Carrier induced magnetic coupling transitions in phthalocyanine-based organometallic sheet†
Jian
Zhou
a and
Qiang
Sun
*ab
aDepartment of Materials Science and Engineering, Peking University, Beijing 100871, China. E-mail: sunqiang@pku.edu.cn
bCenter for Applied Physics and Technology, Peking University, Beijing 100871, China
First published on 7th October 2013
Abstract
A two-dimensional sheet with long range ferromagnetic (FM) order has been hotly pursued currently. The recent success in synthesizing polymerized Fe-phthalocyanine (poly-FePc) porous sheets paves a possible way to achieve this goal. However, the poly-FePc and its analog poly-CrPc structure are intrinsically antiferromagnetic (AFM). Using first principles combined with Monte-Carlo simulations, we study systematically the carrier-induced magnetic coupling transitions in poly-CrPc and poly-FePc sheets. We show that electron doping can induce stable FM states with Curie temperatures of 130–140 K, while hole doping will enhance the stability of the AFM states. Such changes in magnetic couplings depend on the balance of AFM superexchange and FM p–d exchange.
Recently two-dimensional organometallic sheets (2D-OMSs) have received tremendous attention due to their flexibility in synthesis, their well-defined geometry and their promising applications in hydrogen storage, electronic circuits, the quantum Hall effect, and spintronics.1–6 In particular, the feature of regularly and separately distributed transition atoms (TMs) in 2D-OMSs makes this kind of material very special as compared with conventional dilute magnetic semiconductors (DMSs), where the TM atoms tend to form clusters and the microscopic structures are highly dependent on the synthesizing process.7–9 On the other hand, compared with the self-assembled systems on metal surfaces,10,11 these 2D polymerized structures are very stable and can be exfoliated. Different from most of the 2D-OMSs synthesized in solution or on metal surfaces, Abel et al. recently successfully fabricated a 2D Fe-phthalocyanine (denoted as poly-FePc) sheet on an NaCl(100) thin film, where the central Fe can be substituted by other flexible 3d TM atoms.12 This extension of the substrate from the metal surface to the insulating surface provides a new platform to study 2D-OMSs because the insulating NaCl substrate only affects slightly the electronic and magnetic properties of the poly-TMPc sheet. Our previous work has shown that a free-standing poly-MnPc sheet has stable ferromagnetic (FM) order with a Curie temperature estimated to be ∼150 K,13 while most of the other poly-TMPc sheets exhibit weak antiferromagnetic (AFM) order and are not directly suitable for device applications. Recent studies showed that a FM organometallic sheet may be a Chern insulator exhibiting an anomalous quantum Hall effect (AQHE), while an AFM sheet may not possess AQHE.14 Therefore, it is highly desirable to find some strategies for tuning the AFM coupling into a FM coupling.
Recall that in some DMSs15,16 multidecker nanowires,17 and graphene nanoribbons,18 carriers play an important role in tuning the magnetic coupling between magnetic atoms. Following this idea in this study we show that the magnetic couplings in poly-CrPc and poly-FePc sheets will be changed from AFM to FM with high exchange energies under electron doping. The mechanism involved is due to the enhancement of the p–d exchange interactions between the central TM atoms and the Pc network when the p bands are filled with the surplus electrons. Our Monte-Carlo simulations suggest that the Curie temperatures of the poly-CrPc and poly-FePc can be increased to as high as 140 and 130 K, respectively, which are quite comparable with the corresponding value of the poly-MnPc sheet.13
Our first-principles calculations are based on density functional theory with generalized gradient approximation (GGA) to explore the electronic and magnetic properties of the poly-TMPc systems. The projector augmented wave (PAW) method19,20 is used, as implemented in the Vienna ab initio simulation package (VASP).21 The exchange correlation functional is used as the Perdew–Burke–Ernzerhof form.22 The strong correlation interactions between the d electrons of the TM are incorporated by using the GGA+U method,23 where the correlation energy parameter U and exchange energy parameter J take the values of 4 and 1 eV, respectively. Such values have been well-tested in previous studies.13,24–27 We also tested other values (e.g. U = 3 and 5 eV) with similar conclusions. A plane wave energy cutoff of 400 eV and Monkhorst–Pack k point meshes28 of 9 × 9 × 1 and 5 × 5 × 1 for a unit cell and 2 × 2 supercell are adopted, respectively. The Vosko–Wilk–Nusair modification scheme29 is applied to interpolate the correlation energy in the spin polarized calculations. Vacuum space along the z direction of more than 15 Å is used to minimize the interactions between nearest neighbors. A six-layer NaCl(100) slab is used to model the NaCl film in experiments, and the bottom atoms are saturated by H atoms. The lower two layers of the slab are fixed in their bulk structure, while all the other atoms are allowed to relax until all forces are below 0.01 eV Å−1. Grimme's method30 is used for van der Waals interaction corrections between the poly-TMPc sheet and the NaCl surface, and the dipole interaction corrections are also included.31
We use a (2 × 2) NaCl(100) surface with the experimental lattice constant12 to support the poly-TMPc sheets, the lattice mismatch between them is less than 2%. Various initial structures have been relaxed (for detailed structures and their energies, see Fig. S1 in the ESI†), and we find that in the most stable configuration the TM atom prefers the top site of the Na atom (Fig. 1a). The optimized distance between the NaCl top layer and the poly-TMPc is more than 3.1 Å, suggesting weak interactions and nonbonding character between them. In the present case, spin polarized calculations reveal that the magnetic moments of the poly-TMPc sheets are only slightly affected by the NaCl surface as compared with the isolated sheet. This is in contrast with the calculations of TMPc molecules absorbed on a metallic surface where strong interactions exist between them and the magnetism is reduced or even quenched by strong s–d hybridization.32–35 We perform charge analysis based on Bader's method36 on the systems, and find that the poly-TMPc sheets are n-doped. The electron carrier concentrations of the poly-CrPc and poly-FePc are both about 5 × 1013 cm−2. Such high concentrations can be realized experimentally in 2D materials.37,38 Detailed analysis reveals that most injected electrons are from the Na atoms of the NaCl surface. The charge transfer between them is due to their electrostatic potential difference.
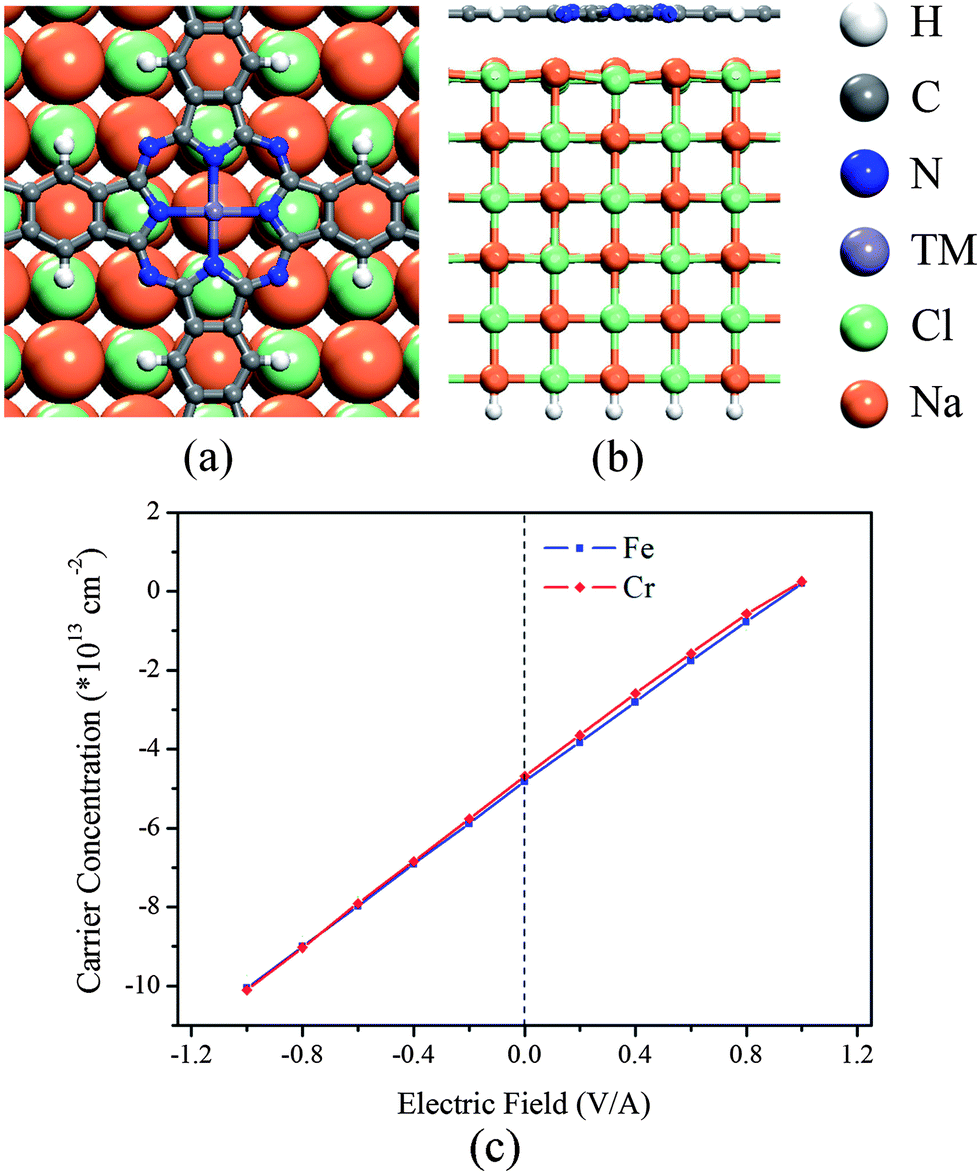 | ||
| Fig. 1 (a) Top-view and (b) side-view of the geometric structure of the poly-TMPc adsorbed on a NaCl(100) film. (c) Electric-field-induced carrier concentration variation of the poly-TMPc sheets. | ||
Next we apply an external electric field to the systems in order to modulate the carrier concentrations of the poly-TMPc sheets (Fig. 1c). The positive electric field direction is defined as pointing out of the surface. The induced carriers show a linear response to the electric field. A negative electric field will inject more electrons into the poly-TMPc sheets, while a positive electric field will make the sheet neutral-like or even p-doped-like. Within the range of the electric field (−1.0 V Å−1 to +1.0 V Å−1), we find that the distances between the poly-TMPc sheets and the NaCl slab change marginally, and the weak interactions and nonbonding character are still kept. We find that the behaviors of the poly-CrPc and poly-FePc systems are almost the same. This is due to their valence and conductance bands being contributed by the ligand Pc,13 so that the responses to the field are not sensitive to the central TM elements.
Up to now we have shown that the carrier concentrations of the poly-TMPc sheets can be tuned effectively via applying a vertical external electric field; it is interesting to check whether the intrinsic magnetic behaviors can be further modulated. A (2 × 2) supercell of the poly-TMPc is used to study the magnetic couplings. Carrier doping to the poly-TMPc sheets can be simulated by using a uniform jellium countercharge background to remain the charge neutrality level. However, under periodic boundary conditions this will lead to additional electrostatic interactions, so that the total energy converges very slowly with respect to the supercell size. Point and dipole interaction corrections are included, and the convergence of O(L−5) can be achieved.39 We plot the exchange energies Eex (= EAFM − EFM) per supercell variation with respect to different carrier concentrations in Fig. 2. A positive (negative) value of Eex indicates an FM (AFM) magnetic coupling ground state. It can be found that, although the magnetic couplings of them in their neutral states are both weak AFM,13 electron doping can increase their exchange energies to high positive values. In detail, when the injected electron concentration reaches 4.2 × 1013 cm−2 in the poly-CrPc, the Eex becomes 132 meV per supercell. Similarly for the poly-FePc sheet, the Eex can also be as high as 102 meV when the electron concentration is about 4.2 × 1013 cm−2. On the contrary, when holes are doped, both the Eex values are still negative, indicating that the ground states remain as AFM.
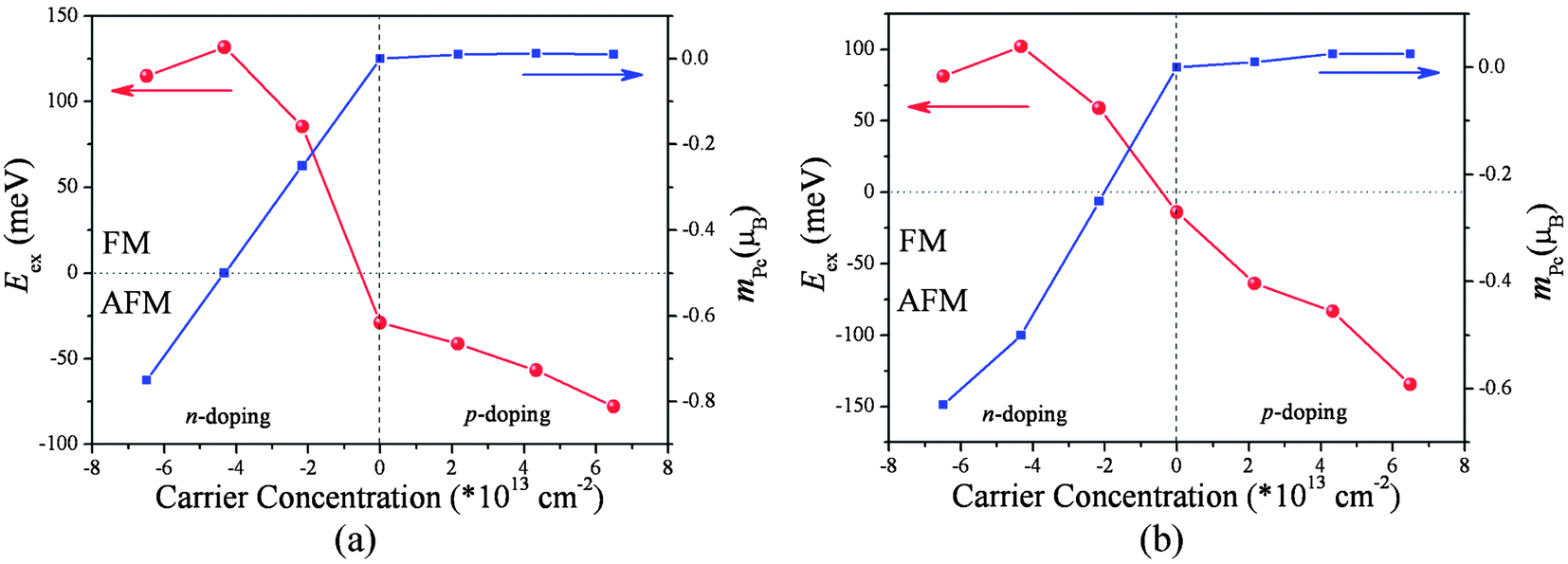 | ||
| Fig. 2 Exchange energies and magnetic moment of the ligand Pc variations with respect to the carrier concentration of the (a) poly-CrPc and (b) poly-FePc. | ||
We take the experimentally synthesized poly-FePc system as an example to explain the carrier induced magnetic coupling variation in detail. The spin densities of the poly-FePc in its neutral and n-doped (with electron carrier concentration of 4.2 × 1013 cm−2) states are plotted in Fig. 3a and b, respectively. It is clearly observed that for the neutral poly-FePc, the ligand Pc carries both spin up and spin down electrons on the N and some C atoms, respectively. This leads to very small total magnetic moments on the Pc. When electrons are injected, the magnetic moment of the central Fe atom does not show any apparent changes, while the ligand Pc exhibits more spin-down polarizations. This suggests that the injected electrons delocalize on the Pc framework instead of on the central Fe atom. This can also be seen from the charge deformation density, ρdeform, (Fig. 3c) which is defined as ρdeform = ρn-doped − ρneutral. We can see that the surplus electrons mainly accumulate on the N atoms and the C–C bonds of the pyrroles.
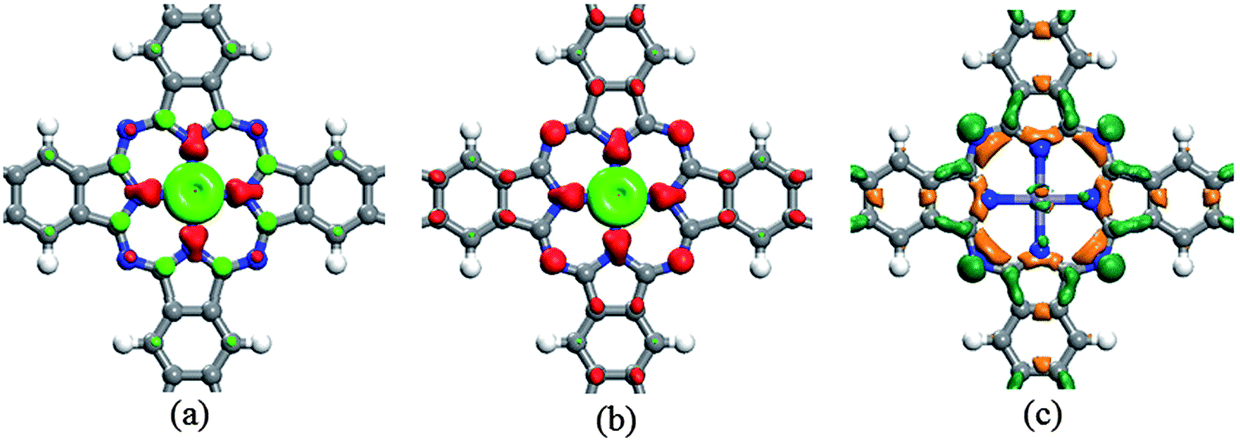 | ||
| Fig. 3 Spin densities of (a) neutral and (b) n-doped (concentration of 4.2 × 1013 cm−2) poly-FePc sheets, with light green and red representing spin-up and spin-down electrons, respectively. In (c) we show the surplus electron density, with dark green and orange as electron accumulation and deficiency, respectively. All the iso-surface values are set as 0.01 e Å−3. | ||
To understand further such behavior, we plot the partial density of states (PDOS) of the neutral and n-doped states as shown in Fig. 4a and b. We can find that the neutral poly-FePc is a semiconductor with the Fermi level in the gap of p orbitals. Besides, the p and d orbitals are strongly hybridized, which makes the p orbitals also spin polarized. However, the integrations of the spin up and spin down PDOS of the p orbitals below the Fermi level are almost the same, indicating the total magnetic moments contributed by the Pc ligand are very small. Based on the Ising model, the magnetic coupling between two Fe atoms can be written as
| Eex = JFe1-PcmFe1mPc + JFe2-PcmFe2mPc + JFe1-Fe2mFe1mFe2 | (1) |
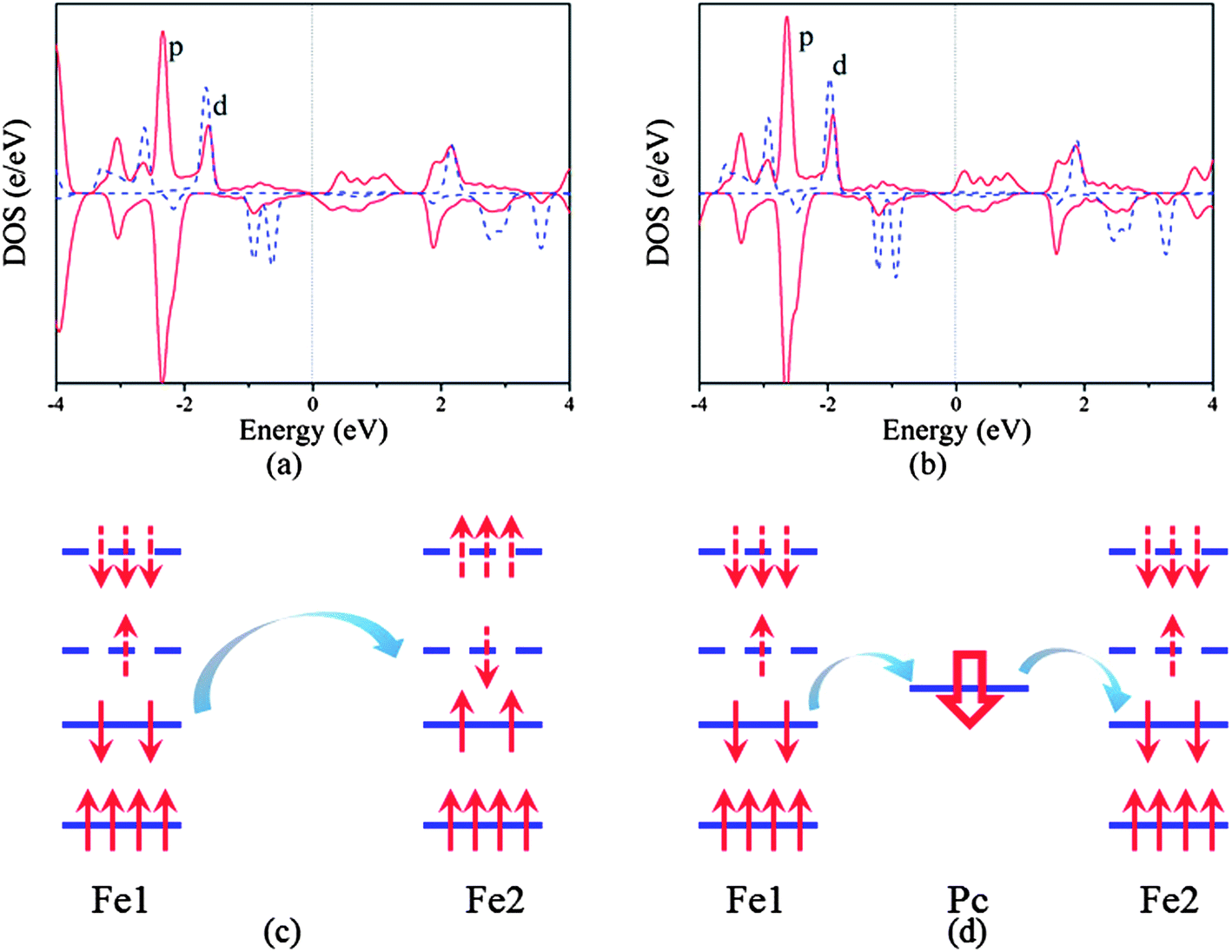 | ||
| Fig. 4 Partial density of states of the (a) neutral and (b) n-doped (concentration of 4.2 × 1013 cm−2) poly-FePc. Solid and dashed curves represent p and d orbitals, respectively. Energy occupations and the magnetic couplings between two Fe atoms are shown of the (c) neutral and (d) n-doped states. Solid and dashed energy levels represent occupied and unoccupied electron bands, respectively. | ||
Such analysis can also explain the p-doped situations. If holes are introduced to the poly-FePc, the Fermi level will move towards lower energies. We can observe that both spin-up and spin-down p electrons will be simultaneously removed, then the total magnetic moments of the ligand Pc remain small (Fig. 2b). The superexchange between the two Fe atoms still dominates, and the AFM state favors the FM state as before. This is consistent with our calculation results (Fig. 3b).
Similar mechanism is also valid for the poly-CrPc sheet. As shown in Fig. 5a, in the neutral state, the highest occupied magnetic d orbitals (dπ) locate in the spin-up channel, while the lowest unoccupied magnetic d orbital (dz2) is in the spin-down channel. This leads to superexchange coupling between two Cr atoms and results in the AFM ground state. When the system is n-doped (Fig. 5b), p–d exchange dominates, and the FM coupling favors the AFM order.
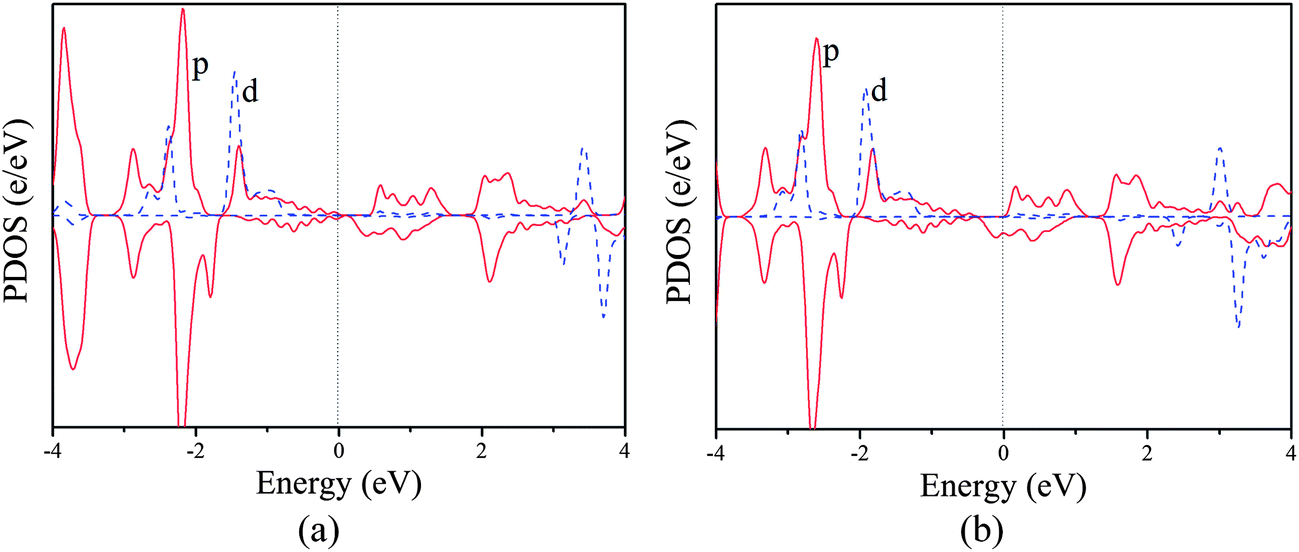 | ||
| Fig. 5 Partial density of states of the (a) neutral and (b) n-doped (concentration of 4.2 × 1013 cm−2) poly-CrPc. | ||
To understand better the results we briefly compare the electronic structures and the doping effects of the isolated FePc molecule with those of the 2D poly-FePc sheet. A similar argument holds for the CrPc and poly-CrPc sheet. In a FePc molecule, the frontier (the highest occupied and the lowest unoccupied) orbitals are contributed by not only ligand-p electrons but also Fe-d electrons.40–42 As a consequence, when electrons (holes) are injected, the magnetic d orbitals of the FePc molecule will be affected. We can expect that the total magnetic moment of the Fe atom changes dramatically.43 However, as the 2D poly-FePc sheet has an infinite extended Pc network, the ligand p orbitals hybridize strongly and form dispersed valence and conductance bands. Then the frontier bands are contributed almost by the ligand p electrons. The carrier doping will only influence the p bands of the Pc ligands and the magnetic moment of the Fe atom keeps constant.
Since the n-doped poly-TMPc sheets could exhibit high exchange energies larger than 100 meV, it is interesting to explore their magnetic behaviors under finite temperature, especially to estimate their Curie temperatures. Therefore, we perform Monte-Carlo simulations to calculate their average total magnetic moments per unit cell for each temperature. We use a (100 × 100) supercell to decrease the periodic boundary constraints. The simulation lasts for 5 × 109 loops. A longer simulation time and larger supercell will make marginal changes to the final results. In each loop, we select a TM atom, and change its magnetic moment randomly to another value according to the spin multiplicity of the TM atom, and then calculate the corresponding energy change ΔE, if ΔE is negative, the new state will be accepted; while if ΔE is positive, the new state will be accepted with the corresponding probability. After 4 × 109 loops, the system reaches an equilibrium magnetic state and another 1 × 109 loops are performed for data collections.
Our simulation results are shown in Fig. 6. We find that the Curie temperatures of the poly-CrPc and poly-FePc are about 140 and 130 K, respectively. These values are comparable with that of the neutral poly-MnPc system, and also comparable with the highest experimental value in (Ga,Mn)As dilute magnetic semiconductors.24
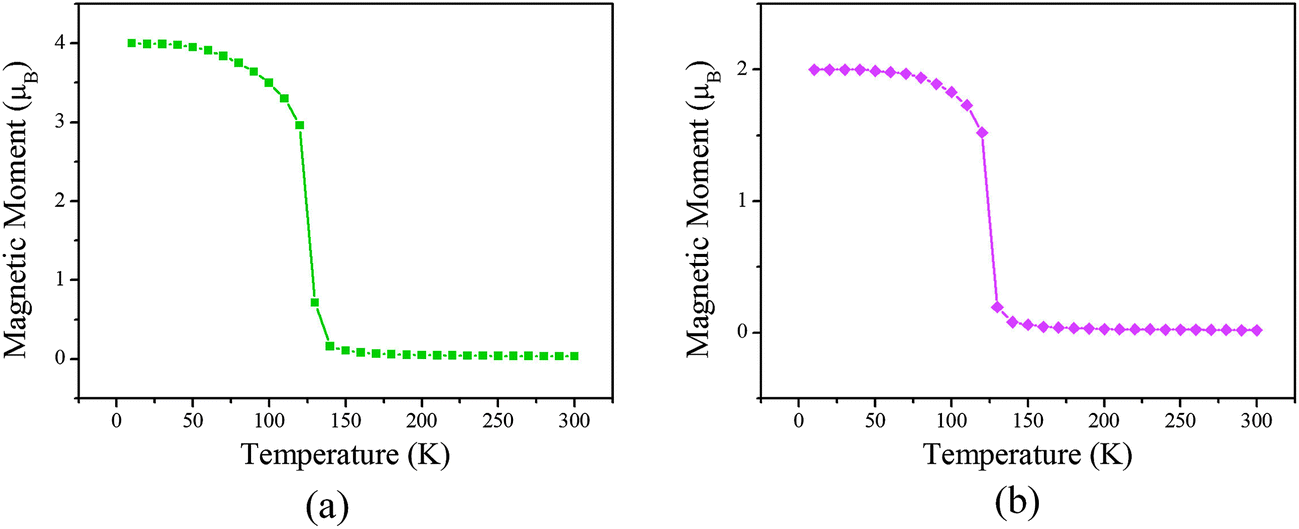 | ||
| Fig. 6 Total magnetic moments per unit cell variation with respect to temperature for the (a) poly-CrPc and (b) poly-FePc. | ||
In conclusion, using first principles calculations, we have found that the magnetic couplings in 2D organometallic porous poly-TMPc (TM = Cr and Fe) sheets can be effectively tuned with carrier doping through the substrate in an applied electric field. The following main conclusions are obtained. (1) Experimentally synthesized poly-TMPc sheets supported on NaCl(100) surface have very weak interactions with the substrate, and their magnetic moments are almost retained. (2) When applying an electric field, we can tune effectively the carrier concentrations of the poly-TMPc sheets. The carriers only influence the occupations of the ligand p bands around the Fermi level, which makes the magnetic moments of the central TM atoms nearly unchanged. This is different from the situations in poly-TMPc molecules. (3) When electrons are injected to the poly-TMPc sheets, the magnetic coupling can be changed from AFM to FM with large exchange energies, which are mainly related to AFM superexchange and FM p–d exchange. (4) Based on Monte-Carlo simulations, the Curie temperatures are found to be about 130–140 K. The high Curie temperatures suggest that these sheets have good potentials for spintronic devices. We hope that the present work can stimulate further experimental effort in this direction.
Acknowledgements
This work is partially supported by grants from the National Natural Science Foundation of China (NSFC-21173007 and 11274023), and from the National Grand Fundamental Research 973 Program of China (2012CB921404).References
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228–231 CrossRef CAS PubMed.
- E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef CAS PubMed.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- T. Kambe, R. Sakamoto, K. Hoshiko, K. Takada, M. Miyachi, J.-H. Ryu, S. Sasaki, J. Kim, K. Nakazato, M. Takata and H. Nishihara, J. Am. Chem. Soc., 2013, 135, 2462–2465 CrossRef CAS PubMed.
- Z. F. Wang, Z. Liu and F. Liu, Nat. Commun., 2013, 4, 1471–1475 CrossRef CAS PubMed.
- T. Dietl, Nat. Mater., 2010, 9, 965 CrossRef CAS PubMed.
- T. Dietl, H. Ohno, F. Matsukura, J. Cibert and D. Ferrand, Science, 2000, 287, 1019–1022 CrossRef CAS.
- Q. Wang, Q. Sun and P. Jena, Phys. Rev. Lett., 2005, 95, 167202–167205 CrossRef CAS.
- D. Hao, C. Song, Y. Ning, Y. Wang, L. Wang, X.-C. Ma, X. Chen and Q.-K. Xue, J. Chem. Phys., 2011, 134, 154703–154706 CrossRef PubMed.
- Y.-L. Wang, J. Ren, C.-L. Song, Y.-P. Jiang, L.-L. Wang, K. He, X. Chen, J.-F. Jia, S. Meng, E. Kaxiras, Q.-K. Xue and X.-C. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245420–245424 CrossRef.
- M. Abel, S. Clair, O. Ourdjini, M. Mossoyan and L. Porte, J. Am. Chem. Soc., 2011, 133, 1203–1205 CrossRef CAS PubMed.
- J. Zhou and Q. Sun, J. Am. Chem. Soc., 2011, 133, 15113–15119 CrossRef CAS PubMed.
- Z. F. Wang, Z. Liu and F. Liu, Phys. Rev. Lett., 2013, 110, 196801–196805 CrossRef CAS.
- K. Sato, P. H. Dederichs, H. Katayama-Yoshida and J. Kudrnovsky, J. Phys.: Condens. Matter, 2004, 16, S5491–S5497 CrossRef CAS.
- H. Raebiger, S. Lany and A. Zunger, Phys. Rev. Lett., 2008, 101, 027203–027206 CrossRef.
- Z. Zhang, X. Wu, W. Guo and X. C. Zeng, J. Am. Chem. Soc., 2010, 132, 10215–10217 CrossRef CAS PubMed.
- K. Sawada, F. Ishii, M. Saito, S. Okada and T. Kawai, Nano Lett., 2009, 9, 269–272 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- K. Sato, L. Bergqvist, J. Kudrnovsky, P. H. Dederichs, O. Eriksson, I. Turek, B. Sanyal, G. Bouzerar, H. Katayama-Yoshida, V. A. Dinh, T. Fukushima, H. Kizaki and R. Zeller, Rev. Mod. Phys., 2010, 82, 1633–1690 CrossRef CAS.
- J. Okabayashi, J. Okabayashi, O. Rader, T. Mizokawa, A. Fujimori, T. Hayashi and M. Tanaka, Phys. Rev. B: Condens. Matter, 1998, 58, R4211–R4214 CrossRef CAS.
- P. M. Panchmatia, B. Sanyal and P. M. Oppeneer, Chem. Phys., 2008, 343, 47–60 CrossRef CAS PubMed.
- J. Zhou, Q. Wang, Q. Sun, Y. Kawazoe and P. Jena, J. Phys. Chem. Lett., 2012, 3, 3109–3114 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- G. Makov and M. C. Payne, Phys. Rev. B: Condens. Matter, 1995, 51, 4014–4022 CrossRef CAS.
- Z. Hu, B. Li, A. Zhao, J. Yang and J. G. Hou, J. Phys. Chem. C, 2008, 112, 13650–13655 CAS.
- Y. Y. Zhang, S. X. Du and H.-J. Gao, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 125446–125453 CrossRef.
- H. Wende, M. Bernien, J. Luo, C. Sorg, N. Ponpandian, J. Kurde, J. Miguel, M. Piantek, X. Xu, Ph. Eckhold, W. Kuch, K. Baberschke, P. M. Panchmatia, B. Sanyal, P. M. Oppeneer and O. Eriksson, Nat. Mater., 2007, 6, 516–520 CrossRef CAS PubMed.
- M. Bernien, J. Miguel, C. Weis, Md. E. Ali, J. Kurde, B. Krumme, P. M. Panchmatia, B. Sanyal, M. Piantek, P. Srivastava, K. Baberschke, P. M. Oppeneer, O. Eriksson, W. Kuch and H. Wende, Phys. Rev. Lett., 2009, 102, 047202–047205 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204–084210 CrossRef CAS PubMed.
- N. Reyren, S. Thiel, A. D. Caviglia, L. Fitting Kourkoutis, G. Hammerl, C. Richter, C. W. Schneider, T. Kopp, A.-S. Rüetschi, D. Jaccard, M. Gabay, D. A. Muller, J.-M. Triscone and J. Mannhart, Science, 2007, 317, 1196–1199 CrossRef CAS PubMed.
- K. V. Emtsev, A. Bostwick, K. Horn, J. Jobst, G. L. Kellogg, L. Ley, J. L. McChesney, T. Ohta, S. A. Reshanov, J. Röhrl, E. Rotenberg, A. K. Schmid, D. Waldmann, H. B. Weber and T. Seyller, Nat. Mater., 2009, 8, 203–207 CrossRef CAS PubMed.
- G. Makov and M. C. Payne, Phys. Rev. B: Condens. Matter, 1995, 51, 4014–4022 CrossRef CAS.
- J. Hu and R. Wu, Phys. Rev. Lett., 2013, 110, 097202–097206 CrossRef.
- T. Suzuki, M. Kurahashi, X. Ju and Y. Yamauchi, J. Phys. Chem. B, 2002, 106, 11553–11556 CrossRef CAS.
- M.-S. Liao, J. D. Watts, M.-J. Huang, S. M. Gorun, T. Kar and S. Scheiner, J. Chem. Theory Comput., 2005, 1, 1201–1210 CrossRef CAS.
- M.-S. Liao and S. Scheiner, J. Chem. Phys., 2001, 114, 9780–9791 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Different stacking patterns and their relative corresponding energies of the poly-FePc and NaCl(100) surface. See DOI: 10.1039/c3nr04041k |
| This journal is © The Royal Society of Chemistry 2014 |