Quantitative determination of fragmentation kinetics and thermodynamics of colloidal silver nanowires by in situ high-energy synchrotron X-ray diffraction†
Zheng
Li
a,
John S.
Okasinski
b,
Jonathan D.
Almer
b,
Yang
Ren
b,
Xiaobing
Zuo
*b and
Yugang
Sun
*a
aCenter for Nanoscale Materials, Argonne National Laboratory, Argonne, Illinois 60439, USA. E-mail: ygsun@anl.gov; Fax: +1-630-252-4646
bX-Ray Science Division, Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois 60439, USA. E-mail: zuox@aps.anl.gov; Fax: +1-630-252-0365
First published on 22nd October 2013
Abstract
Colloidal silver nanowires become instable and tend to fragment into shortened nanorods and nanoparticles at elevated temperatures. Such morphological variations are associated with the transformation of crystalline structures from the body-centered tetragonal (b.c.t.) lattices into the face-centered cubic (f.c.c.) ones. The crystalline phase transformation has been probed in real time with an in situ technique based on time-resolved high-energy synchrotron X-ray diffraction. Comprehensive analysis of the in situ measurements provides, for the first time, the quantitative understanding of kinetics and thermodynamics involved in the fragmentation of the colloidal silver nanowires.
1. Introduction
Silver (Ag) nanowires represent a class of very interesting materials due to their intriguing electrical/optical properties and applications.1–7 For example, Ag nanowire networks have been used as flexible, transparent conducting films in optoelectronic devices to replace the traditional doped metal oxides, most commonly tin-doped indium oxide (ITO).8 The performance of Ag nanowires as conducting films9–12 and in many other applications13–17 (e.g., plasmonic waveguides) is usually determined by the high aspect ratios of the nanowires. Therefore, synthesis and stabilization of long Ag nanowires are crucial to promote these important applications. In general, the quality of the synthesized Ag nanowires is sensitive to reaction conditions. For instance, a previous report shows that the reaction temperature could significantly influence aspect ratios of the nanowires, i.e., shorter nanowires formed at higher temperatures,18 indicating that annealing colloidal Ag nanowires at high temperatures may fragment the Ag nanowires to lower their aspect ratios. Therefore, understanding the fragmentation mechanism involved in shortening the Ag nanowires at different temperatures is important to improve the stability of Ag nanowires with high aspect ratios. The ideal strategy for this purpose is to monitor in real-time the variation of Ag nanowires in colloidal solutions. Until now no in situ investigation of the degradation of colloidal Ag nanowires has been reported due to the lack of appropriate probes that can penetrate the reaction vessels and liquid solutions to provide unique signals of the Ag nanowires and the corresponding fragmented Ag nanorods/nanoparticles. Herein we report, for the first time, the use of a time-resolved, high-energy synchrotron X-ray diffraction (XRD) technique to probe the kinetics and thermodynamics involved in the fragmentation of colloidal Ag nanowires at elevated temperatures, from which the mechanism for transforming the Ag nanowires into nanoparticles has been concluded.2. Experimental section
Silver nanowires were synthesized through the approach reported elsewhere.19 In a typical synthesis, 2.344 g poly(vinyl pyrrolidone) (PVP, Mw ≈ 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich) was dissolved in 76 mL glycerol (Sigma-Aldrich) at 160 °C with assistance of vigorous stirring followed by cooling down to room temperature. 0.632 g AgNO3 powder (Strem Chemicals) and a solution of 0.0236 g NaCl (Fisher Scientific) dissolved in 0.2 mL of deionized water and 4 mL of glycerol were sequentially added to the PVP solution. The solution was then heated from room temperature to 210 °C at a ramp of 5.5 °C min−1 with the assistance of slow magnetic stirring (9.5 × 19 mm egg-shaped stir bar in a 500 mL round-bottom flask) of ∼50 rpm. Once the temperature reached 210 °C, the reaction was stopped by removing the heating mantle and gradually cooling down the reaction solution to room temperature in an ambient environment. In an experiment of thermal annealing, 10 mL of the reaction solution was mixed with 40 mL isopropanol (Fisher scientific) followed by centrifugation at 8500 rpm for 5 min. After removal of the supernatant, 20 mL isopropanol was added to re-disperse the precipitate and the dispersion was then centrifuged at 8500 rpm for 5 min. The resulting precipitate was re-dispersed in 10 mL tetraethylene glycol (TEG, Aldrich) for the in situ thermal annealing experiment.
000, Sigma-Aldrich) was dissolved in 76 mL glycerol (Sigma-Aldrich) at 160 °C with assistance of vigorous stirring followed by cooling down to room temperature. 0.632 g AgNO3 powder (Strem Chemicals) and a solution of 0.0236 g NaCl (Fisher Scientific) dissolved in 0.2 mL of deionized water and 4 mL of glycerol were sequentially added to the PVP solution. The solution was then heated from room temperature to 210 °C at a ramp of 5.5 °C min−1 with the assistance of slow magnetic stirring (9.5 × 19 mm egg-shaped stir bar in a 500 mL round-bottom flask) of ∼50 rpm. Once the temperature reached 210 °C, the reaction was stopped by removing the heating mantle and gradually cooling down the reaction solution to room temperature in an ambient environment. In an experiment of thermal annealing, 10 mL of the reaction solution was mixed with 40 mL isopropanol (Fisher scientific) followed by centrifugation at 8500 rpm for 5 min. After removal of the supernatant, 20 mL isopropanol was added to re-disperse the precipitate and the dispersion was then centrifuged at 8500 rpm for 5 min. The resulting precipitate was re-dispersed in 10 mL tetraethylene glycol (TEG, Aldrich) for the in situ thermal annealing experiment.
The purified Ag nanowires dispersed in TEG were then loaded into a custom-made reaction vessel20 that was wrapped with a heating tape followed by heating to a desirable temperature under magnetic stirring. Meanwhile, high-energy (70 keV) synchrotron XRD patterns were continuously recorded from the Ag nanowire dispersion. The X-ray beam size was 0.30 mm × 0.30 mm. The XRD patterns were recorded with four well-aligned GE 41RT area detectors. The position and orientation of the detectors with respect to the sample were calibrated using a LaB6 powder dispersed in ethanol in the same reaction vessel. The setup allowed us to collect scattered X-ray signals up to ∼13° in the scale of 2θ and over a full range of orientations perpendicular to the X-ray beam. The experimental setup was the same as that used in our previous work20 and the corresponding schematic illustration is presented in Scheme S1.† For the data collection, a 10 s exposure was taken consecutively 5 times, and averaged with a frequency of 1 frame per minute. The two-dimensional diffraction patterns were reduced with the fit2d program and with a correction for dark current, gain, and dead pixels.21 The background was determined by measuring the X-ray scattering from the TEG solvent in the reaction vessel and was subtracted from all diffraction patterns. Rietveld refinements were performed with the General Structure Analysis System (GSAS) program22,23 over 2θ in the 3–13° range to determine the compositional concentration of the Ag f.c.c and Ag b.c.t phases in the samples at different times.
3. Results and discussion
As shown in Fig. 1, the morphology of the colloidal Ag nanowires that are synthesized through a polyol reduction process19 can dramatically change after they are annealed in tetraethylene glycol (TEG) at temperatures ranging from 220 °C to 250 °C. The as-synthesized Ag nanowires exhibit an average diameter of 58 nm and lengths of ∼10–20 μm (i.e., high aspect ratios of ∼170–350) (Fig. 1a). Close observation of the samples reveals the existence of a small fraction of cubic nanoparticles that correspond to the residual AgCl particles formed from the precipitation reaction of AgNO3 and NaCl used in the synthesis.19 The as-synthesized Ag nanowires are separated from the highly viscous glycerol solution that is used for synthesis and they are then re-dispersed in low-viscosity TEG (58.3 and 1499 cP for TEG and glycerol at 20 °C, respectively) for high-temperature annealing by taking advantage of the high boiling point (i.e., 314 °C) of TEG. Heating a TEG dispersion of the Ag nanowires at 220 °C for 2 hours results in a significant decrease in the aspect ratio of the nanowires, leading to the formation of nanorods with lengths less than 4 μm and a small fraction of nanoparticles (Fig. 1b). Increasing the annealing temperature can further reduce the length of the resultant nanorods (e.g., the product formed at 240 °C as shown in Fig. 1c). When the annealing temperature is high enough (e.g., 250 °C), the product is dominated by nanoparticles (Fig. 1d). It is important to directly track the morphological transformation of the Ag nanowires into short nanorods and nanoparticles in the solution phase in order to understand the fragmentation mechanism. Although electron microscopy is straightforward and powerful to image nanostructures, it is difficult (or impossible) to image the Ag nanowires and the corresponding degradation products in TEG solutions at high temperatures.24,25 As the results shown in Fig. 2 and our previous report,26 the Ag nanostructures (i.e., long nanowires, short nanorods, and nanoparticles) exhibit different crystalline phases. Therefore, we propose to use the in situ, high-energy synchrotron XRD technique that provides crystalline information of the Ag nanowires and their corresponding fragmentation products to follow the morphological variation of the colloidal Ag nanowires. The extraordinary penetration power of high-energy, high flux, synchrotron X-rays in liquid solutions and reaction vessels enables sensitive probing of nanocrystals in large volume dispersions.20,27–29 Furthermore, the good q-resolution of this technique enables one to unambiguously distinguish minor differences in XRD patterns. | ||
| Fig. 1 SEM images of (a) the synthesized Ag nanowires and the corresponding products formed after they had been heated for 2 h at different temperatures: (b) 220 °C, (c) 240 °C, and (d) 250 °C. | ||
 | ||
| Fig. 2 (a) Angle-integrated XRD patterns of the samples presented in Fig. 1a–d. The asterisk in the XRD pattern denotes the diffraction peak of AgCl crystals. The diffraction peaks of the synthesized Ag nanowires (Fig. 1a) are asymmetric while the diffraction peaks of the products formed from annealing of the colloidal Ag nanowires are symmetric and correspond to the face-centered cubic (f.c.c.) Ag. (b) Individual XRD peaks (black curves) of the synthesized Ag nanowires (Fig. 1a). Rietveld refinement revealed that the Ag nanowires were composed of both body-centered tetragonal (b.c.t., green curves) and f.c.c. (red curves) crystalline phases. The fitting results are highlighted with the blue dotted curves that represent the sum of intensity of the b.c.t. and f.c.c. phases. The peaks are assigned according to the standard Miller indices of b.c.t. and f.c.c. crystalline lattices for the green and red curves, respectively. The X-ray wavelength was 0.1771 Å. | ||
Our recent results have shown that the synthesized Ag nanowires exhibit a core–shell structure with highly strained cores and less strained sheaths due to the existence of the fivefold twinning.26 The strains in the Ag nanowire cores distort the common face centered cubic (f.c.c.) crystalline lattices to exhibit a body centered tetragonal (b.c.t.) lattice symmetry. In contrast, the crystalline lattices near the nanowire surfaces still retain the f.c.c. symmetry. As shown by the black curve in Fig. 2a and the magnified peaks in Fig. 2b, the colloidal Ag nanowires contain both f.c.c. (24.7 ± 1.1%) and b.c.t. (75.3 ± 1.3%) crystalline lattices. When the Ag nanowires dispersed in TEG are fragmented to short nanorods and nanoparticles under annealing conditions, more surfaces are exposed to facilitate the release of lattice strains inside the nanowires, leading to a crystalline phase transition from b.c.t. to f.c.c. lattices (red, green, and blue curves in Fig. 2a). As a result, we can monitor the change of crystalline phase in the Ag nanowires by using in situ high-energy XRD to evaluate the morphological variation of the colloidal nanowires when they are annealed at different temperatures (e.g., 220 °C, 240 °C, and 250 °C).
Fig. 3a presents the two-dimensional (2D) contour plot of the XRD patterns recorded at different times for the Ag nanowires dispersed in TEG when they are annealed at 240 °C. The asymmetric XRD peaks of the Ag nanowires gradually develop into symmetric peaks as the thermal annealing proceeds. Four representative XRD patterns at different times are enlarged to highlight the first four major peaks as shown in Fig. 3b. With the increase of annealing time (from bottom to top), the peak width decreases, indicating an increase of lateral crystalline domain size and/or release of lattice strains in the annealing products in comparison with the original Ag nanowires. Meanwhile, the 2nd to 4th peaks gradually change from highly asymmetric to symmetric profiles along with the decrease of their widths, agreeing with the b.c.t. → f.c.c. phase transition (Fig. S1†). Each asymmetric peak has a shoulder on the higher angle side of the peak position before the annealing temperature reaches 240 °C (e.g., the green curve in Fig. 3b with the time stamp of 369 s). At the time of 1335 s, the third peak corresponding to the f.c.c. Ag (220) reflections becomes more symmetric while the second and fourth peaks are still asymmetric, indicating a phase change and reduction of lattice strain. All the diffraction peaks become symmetric to be in accordance with the standard diffraction pattern of f.c.c. Ag when the annealing time is long enough (e.g., 3389 s). The difference in thermal response along different crystalline orientations in the Ag nanowires may be due to the anisotropic morphology of the nanowires. Trace amounts (<5%) of AgCl particles remain in the nanowire sample as reflected by the weak XRD peaks of the f.c.c. AgCl. These AgCl peaks quickly disappear due to the reduction of AgCl to Ag by hot TEG within the first several minutes.
 | ||
| Fig. 3 (a) 2D contour of the XRD patterns recorded from the Ag nanowires dispersed in TEG when the dispersion was heated at 240 °C. The white dotted curve presents the solution temperature (top axis) as a function of time. Standard powder XRD patterns of the f.c.c. Ag (JCPDS no. 89-3722) and f.c.c. AgCl (JCPDS no. 85-1355) are included for reference. (b) Enlarged individual XRD peaks recorded at some critical times: 369 s (green curves), 1335 s (red curves), 2120 s (orange curves), and 3389 s (blue curves). For visual clarity the intensities of the 2nd, 3rd and 4th peak were multiplied by a factor of 2.5, 3.5, and 3.5, respectively. The assigned Miller indices correspond to the f.c.c. Ag. | ||
Rietveld refinement analysis (Fig. S2†) of the recorded XRD patterns is used to quantitatively study the crystalline phase transformation in the Ag nanowires. Results for determining the molar ratio of the b.c.t. and f.c.c. phases in the Ag nanowires are obtained by carefully fitting the XRD patterns. Fig. 4a presents the variation of different crystalline phases in the degradation products as a function of the annealing time. During the initial temperature ramping to 240 °C period (i.e., 0–730 s), the compositional ratio between the b.c.t. and f.c.c. crystalline phases remains essentially constant, indicating that no apparent phase transition occurs. After the solution temperature is stabilized at the desired annealing temperature (i.e., 240 °C), the percentage of the Ag f.c.c. crystalline phase continuously increases at the expense of the b.c.t. phase, indicating the b.c.t. → f.c.c. phase transition in the Ag nanowires. A similar phase transition is also observed for the Ag nanowire dispersions annealed at other temperatures (e.g., 220 °C and 250 °C, shown in Fig. S3†). Although the heating ramp rates differ for the three isothermal annealing temperatures, the threshold temperature for initiating the crystalline phase transformation is always observed to be ∼213 °C.
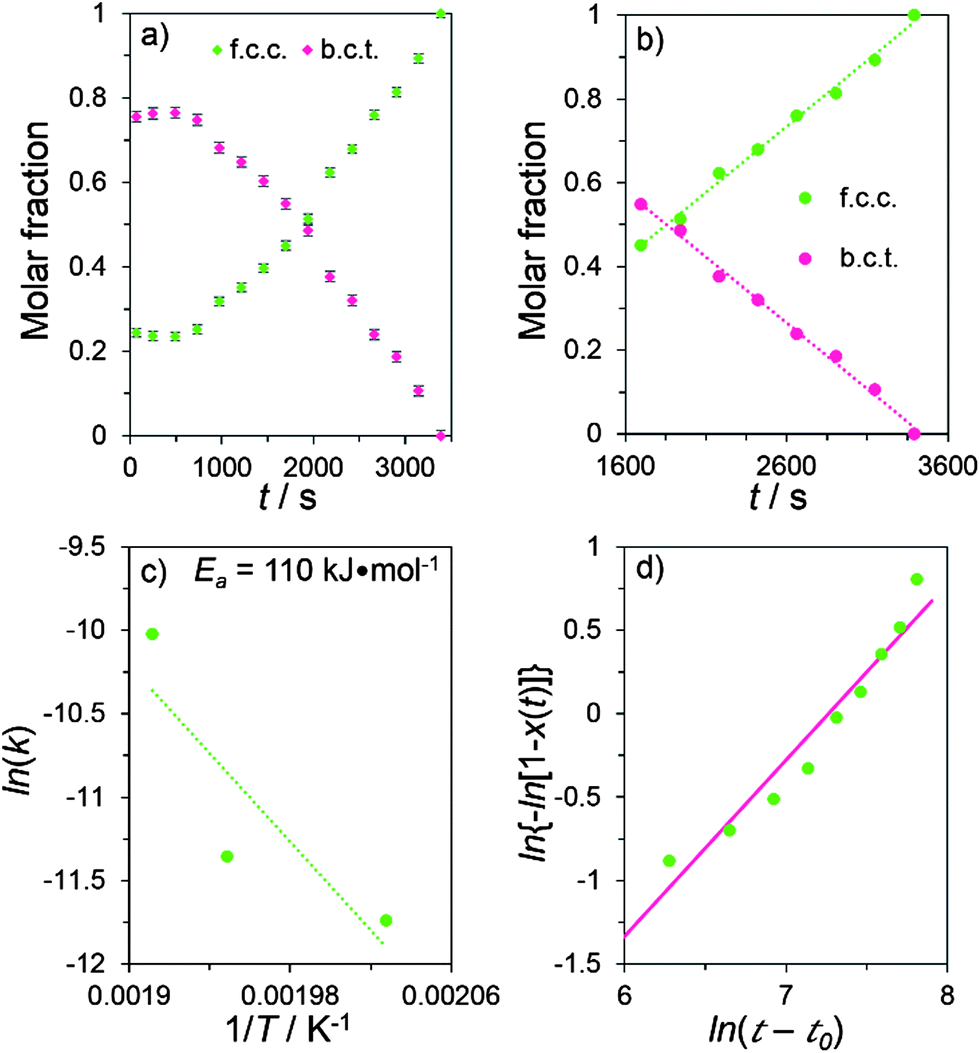 | ||
| Fig. 4 (a) Variation of the molar fraction of the f.c.c. and b.c.t. crystalline phases in the Ag nanowires and the corresponding degradation products as a function of annealing time. (b) Linear dependence of the compositional change of either f.c.c. or b.c.t. crystalline phase with time, indicating a zeroth order reaction kinetics with a reaction constant of 1.17 × 10−5 M s−1. (c) Plot of the phase transition reaction constants (k) at different temperatures (T). Linear fitting according to the Arrhenius equation results in the activation energy of 110 kJ mol−1. (d) Avrami fitting towards the reaction extent x(t) involved in the b.c.t. → f.c.c. phase transition, leading to a linear fitting described by ln{−ln[1 − x(t)]} = 1.06 × ln(t − 682) − 7.67. The annealing temperature was 240 °C for the data shown in (a), (b), and (d). | ||
For a constant temperature, the amount of b.c.t. crystalline phase in the annealed products decreases linearly as a function of time (Fig. 4b and S4†), indicating that the phase transition follows the zeroth order reaction kinetics
| −d[Agb.c.t.]/dt = d[Agf.c.c.]/dt = k, |
the activation energy, Ea, of the b.c.t. → f.c.c. crystalline phase transition can be determined to be 110 kJ mol−1 by linearly fitting ln(k) as a function of 1/T (Fig. 4c). This value of Ea is much lower (by ∼42%) than the self-diffusion activation energy of Ag atoms in bulk Ag metals (∼190 kJ mol−1),30 indicating that the diffusion of Ag atoms involved in the phase transition in the Ag nanowires mainly occurs on the surfaces of the nanowires. In general, the Ag atoms on nanostructure surfaces exhibit higher energy than the Ag atoms buried in the nanostructures and thus the surface Ag atoms require a lower activation energy to drive diffusion than bulk Ag atoms. Once the diffusion of surface Ag atoms in the Ag nanowires exposes the highly strained nanowire cores to the liquid environment, the metastable b.c.t. crystalline lattices in the nanowire cores could spontaneously transform into the more stable f.c.c. lattices through the Bain transformation path.31,32 The results indicate that the events (e.g., Ag atom diffusion, creation of crystalline defects, etc.) on the surfaces of the Ag nanowires represent the determining step to drive the fragmentation of the colloidal Ag nanowires and the corresponding crystalline phase transition.
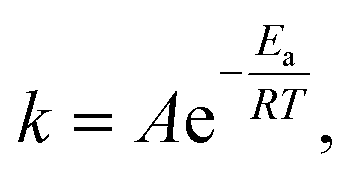
The characterization results presented in Fig. 1–3 clearly show that the b.c.t. → f.c.c. crystalline phase transition occurs in the Ag nanowires along with the morphological transformation from nanowires to nanorods to nanoparticles. As a result, the phase transition kinetics can be fitted with the Johnson–Mehl–Avrami–Kolmogorov equation (also called the Avrami equation)33–36 that represents the typical model describing solid phase transitions
| ln{−ln[1 − x(t)]} = lnkA + n × ln(t − t0), |
| T (°C) | n | k A | t 0 |
|---|---|---|---|
| 220 | 1.08 | 3.00 × 10−4 | 895 |
| 240 | 1.06 | 4.65 × 10−4 | 682 |
| 250 | 1.03 | 3.03 × 10−3 | 578 |
The results and discussions indicate that the surface instability of the colloidal Ag nanowires in hot TEG plays the most important role in the b.c.t. → f.c.c. crystalline phase transition that is associated with the fragmentation of the nanowires (Scheme S2†). At room temperature the as-synthesized Ag nanowires are stable because the less strained surfaces stabilize the inner highly strained b.c.t. lattices. When the Ag nanowires are heated in TEG solution, the mobility of the Ag atoms on the nanowire surfaces increases significantly, in particular, at surface defects (e.g., steps, stacking faults, twins, etc.). The diffusion and reconstruction of the surface Ag atoms at the defect sites can result in inner metastable b.c.t. lattices in the Ag nanowires that are exposed to the surrounding liquid environment. The exposed b.c.t. lattices can then spontaneously transform into the more stable f.c.c. lattices along with the creation of new stable surfaces, leading to the breaking of nanowires into shorter nanorods. The higher annealing temperature can activate more surface defects to expose the inner metastable b.c.t. lattices and to further decrease the aspect ratios of the nanorods derived from the Ag nanowires. The Ostwald ripening process can transform the short nanorods into prolate nanoparticles with sizes larger than the diameters of the Ag nanowires (Fig. 1d). The increased lateral dimension of the resulting nanoparticles is consistent with the narrowed XRD peaks shown in Fig. 2a and 3b after annealing of the Ag nanowires. The dependence of nanowire fragmentation on the exposure of crystalline surface defects is consistent with the observations from the annealing products of the Ag nanowires that were washed with isopropanol different times. Washing the Ag nanowires with a more polar solvent, such as isopropanol, can partially remove the PVP molecules adsorbed on the surfaces of the Ag nanowires to expose the surface crystalline defects. The more times we wash the Ag nanowires, the more PVP surfactant molecules can be removed. As shown in Fig. 1b, the synthesized Ag nanowires that had been washed 2 times were transformed into short nanorods (higher percentage) and nanoparticles (lower percentage) after they had been annealed at 220 °C for 2 h. When the synthesized Ag nanowires had been washed 10 times, more PVP surfactant molecules were removed to expose more surface defects on the Ag nanowires, leading to an acceleration of the fragmentation process under annealing conditions. As shown in Fig. S6a,† annealing these cleaner Ag nanowires leads to the formation of nanoparticles with high yield. In contrast, the synthesized Ag nanowires that are stored in the original synthesis solution can survive even more harsh annealing conditions without the observation of apparent fragmentation. Fig. S6b† presents the products formed by annealing the originally synthesized Ag nanowires at 250 °C for 2 h, clearly showing that the long Ag nanowires still dominate the sample. The differences in the annealing products formed from the Ag nanowires washed with isopropanol different times provide additional evidence showing that the fragmentation of the Ag nanowires initiates at the surface defects on the Ag nanowires.
4. Conclusions
In conclusion, we have probed the crystalline phase transition of the colloidal Ag nanowires annealed at elevated temperatures in real time with the use of an in situ high-energy synchrotron XRD technique. Due to the direct correlation of the phase transition with the morphological transformation of the Ag nanowires, analysis of the phase transition kinetics and reaction thermodynamics reveals that the fragmentation of the nanowires originates from the instability of the nanowire surface lattices at high temperatures. This understanding sheds light on improvement of the stability of colloidal Ag nanowires. For example, coating the Ag nanowires with inert materials (e.g., SiO2) that can prevent the diffusion of surface Ag atoms at high temperature may increase their thermal stability.39 Similarly, synthesizing Ag nanowires through a high-temperature recipe may need a high concentration of surfactants to form thick protection layers on the resulting Ag nanowires to stabilize their surfaces.Acknowledgements
This work was performed at the Center for Nanoscale Materials, a U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences User Facility under Contract no. DE-AC02-06CH11357. Use of Advanced Photon Source (1-ID-C, 12-ID-B) and Electron Microscopy Center for Materials Research at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357.Notes and references
- A. V. Akimov, A. Mukherjee, C. L. Yu, D. E. Chang, A. S. Zibrov, P. R. Hemmer, H. Park and M. D. Lukin, Nature, 2007, 450, 402–406 CrossRef CAS PubMed.
- Y. Sun, Nanoscale, 2010, 2, 1626–1642 RSC.
- L. Hu, H. S. Kim, J. Y. Lee, P. Peumans and Y. Cui, ACS Nano, 2010, 4, 2955–2963 CrossRef CAS PubMed.
- Z. Yu, L. Li, Q. Zhang, W. Hu and Q. Pei, Adv. Mater., 2011, 23, 4453–4457 CrossRef CAS PubMed.
- Z. Yu, Q. Zhang, L. Li, Q. Chen, X. Niu, J. Liu and Q. Pei, Adv. Mater., 2011, 23, 664–668 CrossRef CAS PubMed.
- P. Lee, J. Lee, H. Lee, J. Yeo, S. Hong, K. H. Nam, D. Lee, S. S. Lee and S. H. Ko, Adv. Mater., 2012, 24, 3326–3332 CrossRef CAS PubMed.
- S. Yun, X. Niu, Z. Yu, W. Hu, P. Brochu and Q. Pei, Adv. Mater., 2012, 24, 1321–1327 CrossRef CAS PubMed.
- A. Kumar and C. Zhou, ACS Nano, 2010, 4, 11–14 CrossRef CAS PubMed.
- S. De, T. M. Higgins, P. E. Lyons, E. M. Doherty, P. N. Nirmalraj, W. J. Blau, J. J. Boland and J. N. Coleman, ACS Nano, 2009, 3, 1767–1774 CrossRef CAS PubMed.
- L. Hu, J. W. Choi, Y. Yang, S. Jeong, F. La Mantia, L. F. Cui and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21490–21494 CrossRef CAS PubMed.
- F. Xu and Y. Zhu, Adv. Mater., 2012, 24, 5117–5122 CrossRef CAS PubMed.
- J. van de Groep, P. Spinelli and A. Polman, Nano Lett., 2012, 12, 3138–3144 CrossRef CAS PubMed.
- A. L. Pyayt, B. Wiley, Y. Xia, A. Chen and L. Dalton, Nat. Nanotechnol., 2008, 3, 660–665 CrossRef CAS PubMed.
- M. Pelton, J. Aizpurua and G. Bryant, Laser Photonics Rev., 2008, 2, 136–159 CrossRef CAS.
- R. Yan, P. Pausauskie, J. Huang and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21045–21050 CrossRef CAS PubMed.
- Z. Li, F. Hao, Y. Huang, Y. Fang, P. Nordlander and H. Xu, Nano Lett., 2009, 9, 4383–4386 CrossRef CAS PubMed.
- S. Lal, J. H. Hafner, N. J. Halas, S. Link and P. Nordlander, Acc. Chem. Res., 2012, 45, 1887–1895 CrossRef CAS PubMed.
- Y. Sun, Y. Yin, B. T. Mayers, T. Herricks and Y. Xia, Chem. Mater., 2002, 14, 4736–4745 CrossRef CAS.
- C. Yang, H. Gu, W. Lin, M. M. Yuen, C. P. Wong, M. Xiong and B. Gao, Adv. Mater., 2011, 23, 3052–3056 CrossRef CAS PubMed.
- (a) S. Peng, J. S. Okasinski, J. D. Almer, Y. Ren, L. Wang, W. Yang and Y. Sun, J. Phys. Chem. C, 2012, 116, 11842–11847 CrossRef CAS; (b) Y. Sun and Y. Ren, Part. Part. Syst. Charact., 2013, 30, 399–419 CrossRef CAS.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Hausermann, High Pressure Res., 1996, 14, 235–248 CrossRef.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, GSAS, Los Alamos National Laboratory Report LAUR 86-748, 2004.
- H. Zheng, R. K. Smith, Y. W. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Science, 2009, 324, 1309–1312 CrossRef CAS PubMed.
- Y. Liu, X. Lin, Y. Sun and T. Rajh, J. Am. Chem. Soc., 2013, 135, 3764–3767 CrossRef CAS PubMed.
- Y. Sun, Y. Ren, Y. Liu, J. Wen, J. S. Okasinski and D. J. Miller, Nat. Commun., 2012, 3, 971 CrossRef PubMed.
- H. Jensen, M. Bremholm, R. P. Nielsen, K. D. Joensen, J. S. Pedersen, H. Birkedal, Y. S. Chen, J. Almer, E. G. Sogaard, S. B. Iversen and B. B. Iversen, Angew. Chem., Int. Ed., 2007, 46, 1113–1116 CrossRef CAS PubMed.
- M. Bremholm, J. Becker-Christensen and B. Iversen, Adv. Mater., 2009, 21, 3572–3575 CrossRef CAS.
- M. Bremholm, M. Felicissimo and B. B. Iversen, Angew. Chem., Int. Ed., 2009, 48, 4788–4791 CrossRef CAS PubMed.
- C. T. Tomizuka and E. Sonder, Phys. Rev., 1956, 103, 1182–1187 CrossRef CAS.
- E. C. Bain, Trans. Am. Inst. Min., Metall. Pet. Eng., 1924, 70, 25–46 Search PubMed.
- D. A. Porter, K. E. Easterling and M. Y. Sherif, Phase transformations in metals and alloys, CRC Press, Boca Raton, FL, 2009 Search PubMed.
- M. Avrami, J. Chem. Phys., 1939, 7, 1103–1122 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1940, 8, 212–224 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1941, 9, 177–184 CrossRef CAS.
- C. H. Bamford, Comprehensive chemical kinetics. 22, Reactions in the solid state, Elsevier, Amsterdam, USA, 1980 Search PubMed.
- J. O. Eckert, C. C. HungHouston, B. L. Gersten, M. M. Lencka and R. E. Riman, J. Am. Ceram. Soc., 1996, 79, 2929–2939 CrossRef CAS.
- P. Papon, J. Leblond and P. H. E. Meijer, The physics of phase transitions concepts and applications, Springer-Verlag, Berlin, New York, 2006 Search PubMed.
- S. H. Joo, J. Y. Park, C. K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization data. See DOI: 10.1039/c3nr04368a |
| This journal is © The Royal Society of Chemistry 2014 |