPs were synthesized by replacing the HAuCl4 solution with a H2PdCl4 solution of the same molar concentration,30 and instead of boiling for 2 min, the mixed solution was allowed to boil for 25 min. Pd-on-Au bimetallic NPs were prepared by adding specific amounts of H2PdCl4 solution to the as-synthesized Au NPs. The various surface coverage percentages (sc%) (0, 10, 30, 60, 80, 100, 150, 300 sc%) of Pd-on-Au NPs were obtained by mixing 9, 18, 55, 73, 92, 150, 351 μL H2PdCl4 solution (2.47 mM) and 2 mL of the as-synthesized Au NPs.30 The mixed solution was stirred for 1 min and then bubbled with hydrogen gas for 2 min at a flow rate of ∼200 mL min−1. For the calculation of surface coverage percentages (sc%), we modeled the 4 nm Pd-on-Au NPs as magic clusters, where the Au NP is a magic cluster of 7 shells, and the Pd forms the 8th shell or more (Scheme 2).30
2.3. Characterization
UV-vis absorbance spectra of the NP solutions were measured on a Shimadzu UV-2401 PC spectrophotometer. Transmission electron microscopy (TEM) images were obtained using a JEOL 2010 transmission electron microscope operating at an accelerating voltage of 200 kV. The particle size distribution was calculated by counting around 200 particles. pH measurements were taken using a VWR sympHony SB20 meter with a standard pH electrode.Nitrite ions were analyzed using the Griess test.34 A stock solution of the Griess reagent was prepared by dissolving 10 g of the powder (Sigma-Aldrich) in 250 mL Nanopure water, such that the final concentrations are 0.1 wt% N-(1-naphthyl)ethylenediamine dihydrochloride, 1 wt% sulfanilamide, and 5% H3PO4.35 In a typical colorimetric assay, the Griess reagent solution (0.2 mL), a nitrite-containing solution (0.2 mL), and water (1.6 mL) are mixed together and kept in room temperature for 10 min. The sulfanilamide reacts with a nitrite anion, and the resulting compound further reacts with the amine, forming a colored solution. The absorbance at 540 nm is measured via UV-vis spectroscopy, and the NO2− concentration is determined in the 0 to 2.0 ppm range using a standard curve (Fig. S1†).
Ammonia measurements were made using an ammonium (NH4+) ion selective electrode (Cole-Parmer, detection limit 0.01 ppm, concentration range from 0.01 to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm) and using Nessler's test (concentration range from 0.02 to 2.5 ppm).34,35 In a typical colorimetric assay, a Nessler's reagent solution (1 mL, 0.09 M of potassium tetraiodomercurate and 2.5 M potassium hydroxide, Sigma-Aldrich) and an ammonia-containing sample (1 mL) are mixed together and then kept at room temperature for 1 min. The ammonium reacts with the tetraiodomercurate at high pH, forming a colored solution. The absorbance at 420 nm is measured, and the NH4+ concentration is determined in the 0 to 10.0 ppm range using a standard curve. (Fig. S2†). These concentrations were independently verified using an ammonium ion selective electrode (Cole-Parmer, detection limit 0.01 ppm, concentration range from 0.01 to 17000 ppm) (Fig. S3†).
000 ppm) and using Nessler's test (concentration range from 0.02 to 2.5 ppm).34,35 In a typical colorimetric assay, a Nessler's reagent solution (1 mL, 0.09 M of potassium tetraiodomercurate and 2.5 M potassium hydroxide, Sigma-Aldrich) and an ammonia-containing sample (1 mL) are mixed together and then kept at room temperature for 1 min. The ammonium reacts with the tetraiodomercurate at high pH, forming a colored solution. The absorbance at 420 nm is measured, and the NH4+ concentration is determined in the 0 to 10.0 ppm range using a standard curve. (Fig. S2†). These concentrations were independently verified using an ammonium ion selective electrode (Cole-Parmer, detection limit 0.01 ppm, concentration range from 0.01 to 17000 ppm) (Fig. S3†).
2.4. Catalytic experiments
Batch nitrite reduction experiments were conducted in a three-neck 250 mL round bottomed flask. The amount of Pd-on-Au NPs added was chosen such that the total Pd amount per reaction was 0.0365 mg, the final catalyst charge concentration was 0.365 mg-Pd per L, and the final liquid volume was 99.6 mL (Table S1†). For example, 3.75 mL of a Pd-on-Au NP (80 sc%) sol was combined with 95.85 mL of water in the flask. The solution was then bubbled simultaneously with hydrogen gas (120 mL min−1, to serve as reductant) and carbon dioxide gas (120 mL min−1, to buffer the solution to a pH value of 5–7) for 5 min. For the Pd/Al2O3 case, 36.5 mg was suspended in 10 mL of water, and then 1 mL of suspension was combined with 98.6 mL of water in the flask; the total Pd amount charged to the reactor was 0.0365 mg. For the Au/Al2O3 case, 28 mg was combined with 99.6 mL of water in the flask; the total Au amount charged to the reactor was 0.25 mg. Additionally, a set of catalytic experiments were carried out in the absence of carbon dioxide gas.The catalytic reactions were conducted at room temperature under constant stirring (400 rpm) and continuous hydrogen gas (120 mL min−1) and carbon dioxide gas (120 mL min−1). The NaNO2 solution (0.4 mL, 10 mg mL−1 of NO2−) was injected to start the reaction, such the initial solution NO2− concentration was 40 mg NO2 per L (or 12.2 mg N per L). The reaction was monitored periodically by withdrawing 1 mL aliquots from the reaction flask. By the end of the reaction test, ∼8 to 12 mL of reaction solution was removed from the flask, leaving behind 88–92% of the initial solution. For the Pd-on-Au NP tests, no separation of the particles from the reaction medium was performed. For the Pd/Al2O3 test, the powder was separated from the reaction medium before nitrite and ammonium concentrations were determined.
Because the H2 is excess to nitrite, the observed reaction rate kmeas (with units of min−1) was calculated by assuming first-order dependence on nitrite concentration:36
 | (1) |
To check that the proper amount of catalyst was used, various aliquots (0, 1.25, 2.5, 5.0, 7.5, 10 mL) of 60 sc% Pd-on-Au NP sols were added to the reactor and the total reaction volume was set at 99.6 mL. The corresponding Pd amounts in reactor were 0, 0.00913, 0.0183, 0.0365, 0.0548, 0.073 mg, respectively. Reaction rates were determined as described above.
To determine the effect of chloride, a series of experiments were performed by adding different amounts of 1 M NaCl solution was added to reactor with catalyst before bubbling H2 and CO2 gas, giving the final concentration of chloride from 0 to 0.05 M. The total reaction volume was kept at 99.6 mL. The actual volume of 1 M NaCl and nanopure water added to reactor were shown in Table S2.† The following catalyst was added to the reactor: 5 mL 60 sc% Pd-on-Au NPs solution or 7.2 mg Pd/Al2O3 (1 wt%). The other conditions are the same with the typical catalytic experiment.
3. Results and discussion
3.1. Structure of Pd-on-Au NPs
The synthesis and characterization of Pd-on-Au bimetallic NPs for hydrodechlorination reactions has been previously reported by our group.30,32,37–39Fig. 1 shows an example TEM image of as-prepared Pd-on-Au NPs. These NPs have a relative uniform size distribution, with mean diameter of 4.3 nm and a relative standard deviation of 12%. Our X-ray absorption spectroscopy (XAS) results reported in earlier publications indicated that all Pd atoms were located on the surface of the Au NPs.32,40 At relatively low surface coverage (<30 sc%), Pd atoms were found mostly as scattered atoms on the Au surface. At higher surface coverages, some of Pd atoms formed non-oxidizable two-dimensional (2-D) ensembles, which we concluded to be especially the most active species for the hydrodechlorination (HDC) of trichloroethene (TCE). At surface coverage of 70% and higher, three-dimensional (3-D) ensembles appeared, in which oxidized Pd atoms were found on top of other Pd atoms, and the per-gram-Pd catalytic activity decreased. | ||
| Fig. 1 (a) Transmission electron microscopy (TEM) image and (b) size distribution of Pd-on-Au bimetallic NPs (60 sc%). Each bar represents the total number of NPs of a particle diameter ±0.25 nm. | ||
3.2. Effect of Pd surface coverage
For all catalyst compositions, 100% nitrite conversion was reached within 30 min. Fig. 2 is representative of the evolution of nitrite, ammonia and nitrogen as a function of time. The decrease in nitrite concentration fit well to a pseudo first-order model, and observed reaction rate constant, kmeas, was obtained from the slope of the natural log of nitrite concentration versus reaction time using linear least squares fitting. The selectivity to N2 exceeded 99% for all catalysts (Table 1). These near-100% selectivity values towards N2 are similar to catalytic nitrite reduction studies from other research groups.41 The Pd-on-Au NP catalyst remained active for nitrite reduction after spiking the reactor with additional nitrite solution two more times (Fig. S4†). | ||
| Fig. 2 Concentration–time curves of NO2−, NH4+ and N2. Reaction conditions: 60 sc% Pd-on-Au NPs with 0.365 mg L−1 Pd in reactor, 120 mL min−1 H2, 120 mL min−1 CO2, 400 rpm stirring rate, 1 atm pressure. The initial nitrite/Pd molar ratio was 252/1. | ||
| Sample name | k cat (L gPd−1 min−1) | k ′cat (L gsurface-Pd−1 min−1) | N2 Selectivity (at 90% conversion) |
|---|---|---|---|
| a First-order rate constant kmeas = 0 min−1. | |||
| 0 sc% Pd-on-Au NPs | Not activea | Not activea | Not available |
| 10 sc% Pd-on-Au NPs | 32 | 32 | 99.3 |
| 30 sc% Pd-on-Au NPs | 174 | 174 | 99.5 |
| 60 sc% Pd-on-Au NPs | 479 | 479 | 99.8 |
| 80 sc% Pd-on-Au NPs | 576 | 576 | 99.6 |
| 100 sc% Pd-on-Au NPs | 466 | 466 | 99.5 |
| 150 sc% Pd-on-Au NPs | 421 | 584 | 99.1 |
| 300 sc% Pd-on-Au NPs | 188 | 460 | 99.3 |
| Pd NPs | 40 | 160 | 99.9 |
| Pd/Al2O3 (1 wt%) | 76 | 361 | 99.5 |
| Au/Al2O3 (1.2 wt%) | Not activea | Not activea | Not available |
The observed reaction rates determined for Pd-on-Au NPs (60 sc%) at various catalyst charges showed a linear dependence, showing that the reaction rate increased proportionately to the catalyst amount used (Fig. 3). Here, the implication is that the reaction rate constant determined at the standard catalyst charge using eqn (1) (the middle point of the 5-point line) is equal to the rate constant determined from 5 catalyst amounts (the slope of this 5-point line, which is equivalent to kcat = kmeas/Ccat), This linearity was seen for other catalyst compositions. Fig. 3 gave a slope of 416 L gPd−1 min−1, smaller in value than 479 L gPd−1 min−1 (Table 1, data point circled in Fig. 3), which we attributed to experimental uncertainty. If the reaction rate does not increase proportionately to catalyst amount, then too much catalyst has been added and the mass transfer effect becomes noticeable.42
 | ||
| Fig. 3 Observed reaction rate kmeas determined at different catalyst charges of Pd-on-Au NPs (60 sc%). The circled data point (blue color) indicates the standard Pd amount used for all other catalysts in this study. | ||
The Pd-on-Au NP catalysts with various Pd surface coverage (sc%) were prepared by reducing appropriate amounts of Pd salt precursor in the presence of Au NPs using H2 gas. The Pd surface coverages were calculated by modeling a magic cluster structure for the NPs,30 such that all atoms could be accounted for. A ∼4 nm Au NP with surface coverages of 10, 30, 60, 80, 100, 150, and 300% had Pd weight loadings of 2.4, 5.8, 10.9, 15.5, 18.1, 19.7 wt%, respectively. The convenience and accuracy of the magic cluster model relies on the assumptions that all Pd precursor is fully reduced onto the Au surface and that the Au particles are perfectly monodisperse (which is not the case). Inductively coupled plasma-optical emission spectroscopy results of ∼4 nm Pd-on-Au NPs from an earlier study indicated the metals content was >95% of the expected values.43 We note that the uncertainties in the amount of Pd salt added to the synthesis volume and in the amount of Au NPs used, lead to an uncertainty in the calculated Pd surface coverage of 0.01 × sc% (at one standard deviation).33
Nitrite reduction activity of Pd-on-Au NPs varied significantly with Pd surface coverage (Table 1 and Fig. 4). Monometallic Au NPs (0 sc%) and Au/Al2O3 did not show activity for the reaction. Since Pd was the active metal, catalytic activity was quantified by normalizing kmeas to total Pd content (kcat) or to total surface Pd content (k′cat). The rate constants kcat increased monotonically from 10 sc% to 80 sc% and decreased from monotonically from 80 sc% to 300 sc%, characteristic of a "volcano-shape" structure–activity plot.
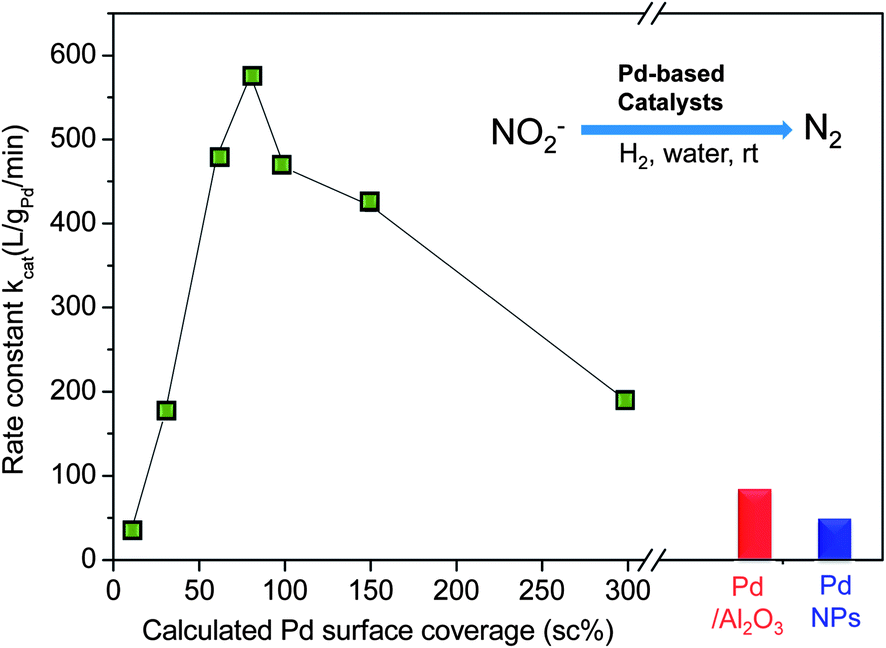 | ||
| Fig. 4 Experimentally determined nitrite reaction rate constants for Pd-on-Au NPs as a function of Pd surface coverage and for monometallic Pd catalysts. | ||
The 80 sc% Pd-on-Au NPs had a rate constant 15x and 7.5x times greater than those of monometallic Pd NPs and Pd/Al2O3, respectively (Table 1). Except for the 10 sc% case, all Pd-on-Au NPs were also more active, indicating a positive and direct interaction of the Au with the Pd metal. The volcano plots and higher catalytic activity of Pd-on-Au NPs observed here are consistent with observations for HDC of TCE and tetrachloroethene (PCE).39 Even though nitrite is not structurally related to TCE (or PCE), the similarity in the volcano-shape plots suggests the reduction reactions occur on a common set of catalytically active sites. We attribute the promotional effect of Au on Pd catalysis of nitrite reduction to the high Pd dispersion on the Au NP surface and the presence of the non-oxidized 2-D Pd ensembles.
At surface coverages of 150 and 300 sc%, the per-gram-Pd catalytic activity was lower due to a lower Pd dispersion and the topmost atoms of the 3-D Pd ensembles being oxidized. Accounting for surface Pd atoms, these Pd-on-Au NPs had higher rate constant k′cat values than those of monometallic Pd NPs and Pd/Al2O3 (which also increased, Table 1). However, the trend in k′cat (which differ from kcat above 100 sc%) was not as clear. This is due to the over-simplication of the metal-on-metal NP structure when using the magic cluster model for calculations, the presence of oxidized Pd and the lack of experimental data quantifying the amount of surface Pd atoms (e.g., through titration methods).32 That the surface-Pd-atom-normalized rate constants were larger than those for Pd NPs and Pd/Al2O3 may be the result of the electronic effect, in which there is change in valence electron density of states in the Pd metal.44–46
In the absence of the CO2 buffer, nitrite reduction was extremely slow. For example, the kcat for 60 sc% Pd-on-Au NPs was 1.0 L gPd−1 min−1 (compared to 479 L gPd−1 min−1). In a sampling of 4 different compositions, the final conversion for all was ∼1 to 2%, and the final pH increased from an initial pH of 5–7 to >7. The low catalytic activity resulted from the alkaline condition, which was observed by other researchers.28,29,47 The reduction of nitrite generates OH− species, which block the active sites of palladium based catalyst. Thus, the use of a buffer like CO2 (ref. 29) and formic acid,15 or a water-solubilized conducting polymer41 with buffering capacity is important to maintain high activities and accurate rate constant measurements.
Recently, Werth and coworkers used isotopically labeled nitrate/nitrite to study the nitrate/nitrite reduction pathways using a Pd–In/Al2O3 catalyst (5 wt% Pd, 0.5 wt% In).27 Surface-bound NO and N2O were found as reaction intermediates, providing greater mechanistic insight into the general reaction pathway of nitrite/nitrate catalytic reduction (Scheme 1). Derived from the further reduction of NO2−, the NO surface intermediate reduces in the presence of H2 to form NH4+.27 The NO intermediate also reacts with another NO to form the nitrous oxide surface intermediate, N2O, in a parallel reduction step. This N2O then reduces to form N2. The high N2 selectivity values observed for all catalysts in this study suggest that the NO-to-N2O surface reaction step dominates the NO-to-NH4+ step under the reaction conditions used, independent of the bimetal nanostructure.27
3.3. Effect of chloride concentration
Chloride is one of the most common ions in the drinking water, with a typical concentration of ∼50 mg L−1 (= ∼50 ppm = ∼0.001408 M).48 Werth and coworkers reported that chloride drastically inhibited the activity of alumina supported Cu–Pd catalysts for nitrate/nitrite reduction.48 We assessed the deactivation resistance of Pd-on-Au NPs to various chloride concentrations, and compared it to that of Pd/Al2O3.In the presence of 35.5 ppm chloride (0.001 M), the observed reaction rate of Pd/Al2O3 decreased by approximately 50%, whereas the reaction rate of Pd-on-Au NPs decreased by ∼2% (Fig. 5). Pd/Al2O3 deactivated completely in the presence of 1775 ppm chloride (0.05 M). In contrast, Pd-on-Au NP activity decreased by ∼20%. Consistent with our previous study on chloride deactivation effects during TCE HDC,38 Pd-on-Au NPs showed significantly enhanced resistance to chloride deactivation for nitrite remediation.
 | ||
| Fig. 5 The reaction rate constants kcat of 60 sc% Pd-on-Au NPs and Pd/Al2O3 at various chloride concentrations. | ||
The significant deactivation of Pd/Al2O3 catalyst was attributed to the chemisorption of chloride to the Pd surface, blocking the active sites from reaction.38 Chloride anions have been observed to oxidatively absorb to a Pd(111) surface in water under acid conditions.49 Pd-on-Au NPs was much less affected by chloride, presumably due to a lesser extent of chloride chemisorption onto the Pd supported on the Au surface.
4. Conclusion
This study demonstrates the application of Pd-on-Au NP catalysts on the reduction of nitrite, a common groundwater contaminant known to cause adverse health effects in humans. While monometallic Au NPs were inactive for the reaction, bimetallic Pd-on-Au NPs exhibited a volcano-shape dependence of activity on Pd surface coverage. 80 sc% Pd-on-Au NPs had maximum activity among all Pd-on-Au compositions, and was more active than monometallic Pd in both NP and Al2O3-supported forms. All catalyst compositions showed near-100% selectivity to N2 over ammonia. The buffered reaction conditions were important to eliminate the pH rise arising from the formation of hydroxide anions during nitrite reduction. The Pd-on-Au catalyst was much more resistant to chloride deactivation compared to Pd/Al2O3, at concentrations typical of groundwater and at higher concentrations. These results suggest that bimetallic Pd-on-Au NPs have catalytic properties amenable for water pollution abatement and that the metal-on-gold nanostructure is a useful direction towards designing improved catalysts for the rapid and selective reduction of NO3− and other oxyanions like perchlorate (ClO4−) and bromate (BrO3−).Acknowledgements
The authors gratefully acknowledge financial support from the J. Evans Attwell-Welch Postdoctoral Fellowship Program of the Smalley Institute of Rice University (to H.Q.), the National Science Foundation (CBET-1134535), the Welch Foundation (C-1676), and GSI Environmental, Inc. We thank Ms. S. Gullapalli for the assistance in TEM characterization.References
- F. T. Wakida and D. N. Lerner, Water Res., 2005, 39, 3–16 CrossRef CAS PubMed.
- G. Jacks and V. Sharma, Environ. Geol., 1983, 5, 61–64 CrossRef CAS.
- E. Lagerstedt, G. Jacks and F. Sefe, Environ. Geol., 1994, 23, 60–64 CrossRef CAS.
- B. R. Scanlon, I. Jolly, M. Sophocleous and L. Zhang, Water Resour. Res., 2007, 43, W03437 Search PubMed.
- H. H. Comly, JAMA, J. Am. Med. Assoc., 1987, 257, 2788–2792 CrossRef CAS.
- S. Shephard, Health aspects of nitrate and its metabolites (particularly nitrite), Proceedings of an international workshop, Bilthoven, Netherlands, 1995 Search PubMed.
- P. Issenberg, Fed. Proc., 1976, 35, 1322–1326 CAS.
- US.EPA, National Primary Drinking Water Regulations and Contaminant Candidate List, ed. US.EPA, 2008 Search PubMed.
- EU, Counicl Directive 98/83/EC, Official Journal of the European Communities, Brussel, 1998 Search PubMed.
- WHO, Guidelines for drinking-water quality, 3rd, World Health Organization 2006 Search PubMed.
- A. Kapoor and T. Viraraghavan, J. Environ. Eng., 1997, 123, 371–380 CrossRef CAS.
- B. P. Chaplin, M. Reinhard, W. F. Schneider, C. Schuth, J. R. Shapley, T. J. Strathmann and C. J. Werth, Environ. Sci. Technol., 2012, 46, 3655–3670 CrossRef CAS PubMed.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 CAS.
- K.-D. Vorlop and T. Tacke, Chem. Ing. Tech., 1989, 61, 836–837 CrossRef CAS.
- U. Prusse, M. Hahnlein, J. Daum and K.-D. Vorlop, Catal. Today, 2000, 55, 79–90 CrossRef CAS.
- I. Dodouche and F. Epron, Appl. Catal., B, 2007, 76, 291–299 CrossRef CAS PubMed.
- J. Sa and H. Vinek, Appl. Catal., B, 2005, 57, 247–256 CrossRef CAS PubMed.
- A. Pintar and J. Batista, Appl. Catal., B, 2006, 63, 150–159 CrossRef CAS PubMed.
- F. Zhang, S. Miao, Y. Yang, X. Zhang, J. Chen and N. Guan, J. Phys. Chem. C, 2008, 112, 7665–7671 CAS.
- A. Garron and F. Epron, Water Res., 2005, 39, 3073–3081 CrossRef CAS PubMed.
- F. A. Marchesini, S. Irusta, C. Querini and E. Miro, Appl. Catal., A, 2008, 348, 60–70 CrossRef CAS PubMed.
- A. Kapoor and T. Viraraghavan, J. Environ. Eng., 1997, 123, 371–380 CrossRef CAS.
- N. Barrabes and J. Sa, Appl. Catal., B, 2011, 104, 1–5 CrossRef CAS PubMed.
- U. Prusse, S. Horold and K.-D. Vorlop, Chem. Ing. Tech., 1997, 69, 93–97 CrossRef.
- A. Pintar, J. Batista, J. Levec and T. Kajiuchi, Appl. Catal., B, 1996, 11, 81–98 CrossRef CAS.
- K. Daub, G. Emig, M. J. Chollier, M. Callant and R. Dittmeyer, Chem. Eng. Sci., 1999, 54, 1577–1582 CrossRef CAS.
- R. Zhang, D. Shuai, K. A. Guy, J. R. Shapley, T. J. Strathmann and C. J. Werth, ChemCatChem, 2012, 5, 313–321 CrossRef.
- K. A. Guy, H. Xu, J. C. Yang, C. J. Werth and J. R. Shapley, J. Phys. Chem. C, 2009, 113, 8177–8185 CAS.
- U. Prusse and K.-D. Vorlop, J. Mol. Catal. A: Chem., 2001, 173, 313–328 CrossRef CAS.
- M. O. Nutt, K. N. Heck, P. Alvarez and M. S. Wong, Appl. Catal., B, 2006, 69, 115–125 CrossRef CAS PubMed.
- H. Qian, L. A. Pretzer, J. C. Velazquez, Z. Zhao and M. S. Wong, J. Chem. Technol. Biotechnol., 2013, 88, 735–741 CrossRef CAS.
- L. A. Pretzer, H. J. Song, Y.-L. Fang, Z. Zhao, N. Guo, T. Wu, I. Arslan, J. T. Miller and M. S. Wong, J. Catal., 2013, 298, 206–217 CrossRef CAS PubMed.
- Z. Zhao, Y.-L. Fang, P. J. J. Alvarez and M. S. Wong, Appl. Catal., B, 2013, 140–141, 468–477 CrossRef CAS PubMed.
- D. F. Boltz and J. A. Howell, Colorimetric Determination of Nonmetals, John Wiley & Sons, New York, 1978 Search PubMed.
- L. C. Green, D. A. Wagner, J. Glogowski, P. L. Skipper, J. S. Wishnok and S. R. Tannenbaum, Anal. Biochem., 1982, 126, 131–138 CrossRef CAS.
- D. Shuai, J. K. Choe, J. R. Shapley and C. J. Werth, Environ. Sci. Technol., 2012, 46, 2847–2855 CrossRef CAS PubMed.
- M. O. Nutt, J. B. Hughes and M. S. Wong, Environ. Sci. Technol., 2005, 39, 1346–1353 CrossRef CAS.
- K. N. Heck, M. O. Nutt, P. Alvarez and M. S. Wong, J. Catal., 2009, 267, 97–104 CrossRef CAS PubMed.
- M. S. Wong, P. J. J. Alvarez, Y. L. Fang, N. Akcin, M. O. Nutt, J. T. Miller and K. N. Heck, J. Chem. Technol. Biotechnol., 2009, 84, 158–166 CrossRef CAS.
- Y.-L. Fang, J. T. Miller, N. Guo, K. N. Heck, P. J. J. Alvarez and M. S. Wong, Catal. Today, 2011, 160, 96–102 CrossRef CAS PubMed.
- I. Dodouche, D. P. Barbosa, M. d. C. Rangel and F. Epron, Appl. Catal., B, 2009, 93, 50–55 CrossRef CAS PubMed.
- B. C. Gates, Catalytic Chemistry, Wiley, New York, 1992 Search PubMed.
- Y.-L. Fang, K. N. Heck, P. J. J. Alvarez and M. S. Wong, ACS Catal., 2011, 1, 128–138 CrossRef CAS.
- B. E. Koel, A. Sellidj and M. T. Paffett, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 7846–7856 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- L. Kesavan, R. Tiruvalam, M. H. A. Rahim, M. I. bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195–199 CrossRef CAS PubMed.
- A. Pintar, M. Vetinc and J. Levec, J. Catal., 1998, 174, 72–87 CrossRef CAS.
- B. P. Chaplin, E. Roundy, K. A. Guy, J. R. Shapley and C. J. Werth, Environ. Sci. Technol., 2006, 40, 3075–3081 CrossRef CAS.
- A. Carrasquillo, J. J. Jeng, R. J. Barriga, W. F. Temesghen and M. P. Soriaga, Inorg. Chim. Acta, 1997, 255, 249–254 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nr04540d |
| This journal is © The Royal Society of Chemistry 2014 |