1,4-Dialkyl-1,4-diazabutadienes: their reactions with aluminum and indium halides†
Héctor
Rojas-Sáenz
,
Galdina V.
Suárez-Moreno
,
Iris
Ramos-García
,
Angélica M.
Duarte-Hernández
,
Edgar
Mijangos
,
Adrián
Peña-Hueso
,
Rosalinda
Contreras
and
Angelina
Flores-Parra
*
Department of Chemistry, Cinvestav, AP 14-740, CP 07000, Mexico DF. E-mail: aflores@cinvestav.mx
First published on 5th November 2013
Abstract
Reactions of 1,4-bis[(S) methylbenzyl]-1,4-diazabutadiene (1), 1,4-bis[(R) 1′-chlorobutan-2′-yl]-1,4-diazabutadiene (2), 1,4-bis[(S,S) 1′-chloro-1′-phenyl-propan-2′-yl]-1,4-diazabutadiene (3), and 1,4-di-tert-butyl-1,4-diazabutadiene (4) with aluminum and indium halides were performed. Reaction of 1,4-diazabutadienes 1–4 with two equivalents of aluminum halides in toluene afforded ionic aluminum coordination compounds (5–10) which in THF solution are transformed into 1,3-dialkyl-2-methaneiminealkyl imidazolium heterocycles (11–14) by condensation of two 1,4-diazabutadienes and elimination of amine hydrochloride. Equimolar reactions of 1–4 with InCl3 in dry acetonitrile at −78 °C afforded the neutral and diamagnetic InCl3 coordination compounds (15–18), which are stable in the solid state under dry conditions, but in THF solution are slowly transformed into the corresponding 1,3-dialkyl-imidazolium heterocycles 19–22. The X-ray diffraction analyses of compounds 2, 3, 10, 11, 14 and 17 are described. Quantum mechanical calculations were performed in order to find the minimum energy conformers of 1,4-diazabutadienes, 1,3-dialkyl-2-methaneiminealkyl imidazolium and 1,3-dialkyl-imidazolium derivatives as well as the aluminum and indium coordination compounds.
Introduction
Enantiomerically pure ligands are relevant for the preparation of coordination compounds which may be used as reagents or catalyzing agents in enantioselective reactions.1–4 In our research program concerning enantiomerically pure metal coordination compounds,5–9 we are interested in the synthesis of optically active aluminum and indium derivatives and therefore we have investigated the reactions of aluminum and indium halides with 1,4-dialkyl-1,4-diazabutadienes. These ligands have two sp2 nitrogen atoms as metal coordination sites and their N-substituents may have stereogenic centers. 1,4-Diazabutadienes can be found in three different oxidation states: neutral, a π radical monoanion or a reduced dianion, Scheme 1.10,11 Coordination compounds formed with the radical anion or the dianion have been widely explored. Reactions of group 13 elements in the low oxidation state give paramagnetic products.12–17 The synthesis of coordination compounds could be complicated by competition between addition and coupling processes.18 The coordination chemistry of neutral diazabutadienes is less known, reactions give diamagnetic ionic coordination derivatives,19 as was described for the reaction of di-tert-butyl-diazabutadiene with GaCl3.20 | ||
| Scheme 1 Neutral, anion and dianionic oxidation states of 1,4-diazabutadienes. | ||
Herein, we described the reactions of aluminum and indium halides with 1,4-bis[(S) methylbenzyl]-1,4-diazabutadiene (1), 1,4-bis[(R) 1′-chlorobutan-2′-yl]-1,4-diazabutadiene (2), 1,4-bis[(S,S) 1′-chloro-1′-phenylpropan-2′-yl]-1,4-diazabutadiene (3) and 1,4-di-tert-butyl-1,4-diazabutadiene (4). Ligands 1–3 have tertiary N-substituents whereas those of 4 are quaternary, Scheme 2. Compounds 1–3 have phenyl groups and/or chlorine atoms which could give rise to intra- and intermolecular weak interactions promoting crystallization or supramolecular arrangements of their derivatives. The chloro substituents could be used to add other coordinating functional groups, such as phosphines.21 It is known that diazabutadiene 4 is able to stabilize metallic derivatives because of the steric hindrance that inhibits subsequent reactions once the coordination compound is formed.22,23
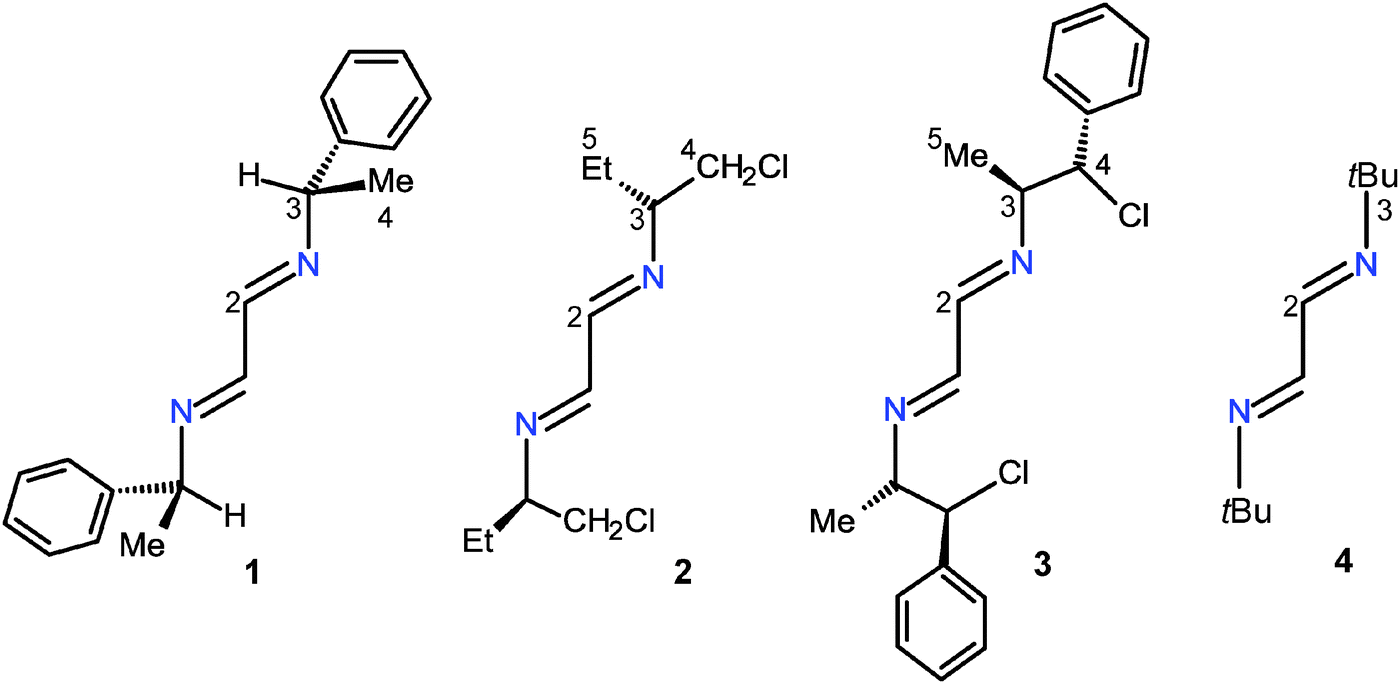 | ||
| Scheme 2 1,4-Dialkyl-1,4-diazabutadiene compounds 1–4. | ||
Results and discussion
Synthesis and structures of 1,4-diazabutadienes 1–4
Diazabutadienes 1 and 4 were prepared by reactions of aqueous glyoxal with two equivalents of the primary amines: (S)-methylbenzylamine and tert-butylamine. Compound 2 was prepared in two steps. First, (R)-2-amino-butan-1-ol was chlorinated with SOCl2 in THF to give the (R)-1-chloro-butan-2-amine hydrochloride, which in turn reacted with aqueous glyoxal and Na2CO3. It was crystallized from CHCl3 and the X-ray diffraction structure was obtained. Compound 3 was synthesized by the condensation reaction of (1S,2S)-1-chloro-1-phenyl-propan-2-amine hydrochloride24 with aqueous glyoxal and Na2CO3, Scheme 3. 1,4-dialkyl-1,4-diazabutadiene 1 is a liquid, whereas 2–4 are crystalline solids. | ||
| Scheme 3 Synthesis of diazabutadienes 1–4. | ||
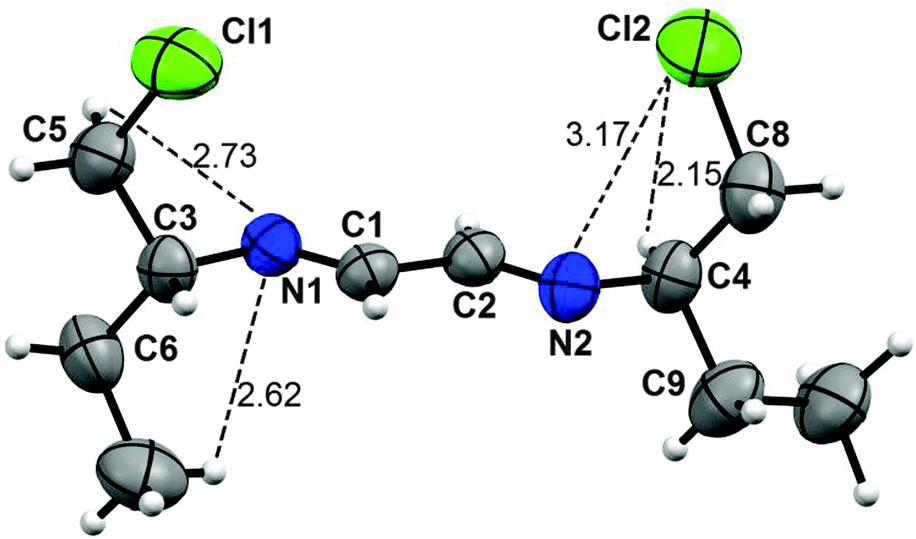
Compounds 2 and 3 are new. Their X-ray diffraction analyses were performed. They have in common a planar arrangement for the CH–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CHN–CH chain in trans conformation and imines in E configuration. Selected bond lengths and angles are in Tables 1 and 2. The C3 substituents are perpendicular to the diazabutadiene plane with the chlorine atoms pointing to the same side.
CH–CHN–CH chain in trans conformation and imines in E configuration. Selected bond lengths and angles are in Tables 1 and 2. The C3 substituents are perpendicular to the diazabutadiene plane with the chlorine atoms pointing to the same side.
|
|
|||
|---|---|---|---|
| Bond lengths | Bond angles | ||
| N1–C1 | 1.259(3) | N1–C1–C2 | 120.3(3) |
| N2–C2 | 1.264(4) | N2–C2–C1 | 119.4(3) |
| C1–C2 | 1.471(4) | C1–N1–C3 | 118.4(2) |
| N1–C3 | 1.457(3) | C2–N2–C4 | 117.6(2) |
| N2–C4 | 1.451(4) | C4–C8–Cl2 | 111.4(2) |
| C3–C5 | 1.511(5) | C3–C5–Cl1 | 112.0(3) |
| C4–C8 | 1.495(5) | N2–C4–C8 | 108.8(3) |
| Cl2–C8 | 1.790(4) | N2–C4–C9 | 107.5(3) |
| Cl1–C5 | 1.787(4) | ||
|
|
|||
|---|---|---|---|
| Bond lengths | Bond angles | ||
| N1–C1 | 1.270(4) | N1–C1–C2 | 120.5(3) |
| N2–C2 | 1.266(4) | N2–C2–C1 | 119.3(3) |
| C1–C2 | 1.466(4) | C1–N1–C3 | 118.6(3) |
| N1–C3 | 1.456(4) | C2–N2–C4 | 118.2(3) |
| N2–C4 | 1.460(4) | C4–C7–Cl2 | 108.7(2) |
| C3–C5 | 1.538(3) | C3–C5–Cl1 | 110.2(2) |
| C4–C7 | 1.514(5) | Ci–C7–C4 | 114.2(2) |
| Cl2–C7 | 1.815(3) | Ci–C5–C3 | 115.0(2) |
| Cl1–C5 | 1.824(3) | ||

In compound 2, six CH⋯N and two CH⋯Cl hydrogen bonds, together with two contacts of chlorine atoms with the nitrogen π electrons (3.17 Å; ΣvdWr Cl–N = 3.42 Å25–27) stabilize the conformation. In the crystal, the molecules of 2 are arranged in a helix, with a path of four molecules.
In compound 3, the phenyl groups are parallel to the diazabutadiene plane. The chlorine atoms have short distances to the nitrogen atoms (3.08 and 3.09 Å). The Ci–C5–C3 and Ci–C7–C4 angles are wide (115.0 and 114.2°) indicating repulsion between methyl and phenyl groups. One of the chlorine atoms has a C–H⋯Cl hydrogen bond and a Cl⋯π-interaction with another molecule, Fig. 1.
 | ||
| Fig. 1 Intermolecular contacts in the enantiomerically pure compound 3 (S,S). | ||
It was found in the solid state and in solution that the more stable conformation of 1,4-diazabutadienes is the s-trans E,E. The bidentate coordination of these ligands needs rotation of C2–C2′ in order to get the s-cis E,E conformation. The energy barrier for this isomerization in compounds 1–4 was calculated using a quantum mechanical approach,28 ΔG* = 37.0 kJ mol−1. The energy difference between the more stable trans isomers and the cis isomers was also calculated. The values were: 1 (27.1 kJ mol−1), 2 (24.4 kJ mol−1), 3 (25.6 kJ mol−1) and 4 (30.4 kJ mol−1). The energy difference is related to the steric effects of the tertiary or quaternary substituents.
Reactions of diazabutadienes 1–4 with aluminum halides in toluene
Reactions of 1,4-diazabutadienes 1–4 with two equivalents of aluminum halides at −78 °C in toluene and in a N2 atmosphere gave the corresponding ionic aluminum coordination compounds 5–10, Scheme 4. Aluminum compounds were mainly characterized by 27Al NMR, Table 3. Each spectrum presented two resonances: broad signals for the cations and sharp resonances for the anions. Compounds 5–7 are brown solids which precipitate from the reaction mixture. In THF solution, they are transformed into imidazole derivatives as will be later discussed. N-tert-Butyl derivatives 8–10 are more stable than 5–7, they were isolated as yellow solids. The X-ray diffraction analysis of compound 10 was conducted. | ||
| Scheme 4 Ionic aluminum coordination compounds 5–10. | ||
| 5 | 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|
| [AlXY]+ | +34 (1250) | +62 (3036) | +68 (3754) | +97 (3550) | +89 (2050) | +130 (2000) |
| [AlX3Y]− | +107.1 (94) | +100.1 (40) | +100.2 (80) | +102.0 (154) | +20.0 (137) | +102.0 (22) |
1H and 13C NMR spectra of compounds 5 and 8–10 were obtained. Comparison with data of the starting diazabutadienes shows that the C2–H protons in the coordination compounds are shifted to higher frequencies: 5 (Δδ 0.26 ppm); 8 (Δδ 1.26 ppm), 9 (Δδ 1.49 ppm), 10 (Δδ 1.20 ppm), whereas the C2 signals appear at lower frequencies with respect to those of the diazabutadienes, the explanation could be based on the change of the ligand conformation from trans to cis.
The ionic coordination compound 10 crystallized from CHCl3 and the X-ray diffraction analysis was performed, selected bond lengths and angles are in Table 4. The aluminum atom is bound to the nitrogen atoms, to a chlorine atom and to a methyl group, the [AlCl3Me]− anion neutralizes the cation.
|
|
|||
|---|---|---|---|
| Bond lengths | Bond angles | ||
| Al1–N1 | 1.956(5) | N1–Al1–N2 | 84.6(2) |
| Al1–N2 | 1.971(5) | C5–Al1–Cl1 | 121.7(2) |
| N1–C1 | 1.284(7) | C1–N1–Al1 | 110.1(4) |
| N2–C2 | 1.279(8) | C2–N2–Al1 | 110.2(4) |
| C1–C2 | 1.459(9) | N1–C1–C2 | 117.9(5) |
| Al1–C5 | 1.971(5) | N2–C2–C1 | 117.1(6) |
| Cl1–Al1 | 2.077(3) | C1–N1–C3 | 121.3(5) |
| N1–C3 | 1.508(7) | C2–N2–C4 | 121.7(5) |
| N2–C4 | 1.507(8) | ||
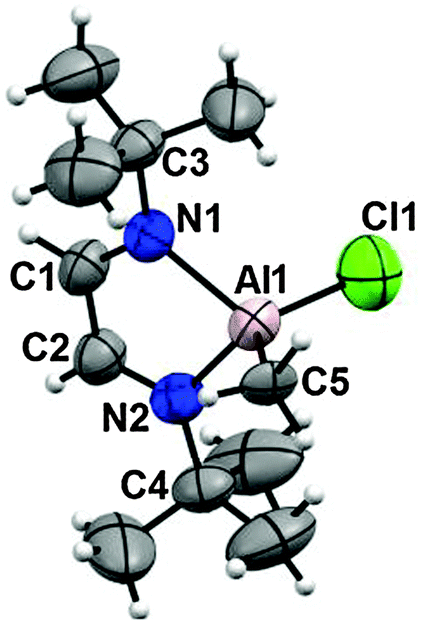
The tert-butyl protons have a short distance to the aluminum atom giving rise to anagostic interactions (2.78 and 2.99 Å, ΣvdWr = 3.3 Å29). The exocyclic N–C bonds of the tert-butyl groups present alternate conformations with respect to the ring plane. Each ring has two intermolecular Cl⋯π interactions, Fig. 2.25–27
 | ||
| Fig. 2 Intermolecular interactions in the crystal of compound 10. The aluminum compounds form chains by Cl⋯CC π contacts (Cl⋯C distances are in the range of 3.17–3.27 Å; ΣvdWr 3.55 Å28). | ||
Due to the fact that we were unable to get crystals for compounds 6 and 7 we decided to calculate their structures in order to get some information about the possible weak interactions of the substituent chlorine atoms. The minimum energy conformers for the aluminum cations in compounds 6 and 7 were calculated,28Fig. 3. The optimized conformer of compound 6 shows that a short contact of a chlorine atom with the aluminum (2.93 Å) atom and an H⋯Cl intramolecular hydrogen bond stabilize the structure. A short C–H⋯Al distance (2.85 Å) was also found. In the calculated structure of compound 7, the chlorine atoms are oriented towards the CN bonds with short distances (3.08 and 3.12 Å; ΣvdWr = 3.55 Å). The methyl protons have short distances to the aluminum atom (3.08 Å).
 | ||
| Fig. 3 Calculated minimum energy structures for cations 6 and 7. | ||
Reactions of 1,4-diazabutadienes 1–4 with aluminum halides in THF
Equimolar reactions of diazabutadienes 1–4 with AlX3 (X = Cl, I) in dry THF at −78 °C for 2 h were performed. Examination of the reaction mixtures by 27Al NMR in THF showed the presence of aluminates [AlCl4]− (δ27Al +101.3, Δ1/2 215 Hz) or [AlI4]− (δ27Al −20.2, Δ1/2 137 Hz). Reactions performed with AlI3 in the presence of CH2Cl2 afforded the corresponding [AlCl4]− anions. Solvent extraction of the reaction products and water washing gave the heterocycles stabilized by chloride anions. Compounds 11, 13 and 14 are brown solids, whereas 12 is a viscous liquid. Compounds were characterized by IR, mass spectrometry and elemental analyses. The 1H and 13C NMR spectra indicate the formation of 1,3-dialkyl-2-methaneiminealkyl imidazolium heterocycles 11–14, Scheme 5. To our knowledge the aluminum compounds’ transformation into the imidazolium heterocycles has not been described before. The new compounds show an imine group attached to C2, as a result of the condensation of one diazabutadiene with half of another diazabutadiene. 1,3-Dialkyl-2-methaneiminealkyl imidazoliums (11–14) are analogues of α,β-unsaturated acyl imidazolium cations, intermediates in polymer syntheses.30 They are also structurally related to imidazolium 2-carboxylates used as pre-catalysts for polyurethane formation.31 | ||
| Scheme 5 Transformation of compounds 5–8 into 11–14. | ||
A possible reaction path for the synthesis of 1,3-dialkyl-2-methaneiminealkyl imidazolium compounds under anhydrous conditions and in HCl was proposed earlier.32 A similar path can be depicted for the AlCl3 reaction. It is assumed that the aluminum coordination compounds 5–10 could suffer a nucleophilic attack from a diazabutadiene to one of the carbon atoms of the metallacycle with elimination of AlCl2NHR, Scheme 6.
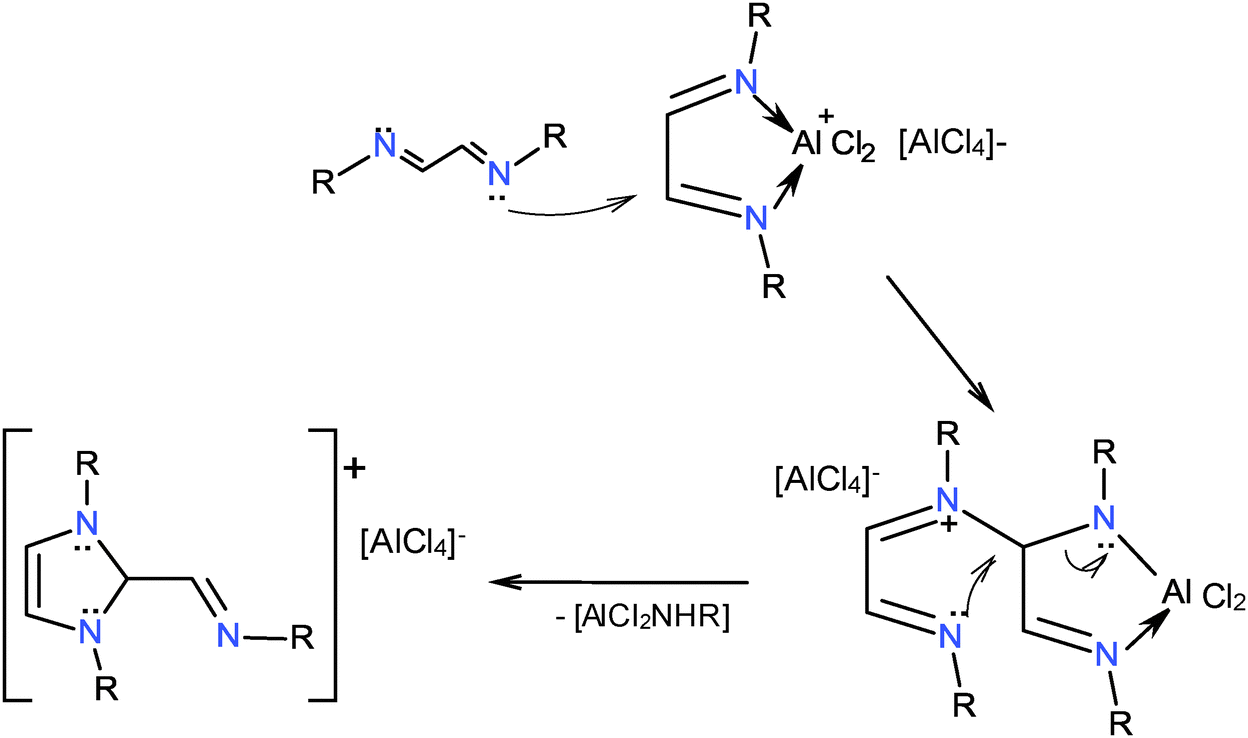 | ||
| Scheme 6 1,3-Dialkyl-2-methaneiminealkyl imidazolium heterocycles could be formed by the nucleophilic attack of diazabutadienes to the aluminum coordination compounds. | ||
Compound 11 crystallized from CHCl3. Selected bond lengths and angles are in Table 5. The exocyclic imine is almost coplanar with the imidazolium ring (the N–C–C–N dihedral angle is 13°). The benzylic carbon atoms of the intracyclic nitrogen atoms are also in the imidazolium plane. The phenyl groups are pointing to the opposite sides of the ring plane. The conformation of one of the endocyclic N-substituents is fixed by a hydrogen bond between the benzylic proton and the exocyclic imine (2.47 Å). The other endocyclic N-substituent presents some disorder due to its free rotation. One of the chlorine atoms of the [AlCl4]− anion has a Cl⋯C π-interaction (3.41 Å) with the imidazolium C2 (not shown).25–27
|
|
|||
|---|---|---|---|
| Bond lengths | Bond angles | ||
| C3–N1 | 1.345(5) | N1–C3–N2 | 107.6(3) |
| C3–N2 | 1.344(5) | C3–N1–C1 | 108.4(3) |
| C1–N1 | 1.373(5) | C3–N2–C2 | 108.5(3) |
| C2–N2 | 1.368(5) | C2–C1–N1 | 107.5(3) |
| C2–C1 | 1.338(6) | C1–C2–N2 | 107.9(3) |
| C3–C4 | 1.465(5) | N3–C4–C3 | 120.8(3) |
| C4–N3 | 1.243(5) | C3–N1–C5 | 127.2(3) |
| C5–N1 | 1.503(5) | C3–N2–C6 | 126.5(3) |
| C6–N2 | 1.483(5) | C4–N3–C7 | 117.6(3) |
| C7–N3 | 1.467(5) | N1–C3–C4 | 124.4(3) |
| N2–C3–C4 | 128.0(3) | ||

The reaction of 4 with AlCl3 afforded crystals of 14 with [AlCl4]− as the anion whereas the AlI3 reaction product treated with water and extracted with CH2Cl2 afforded crystals of 14 with [I3]− as the anion. The two crystals were subjected to the X-ray diffraction analyses; selected bond lengths and angles are in Table 6.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| 14[AlCl4] | 14[I3] | ||||||
| Bond lengths | Bond angles | Bond lengths | Bond angles | ||||
| N1–C3 | 1.339(6) | N2–C3–N1 | 107.9(4) | C3–N1 | 1.28(3) | N1–C3–N2 | 112(2) |
| N2–C3 | 1.333(6) | C3–N1–C1 | 108.7(4) | C3–N2 | 1.34(2) | C3–N1–C1 | 107(2) |
| N1–C1 | 1.391(6) | C3–N2–C2 | 107.8(4) | C1–N1 | 1.39(3) | C3–N2–C2 | 104(2) |
| N2–C2 | 1.372(6) | C2–C1–N1 | 106.1(5) | C2–N2 | 1.41(3) | C2–C1–N1 | 108(2) |
| C2–C1 | 1.315(7) | C1–C2–N2 | 109.5(5) | C2–C1 | 1.29(3) | C1–C2–N2 | 109(2) |
| C3–C4 | 1.482(7) | N3–C4–C3 | 119.1(5) | C3–C4 | 1.49(3) | N3–C4–C3 | 121(2) |
| N3–C4 | 1.240(6) | C3–N1–C5 | 128.9(4) | C4–N3 | 1.26(3) | C3–N1–C5 | 130(2) |
| N1–C5 | 1.518(6) | C3–N2–C6 | 127.2(4) | C5–N1 | 1.52(3) | C3–N2–C6 | 131(2) |
| N2–C6 | 1.522(6) | C4–N3–C7 | 123.9(7) | C6–N2 | 1.47(3) | C4–N3–C7 | 123(2) |
| N3–C7 | 1.50(2) | C7–N3 | 1.48(3) | ||||

The cation has the same conformation in both crystals. The plane of the exocyclic imine is perpendicular to the imidazolium ring, Fig. 4A. This conformation is different from that of 11, and is attributed to the steric effect. Two tert-butyl methyl groups have two C–H⋯C π-interactions (2.32 and 2.34 Å) with the exocyclic imine. The imidazolium ring is aromatic (bond lengths vary from 1.34 to 1.37 Å). In the crystal of 14 [AlCl4]−, one chlorine atom has a π contact25–27 with the imidazolium C2, Fig. 4B. The X-ray diffraction structure of 1,3-di-tert-butyl-2-[N-tert-butylmethanimine]-imidazolium having Cl− as the anion is known. The compound was synthesized by reaction of the diazabutadiene with dry HCl in anhydrous toluene.32
 | ||
| Fig. 4 X-Ray diffraction structure of imidazolium 14. (A) The cation has four hydrogen bonds to the exocyclic CN bond (2.37–2.95 Å). (B) One chlorine atom of the anion has a Cl⋯C2 π-interaction. | ||
Optimization of the minimum energy conformations of 1,3-dialkyl-2-methaneiminealkyl imidazolium compounds 11–14 was performed.28 The calculated structures of cations 11 and 14 are similar to their X-ray diffraction structures; therefore we have concluded that calculated cations 12 and 13 (Fig. 5) could be used to analyze their conformations and possible weak interactions. In both structures the exocyclic imines are coplanar with the imidazolium ring due to the electronic delocalization and formation of a CH⋯N hydrogen bond [C–H⋯N distances: 2.27 Å (12) and 2.17 Å (13)]. The N-tertiary carbon reduces the steric effect and allows formation of a stabilizing hydrogen bond. In cations 12 and 13 some chlorine atoms are oriented towards the π electrons of the ring nitrogen atoms (12 3.15 Å; 13 3.05 Å).
 | ||
| Fig. 5 Optimized minimum energy conformers for imidazolium heterocycles 12 and 13. | ||
In order to know the contribution of the exocyclic imine to the stabilization of the molecule, we have calculated the energy of two conformers for the hypothetical molecule 1,3-di(methyl)-2-[N-methylmethanimine]-imidazolium, where the steric effect has been minimized. We have found that the coplanar conformation of the exocyclic imine is more stable than the perpendicular conformation by 13.6 kJ mol−1, Fig. 6.
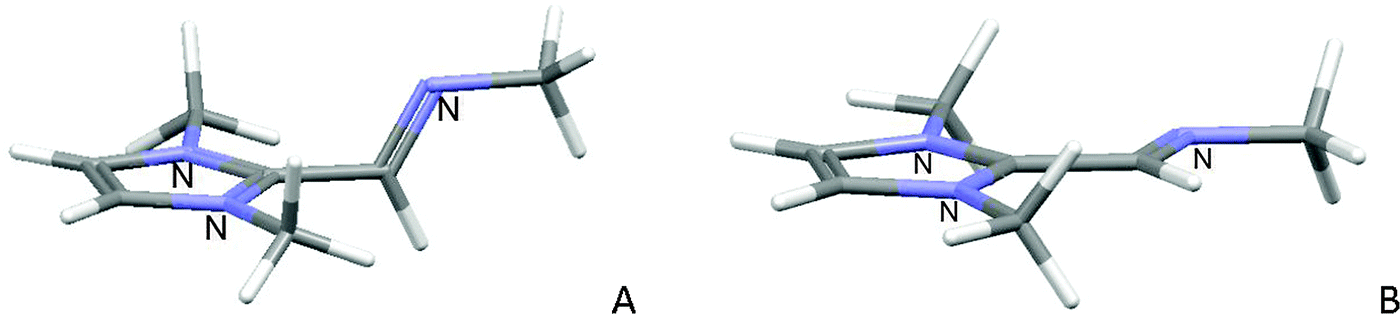 | ||
| Fig. 6 Calculated minimum energy conformers for 1,3-dimethyl-2-[N-methylmethanimine]-imidazolium. | ||
Reactions of 1,4-diazabutadienes 1–4 with InCl3 in acetonitrile
Reactions of compounds 1–4 with one or two equivalents of InCl3 in dry acetonitrile and a N2 atmosphere at −78 °C gave the indium coordination compounds 15–18, Scheme 7. Neutral and diamagnetic indium coordination compounds derived from 1,4-diazabutadienes are not described in the literature. Compounds are stable in the solid state under dry conditions. In the IR spectra the CN bands of the starting diazabutadienes (1633 cm−1) become broad and are shifted towards lower frequencies (1605 cm−1). In the NMR spectra, the C2–H protons signals are shifted to the higher frequencies with respect to the starting diazabutadienes, whereas the chemical shifts of C2 are shielded (between 2–4 ppm) probably due to the diazabutadiene conformational change. A similar behavior was found for the aluminum compounds 5, 8–10.
 | ||
| Scheme 7 Synthesis of indium compounds 15–18. | ||
We have followed the reactions of compound 3 with one and two equivalents of InCl3 in THF by 1H and 13C NMR at low temperature. The spectra of the reaction of 3 with one equivalent of InCl3 in TDF at −65 °C show at least five compounds out of which one was predominant [1H: 8.9 (H1), 5.9 (H4), 4.4 (H3), 1.3 (H5) ppm; 13C: 158.7 (C1), 138.7 (Ci), 75.9 (C4) and 19.0 (C5) ppm]. All signals were very broad, indicating isomers in equilibrium. When the solution was heated at +20 °C, only one set of broad signals was observed [1H: 8.0 (H2), 5.0 (H4), 3.7 (H3), 1.0 (H5) ppm; 13C: 161.7 (C2), 139.1 (Ci), 71.9 (C4), 65.0 (C3) and 19.0 (C5) ppm], which reveals that isomers are in fast equilibrium and that their signals were averaged, Scheme 8. It is assumed that the indium is coordinated by nitrogen and chlorine atoms because, all carbon and hydrogen atoms showed broad signals, Scheme 9.
 | ||
| Scheme 8 Proposed equilibrium between isomers in the InCl3 coordination compound. | ||
 | ||
| Scheme 9 Proposed structure for a dinuclear InCl3 coordination compound. | ||
The reaction of 3 with two equivalents of InCl3 in TDF at −65 °C showed seven sets of broad signals attributed to isomers of coordination compounds. When the solution reached +20 °C, only two sets of broad signals were observed, indicating the existence of fast equilibrium between the isomeric species. One of the signal sets corresponds to that observed in the spectrum of the equimolar reaction (∼35%). Whereas, the second set [1H: 7.8 (H2), 5.6 (H4), 4.4 (C3) and 1.3 (5) ppm; 13C: 159.1 (C2), 138.6 (Ci), 71.9 (C4), 66.0 (C3) and 18.5 (C5) ppm] was attributed to the complex of the 1,4-diazabutadiene with two molecules of InCl3 (∼65%), Scheme 9. Calculation of the optimized structure is shown in Fig. 7. The In⋯Cl distances (3.61 Å, ΣvdWr = 4.0 Å29) indicate that the C–Cl chlorine atoms have stabilizing contacts with the indium atom. A similar fluxional behavior was found for a platinum derivative of ligand 4.33
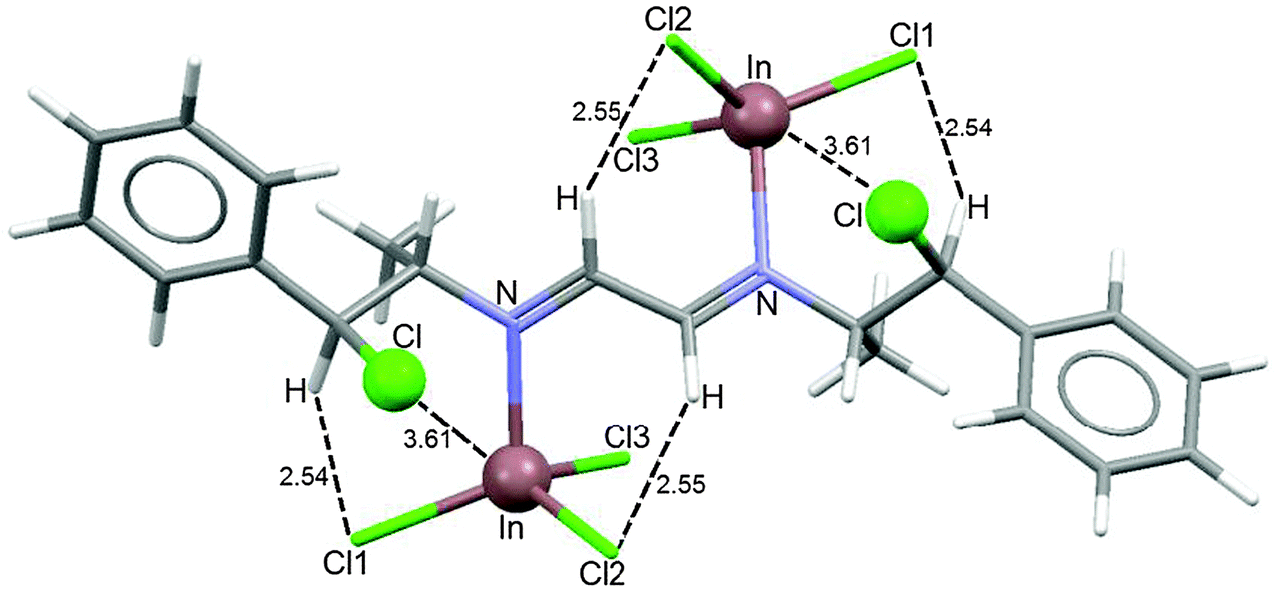 | ||
| Fig. 7 Calculated minimum energy structure for diazabutadiene 3 coordinated to two InCl3. | ||
From the reaction of diazabutadiene 3 with InCl3, a solid was obtained which was dissolved in THF and crystals of compound 17 suitable for X-ray diffraction analyses were obtained. Selected bond lengths and angles are in Table 7.
|
|
|||
|---|---|---|---|
| Bond lengths | Bond angles | ||
| In–N1 | 2.313(7) | N1–In–N2 | 72.2(2) |
| In–N2 | 2.380(6) | Cl4–In–Cl5 | 95.06(8) |
| N1–C1 | 1.242(9) | Cl3–In–O1 | 172.4(2) |
| N2–C2 | 1.26(1) | N1–In–Cl4 | 98.3(2) |
| C1–C2 | 1.50(1) | N2–In–Cl5 | 93.6(2) |
| N1–C3 | 1.453(9) | C1–N1–In1 | 114.8(6) |
| N2–C4 | 1.492(9) | C2–N2–In1 | 112.1(5) |
| Cl1–C5 | 1.817(9) | C1–N1–C3 | 116.3(7) |
| Cl2–C7 | 1.825(9) | C2–N2–C4 | 117.9(7) |
| In–Cl3 | 2.438(2) | C3–C5–Cl1 | 106.9(6) |
| In–Cl4 | 2.434(2) | C4–C7–Cl2 | 108.7(7) |
| In–Cl5 | 2.427(2) | ||
| In–O1 | 2.287(6) | ||

It was found that the indium atom is hexacoordinated with an octahedral geometry. It is chelated by the diazabutadiene and coordinated to three chlorine atoms and to one THF molecule. The THF is located perpendicular to the metallacycle plane. The C–Cl atoms are in opposite faces of the ring with short distances to the nitrogen atoms (3.06 and 3.15 Å). The coordinated oxygen atom has a planar geometry attributed to sp2 hybridization. The chlorine atoms situated in the metallacycle plane have hydrogen bonds with the methyl and benzylic groups Cl4⋯H–C19 (2.643 Å), Cl5⋯H–C6 (2.641 Å). Intermolecular interactions are depicted in Fig. 8.
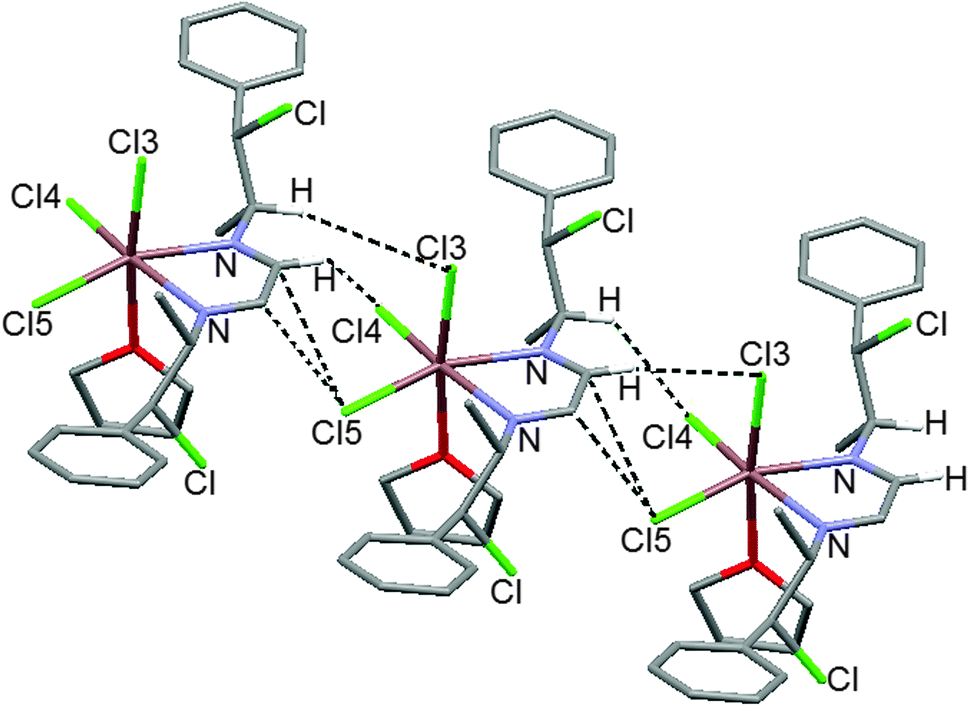 | ||
| Fig. 8 The indium coordination compound 17 forms chains by cooperative C–H⋯Cl hydrogen bonds (Cl3⋯H–C3 2.762 Å, Cl4⋯H–C3 2.865 Å) and Cl5⋯π-interactions [Cl5⋯C3 (3.35 Å); Cl5⋯C4 (3.24 Å)]. | ||
To our knowledge there is only another X-ray diffraction analysis reported for an indium compound where the metal atom is coordinated to a 1,4-diazabutadiene.15 It is a paramagnetic compound of formula LInCl2·THF (L = 1,4-diisopropylphenyl-1,4-diazabutadiene), in the reported compound the indium is pentacoordinated, the THF is in the anti position to one nitrogen atom and the ligand is a radical monoanion.
The optimized conformation for compound 17 is similar to its X-ray diffraction analysis structure; therefore calculations could provide a good approach for determining the structure of coordinated indium compounds that could not be crystallized. The calculated minimum energy conformer29 for compound 16 coordinated to THF is shown in Fig. 9. The chlorine atoms in the structure are oriented towards the imine π electrons.
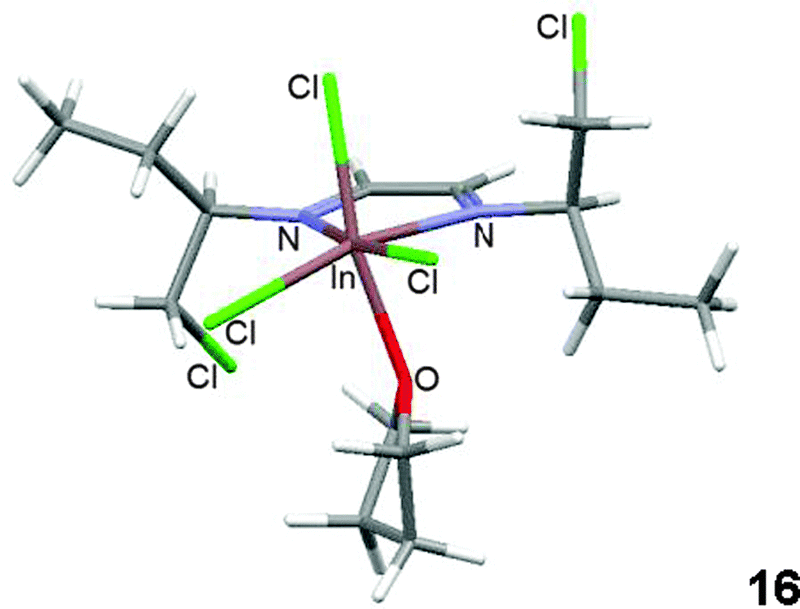 | ||
| Fig. 9 Calculated structure of the coordination compound 16. | ||
Reactions of diazabutadienes 1–4 with InCl3 in THF
The reactions of ligands 1–4 with InCl3 in THF at room temperature were followed by NMR. The formation of the indium coordination compounds was observed, and then the signals for the 1,3-dialkyl-imidazolium tetrachloroindates 19–22 slowly emerged, Scheme 10. The transformations attained after one week were 21% for 19; 98% for 20, 25% for 21 and 25% for 22. It was observed that these reactions slowed down in the dark; therefore they could be photo-induced.34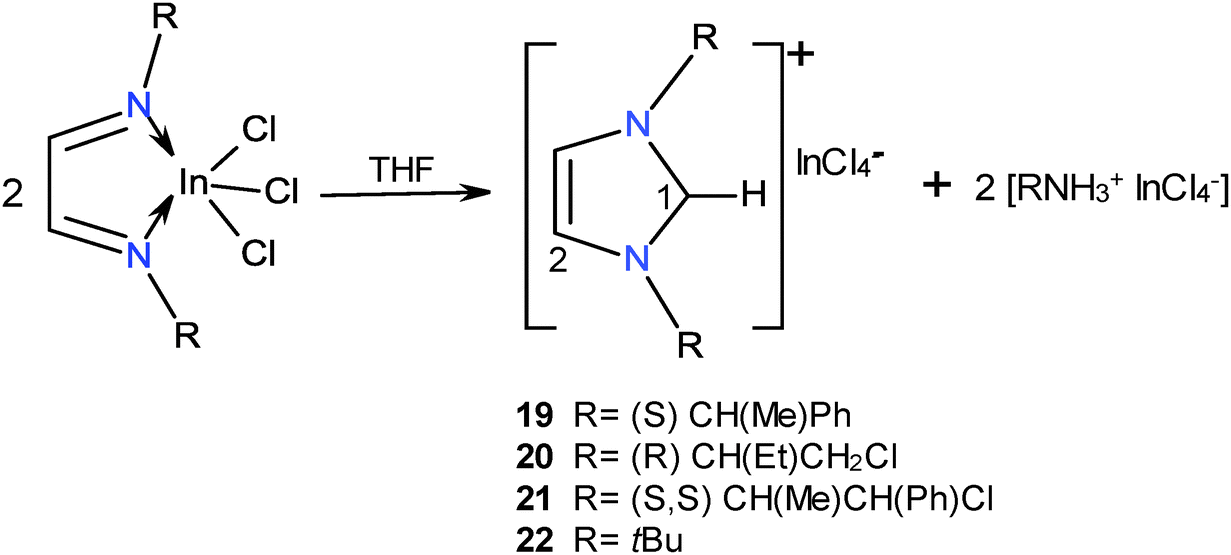 | ||
| Scheme 10 Synthesis of compounds 19–22. | ||
The transformation of the ionic indium coordination compound into the imidazoliums 19–21 is interesting because the only evident source for the carbon introduced into the imidazole ring was the ligand itself. Therefore it is assumed that the C–H imidazolium compounds may come from the 1,3-dialkyl-2-methaneiminealkyl imidazolium derivatives (11–14), which in the presence of InCl3 are transformed into the relatively stable carbene-InCl3.35–37 The latter by reaction with labile protons affords the C–H derivatives, Scheme 11.
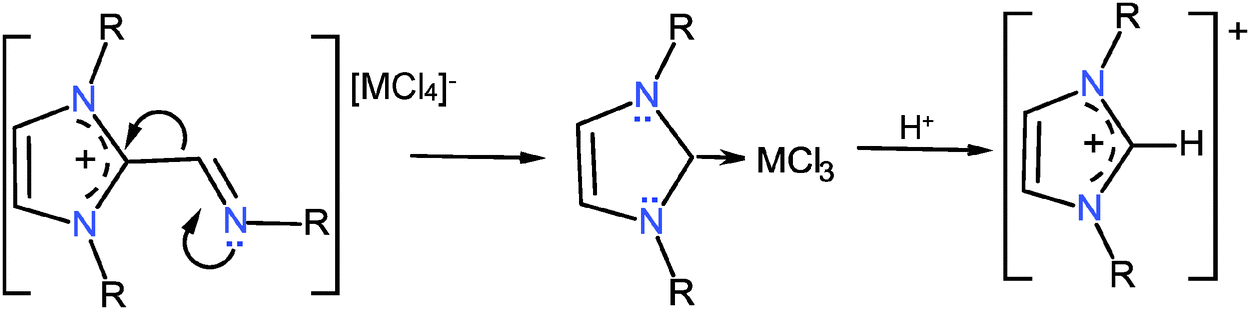 | ||
| Scheme 11 Plausible transformation of 1,3-dialkyl-2-methaneiminealkyl imidazolium into the 1,3-dialkyl-imidazolium, promoted by InCl3 through an intermediate carbene which is protonated. | ||
In order to check the latter statement, we have isolated the chloride of the 1,3-dialkyl-2-methaneiminealkyl imidazolium 12, dissolved it in CH2Cl2 and added one equivalent of InCl3, afterwards the reaction was followed by 1H and 13C NMR. Two weeks later compound 12 was transformed into compound 20 (60%). An inverse reaction is known for the alkylation of an imidazole carbene by an α,β-unsaturated enol ester.38 The observation of these transformations is relevant because 1,3-dialkyl-imidazolium compounds are precursors of imidazole carbenes; important ligands for metal derivatives39 also used as reagents40 and as ionic liquids.41 Optically active carbenes are catalyzing agents for enantiosynthesis.42,43
The structure of compounds 19–22 was determined by NMR. The aromatic character of the ring is denoted by the 1H and 13C NMR chemical shifts. C1–H protons for 19–22 appear in the range of 8.3–10.63 ppm which indicates its positive character, whereas C2–H is observed at 7.14–8.08 ppm. 13C NMR C1 signals are found at 130.2–138.4 ppm and C2 appears between at 120.7 and 122.4 ppm. The 1,3-dialkyl-imidazoliums 19–22 were observed in the (+)TOF mass spectra and the [InCl4]− anion in the (−)TOF spectra.
Calculations of the minimum energy structures of 1,3-dialkylimidazolium heterocycles show that there is no steric effect between the ring and the N-substituents, which freely rotate.28 The rotational barrier for the t-butyl group rotamers in compound 22 was calculated to be 2.57 kJ mol−1.
Conclusions
Two new 1,4-dialkyl-1,4-diazabutadienes bearing two optically active N-substituents: 1-chlorobutan-2-yl (2) and 1-chloro-1-phenyl-propan-2-yl (3) and their X-ray diffraction analyses are reported. The solid state conformation showed that the diazabutadiene system adopts the more stable anti conformation with imines in E configuration.The reactions of diazabutadienes 1–4 with aluminum halides in toluene afforded unstable ionic coordination compounds. The X-ray diffraction of one of them was obtained. The aluminum coordination compounds in THF solution are slowly and completely transformed into 1,3-dialkyl-2-methaneiminealkyl imidazolium heterocycles (11–14). This transformation has not been described before. The X-ray diffraction analysis of compounds 11 and 14 indicated that the conformation of the exocyclic imine was determined by the steric effect of the nitrogen substituents. Calculations showed that the most stable conformations were those found in the solid state.
Reactions of InCl3 with diazabutadienes 1–4 in acetonitrile afforded the corresponding neutral and diamagnetic indium coordination compounds 15–18. To our knowledge these are the first examples of diamagnetic and neutral indium coordination compounds derived from 1,4-diazabutadiene ligands. They are stable in the solid state and could be isolated. The X-ray diffraction analyses of the THF adduct of the indium coordination compound 17 was obtained.
The reactions of InCl3 in THF also gave the neutral diamagnetic InCl3 coordination compounds 15–18, however in the THF solution they are slowly transformed into the 1,3-dialkyl imidazolium heterocycles 19–21. This transformation has not been reported before in the literature. The origin of 1,3-dialkylimidazolium compounds 15–18 could be explained, if it is assumed that 1,3-dialkyl-2-methaneiminealkyl imidazoliums are previously formed and cleaved by the InCl3 to give the corresponding carbenes stabilized by the InCl3. Consequent reactions of the carbenes with labile protons afford the C–H imidazolium heterocycles (19–22).
It was found that in the X-ray diffraction analyses and in calculations of compounds derived from ligands 2 and 3 that the chlorine atoms have stabilizing contacts with the π system of the imines.
In the reactions of AlCl3 and InCl3 with 1,4-dialkyl-1,4-diazabutadienes (1–4) the nature of the metal halides, the nitrogen substituents and the solvent determine the reaction products. The two metal coordination compounds transformations into imidazolium derivatives are of interest in the heterocyclic chemistry.
Experimental section
General remarks
Reagents were purchased from Sigma-Aldrich Chemical, Fluka Chemika and Strem Chemical, and were not purified. Vacuum line techniques were employed for all manipulations of air and moisture sensitive compounds. THF, toluene, CH2Cl2 and acetonitrile were dried prior to use44 Dry CDCl3, DMSO-d6, THF-d8, were purchased from Aldrich and used without further purification. (1S,2S)-1-Chloro-1-phenyl-2-aminepropane hydrochloride was prepared as reported,24 as well as compounds 145,46 and 4.47Melting points were obtained using a Mel-Temp II apparatus and are uncorrected. Mass spectra were obtained by LC/MSD TOF on an Agilent Technologies instrument with ESI as an ionization source. Elemental analyses were performed on Flash (EA) 1112 series equipment. IR spectra were recorded on a KBr disc using a FT Spectrum GX Perkin Elmer spectrometer. NMR spectra were obtained on a Jeol GSX-270, Jeol Eclipse 400 MHz and Bruker Avance 300 MHz. 1H, 13C, 27Al [Ξ 26.077, Al(NO3)3]. Numbering of atoms for identification of the NMR signals is shown in Scheme 2.
Calculations were performed in order to obtain the molecular geometries using the Gaussian 0328 using DFT and the hybrid method B3LYPP/6-31+G(d,p). For indium compounds, the base used was 3-21G. Geometries were checked to be the minimal by the frequency analysis.
Crystallographic data were measured using a Nonius Kappa CCD instrument with a CCD area detector using graphite-monochromated MoKα radiation. Intensities were measured using φ+ω scans. Crystal data are in Tables 8 and 9. Structures were solved using direct methods with SHELX-97,48 Sir 2002 and Sir 2004.49
| Compd | 2 | 3(S,S) | 10 | 11 |
|---|---|---|---|---|
| w = 1/[s2(Fo2) + (aP)2 + bP] where P = (Fo2 + 2Fc2)/3. | ||||
| Empirical formula | C10H18Cl2N2 | C20H22Cl2N2 | C11H23AlClN2CH3AlCl3 | C28H30N3AlCl4 |
| Formula weight | 237.16 | 361.30 | 394.11 | 577.33 |
| Crystal size [mm] | 0.4 × 0.2 × 0.2 | 0.23 × 0.05 × 0.05 | 0.4 × 0.15 × 0.05 | 0.5 × 0.4 × 0.35 |
| Crystal shape | Prism | Needle | Prism | Prism |
| Crystal color | Colorless | Colorless | Colorless | Colorless |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P212121 | P21 | Pbca | P21 |
| a [Å] | 13.2941 (5) | 5.6218 (2) | 11.6141 (3) | 6.988 (10) |
| b [Å] | 4.6884 (2) | 26.6363 (11) | 17.4168 (5) | 23.822 (2) |
| c [Å] | 21.0334 (10) | 6.8310 (2) | 21.0756 (8) | 9.334 (5) |
| α [°] | 90.000 | 90.000 | 90.000 | 90.000 |
| β [°] | 90.000 | 113.898 (2) | 90.000 | 101.68 |
| γ [°] | 90.000 | 90.000 | 90.000 | 90.000 |
| V [Å3] | 1310.97 | 935.20 (6) | 4263.2 (2) | 1521 (9) |
| Z | 4 | 2 | 8 | 2 |
| D x (calcd) (mg m−3) | 1.202 | 1.279 | 1.228 | 1.26 |
| μ [mm−1] | 0.46 | 0.35 | 0.63 | 0.44 |
| F(000) | 504 | 380 | 1648 | 600 |
| Temperature [K] | 293 | 173 | 293 | 293 |
| θ range for data collection | 4.2–27.5 | 1–27.5 | 2.9–27.5 | 0.8–27.5 |
| Index ranges | −17 ≤ h ≤ 17 | −7 ≤ h ≤ 7 | −14 ≤ h ≤ 14 | −8 ≤ h ≤ 8 |
| −6 ≤ k ≤ 5 | −20 ≤ k ≤ 34 | −22 ≤ k ≤ 22 | −29 ≤ k ≤ 30 | |
| −27 ≤ l ≤ 27 | −8 ≤ l ≤ 8 | −27 ≤ l ≤ 27 | −12 ≤ l ≤ 12 | |
| Reflections measured | 5462 | 6597 | 8869 | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 617 617 |
| Independent reflections | 2940 | 2908 | 4677 | 6327 |
| Reflections observed [I > 2σ(I)] | 1788 | 2618 | 1948 | 4427 |
| R(int) | 0.056 | 0.035 | 0.105 | 0.032 |
| Number of parameters | 177 | 305 | 189 | 294 |
| Weighting scheme R/wR | 0.0497/0.1768 | 0.0282/0.4729 | 0.1191/3.1453 | 0.065/0.6828 |
| GOOF | 1.02 | 1.06 | 1.02 | 1.03 |
| R [F > 2σ(F2)] | 0.051 | 0.039 | 0.088 | 0.062 |
| wR (F2) | 0.131 | 0.089 | 0.278 | 0.163 |
| Largest residual peak [e Å−3] | 0.21, −0.16 | 0.24, −0.27 | 0.52, −0.39 | 0.42, −0.28 |
| Compd | 14[AlCl4] | 14[I3] | 17 | Chlorohydrate |
|---|---|---|---|---|
| w = 1/[s2(Fo2) + (aP)2 + bP] where P = (Fo2 + 2Fc2)/3. | ||||
| Empirical formula | C16H31N3AlCl4 | C16H30N3I3 | C24H30Cl5InN2OC4H8O | C4H11ClNCl |
| Formula weight | 433.21 | 645.13 | 726.67 | 144.04 |
| Crystal size [mm] | 0.38 × 0.3 × 0.15 | 0.25 × 0.13 × 0.03 | 0.25 × 0.05 × 0.05 | 0.45 × 0.2 × 0.15 |
| Crystal shape | Prism | Fragment | Needle | Prism |
| Crystal color | Colorless | Red | Colorless | Colorless |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic | Tetragonal |
| Space group | Pbca | P21/c | P212121 | I4 |
| a [Å] | 10.9102 (3) | 13.533 (2) | 6.9619 (2) | 14.5159 (3) |
| b [Å] | 20.2966 (5) | 11.4237 (13) | 13.4095 (5) | 14.5159 (3) |
| c [Å] | 21.7692 (7) | 18.787 (2) | 35.1631 (9) | 7.1671 (2) |
| α [°] | 90.000 | 90.000 | 90.000 | 90.000 |
| β [°] | 90.000 | 125.466 (8) | 90.000 | 90.000 |
| γ [°] | 90.000 | 90.000 | 90.000 | 90.000 |
| V [Å3] | 4820.6 (2) | 2365.5 (5) | 3282.67 (18) | 1510.19 (8) |
| Z | 8 | 4 | 4 | 8 |
| D x (calcd) (mg m−3) | 1.194 | 1.811 | 1.47 | 1.267 |
| μ [mm−1] | 0.53 | 3.97 | 1.15 | 0.76 |
| F(000) | 1824 | 1224 | 1480 | 608.0 |
| Temperature [K] | 293 | 293 | 173 | 293 |
| θ range for data collection | 3.3–27.5 | 0.8–27.5 | 1.0–27.5 | 0.8–27.5 |
| Index ranges | −13 ≤ h ≤ 13 | −16 ≤ h ≤ 15 | −9 ≤ h ≤ 5 | −18 ≤ h ≤ 18 |
| −25 ≤ k ≤ 26 | −13 ≤ k ≤ 14 | −17 ≤ k ≤ 10 | −18 ≤ k ≤ 17 | |
| −28 ≤ l ≤ 28 | −18 ≤ l ≤ 23 | −45 ≤ l ≤ 28 | −9 ≤ l ≤ 9 | |
| Reflections measured | 10069 |
4431 | 14201 |
11708 |
| Independent reflections | 5386 | 3093 | 7082 | 1686 |
| Reflections observed [I > 2σ(I)] | 2246 | 1598 | 3687 | 1490 |
| R(int) | 0.154 | 0.065 | 0.088 | 0.041 |
| Number of parameters | 279 | 209 | 428 | 108 |
| Weighting scheme R/wR | 0.0983/3.5245 | 0.1434/27.4624 | 0.0476 | 0.030/0.1586 |
| GOOF | 1.02 | 1.15 | 0.99 | 1.09 |
| R [F > 2σ(F2)] | 0.088 | 0.109 | 0.071 | 0.026 |
| wR (F2) | 0.251 | 0.366 | 0.144 | 0.059 |
| Largest residual peak [e Å−3] | 0.38, −0.34 | 1.04, −1.60 | 0.90, −0.60 | 0.12, −0.16 |
The refinement for all structures (based on F2 of all data) was performed by full matrix least-squares techniques using Crystals 12.84.50 All non-hydrogen atoms were refined anisotropically. Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre with numbers: 2 (960655), 3(S,S) (960656), 3(meso) (960657), 10(960658), 11 (960659), 14(AlCl4)− (960660), 14(I3)− (960661), 17 (960662), (R)-(−)-1-chloro-butan-2-amine hydrochloride (960663).
Synthesis of the compounds
:1] (100 mL), Na2CO3 (3.71 g, 35 mmol) and then aq. glyoxal 40% (2 mL, 17.5 mmol) were slowly added and stirred for 15 h at rt. The reaction mixture was extracted with CH2Cl2, and dried with Na2SO4, filtered and the solvent evaporated. Compound 2 is a yellow solid (5.8 g, 70%). Mp 56–58 °C. [α]D = + 292.45 (EtOH, 25 °C). NMR (CDCl3, 25 °C; δ ppm), 1H: 7.89 (s, 2H, H2), 3.23 (m, 2H, H3), 3.67 (dd, 3J 4.35 Hz, 2H, H4a), 3.57 (dd, 3J 7.70 Hz, 2H, H4b), 1.60 (qdd, 3J 4.07 Hz, 2H, H5a), 1.77 (qdd, 3J 4.22 Hz, 2H, H5b), 0.83 (t, 3J 7.44 Hz, 6H, H6). 13C:162.5 (C2), 73.6 (C3), 47.5 (C4), 26.5 (C5), 10.5 (C6). IR (KBr, νmax cm−1): 1625 (CN), 729 (C–Cl). Anal. calcd for [C10H18Cl2N2]: C, 50.64; H, 7.65; N, 11.81%; found: C, 50.58; H, 8.08; N, 11.62%.
:1] (100 mL), and 40% aq. glyoxal (1.4 mL, 12 mmol). The reaction mixture was extracted with CH2Cl2, and dried with Na2SO4, filtered and the solvent evaporated. Compound 2 (1R,2R) is a yellow solid, it crystallized from EtOH–CH2Cl2 (8.3 g, 96%). Mp 120–122 °C. [α]D −138 (CH2Cl2, 25 °C). NMR (CDCl3, 25 °C δ ppm), 1H: 8.06 (s, 2H, H2), 3.78 (dq, 3J 7.68 and 6.59 Hz, 2H, H3), 4.92 (d, 3J 7.68 Hz, 2H, H4), 1.05 (d, 3J 6.59 Hz, 6H, H5), 7.36 (m, 10H, H–Ph). 13C: 162.3 (C2), 72.3 (C3), 67.3 (C4), 20.2 (C5), 138.9 (Ci), 128.7 (Co), 128.7 (Cm), 127.9 (Cp). IR (KBr, νmax cm−1): 1632 (CN), 736 (C–Cl). Anal. calcd for [C20H22Cl2N2]: C, 66.49; H, 6.14; N, 7.75%; found: C, 66.11; H, 6.18; N, 7.68%.
N), 1482 (C–N), 1381 (C–C). Anal. calcd for [C10H20N2Al2Cl6]: C, 27.61; H, 4.63; N, 6.44%; found: C, 27.11; H, 4.43; N, 6.25%.
N): Anal. calcd for [C10H20N2Al2I6]: C, 12.21; H, 2.05; N, 2.85%; found: C, 11.88%, H, 2.42; N, 2.96%.
N).
N]. (+)TOF, m/z (amu): calcd for [C28H30N3]+: 408.2434; found: 408.2436. (−)TOF, m/z (amu): calcd. for [AlCl4]− 126.9045; found: 126.9055. Anal. calcd for (C28H38N3AlCl4)5(CHCl3)3: C, 52.28; H, 5.92; N, 6.40%; found: C, 52.33; H, 5.65; N, 6.54%.
N], 1677 [CN], 1554 [C–N]. (+)TOF, m/z (amu): calcd for [C31H33N3Cl3]+, 552.1734; found, 552.1742. Anal. calcd for (C31H33N3Cl4·3H2O): C, 57.86; H, 6.11; N, 6.53%; found: C, 58.54; H, 6.12; N, 6.65%.
Crystallization from methanol affords compound 14 with I3 as the anion. Mp 146–148 °C. (+)TOF, calcd for [C16H30N3]+, m/z (amu): 264.2434; found: 264.2435. (−)TOF, calcd. for [I]−, m/z (amu): 126.9045; found: 126.9048. NMR (CDCl3, 25 °C; δ ppm), 1H: 7.43 (s, 2H, H2), 1.59 (s, 18H, H4), [substituent in C1: 8.37 (s, 1H, H2′), 1.31 (s, 9H, H4′)] 13C: 119.5 (C2), 140.2 (C1), 63.1 (C3), 30.2 (C4), [substituent in C1: 145.4 (C2′), 61.4 (C3′), 27.8 (C4′)]. IR (KBr, νmax cm−1): 1641 [CN], 1596 [C–N]. Anal. calcd for [C16H30N3I3·1/2I]: C, 27.12; H, 4.27; N, 5.93%; found: C, 27.42; H, 4.34; N, 6.33%. Crystals were subjected to X-ray diffraction analyses.
Crystallization from CHCl3 gave compound 14 having AlCl4 as the anion. (+)TOF, m/z (amu): calcd for [C16H30N3]: 264.2435; found: 264.2437. (−)TOF, m/z (amu): calcd for [AlCl4]− 126.9045; found: 126.9055. Crystals were subjected to X-ray diffraction analyses.
N, Ph).
N). Anal. calcd for [C10H18N2Cl5In·1/2CH3CN]: C, 29.15; H, 4.48; N, 5.67%; found: C, 29.43; H, 4.56; N, 5.41%.
N, Ph). Anal. calcd for [C20H22Cl5N2In·1/2(C4H8O)]: C, 42.17%, H, 4.18; N, 4.47%; found: C, 41.97; H, 4.65; N, 4.50%.
N). Anal. calcd for [C10H20N2Cl3In]: C, 30.85; H, 5.14; N, 7.20%; found: C, 30.73; H, 5.66; N, 5.63%.
N], 1611 [Ph]. (+)TOF, m/z (amu): calcd for [C19H21N2]+ 277.1699; found 277.1703. (−)TOF, m/z (amu): calcd for [InCl4]− 254.7798, found 254.7754.
Acknowledgements
H.R-S. and A.M.D-H. thank Conacyt for PhD scholarships. We are grateful to Cinvestav for the facilities of the supercomputer HPC-cluster Xiuhcoatl. We thank Professor Angeles Paz-Sandoval for helpful discussions.References
- G. Chelucci, Coord. Chem. Rev., 2013, 257, 1887 CrossRef CAS PubMed.
- Ch. P. Pradeep and S. K. Das, Coord. Chem. Rev., 2013, 257, 1699 CrossRef CAS PubMed.
- J. F. Larrow and E. N. Jacobsen, Top. Organomet. Chem., 2004, 6, 123 CAS.
- M. Sodeoka and Y. Hamashima, Pure Appl. Chem., 2008, 80, 763 CrossRef CAS.
- V. M. Jiménez-Pérez, A. Ariza-Castolo, A. Flores-Parra and R. Contreras, J. Organomet. Chem., 2006, 691, 1584 CrossRef PubMed.
- R. Contreras, A. Flores-Parra, H. C. López-Sandoval and N. Barba-Behrens, Coord. Chem. Rev., 2007, 251, 1852 CrossRef CAS PubMed.
- G. Vargas-Díaz, H. López-Sandoval, A. B. Vázquez-Palma, M. Flores-Alamo, A. Peña-Hueso, S. A. Sánchez-Ruiz, A. Flores-Parra, R. Contreras and N. Barba-Behrens, J. Chem. Soc., Dalton Trans., 2007, 251, 1852 Search PubMed.
- R. Salas-Coronado, R. Colorado-Peralta, S. A. Sánchez-Ruiz and R. Contreras A. Flores-Parra, J. Organomet. Chem., 2009, 694, 616 CrossRef CAS PubMed.
- R. Ramírez-Trejo, A. Flores-Parra, J. A. Peña-Hueso, E. Mijangos, R. Contreras and N. Barba-Behrens, Polyhedron, 2010, 29, 1007 CrossRef PubMed.
- N. Muresan, K. Chlopek, T. Weyhermüller, F. Neese and K. Wieghardt, Inorg. Chem., 2007, 46, 5327 CrossRef CAS PubMed.
- M. Ghosh, S. Sproules, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2008, 47, 5963 CrossRef CAS PubMed.
- R. J. Baker, R. D. Farley, C. Jones, M. Kloth and D. M. Murphy, J. Chem. Soc., Dalton Trans., 2002, 3844 RSC.
- R. J. Baker and C. Jones, J. Chem. Soc., Dalton Trans., 2005, 1341 RSC.
- V. Lorenz, C. G. Hrib, D. Grote, l. Hilfert, M. Krasnopolski and F. T. Edelmann, Organometallics, 2013, 32, 4636 CrossRef CAS and references cited there.
- R. J. Baker, R. D. Farley, C. Jones, D. P. Mills, M. Kloth and D. M. Murphy, Chem.–Eur. J., 2005, 11, 2972 CrossRef CAS PubMed.
- F. G. N. Cloke, C. I. Dalby, M. J. Henderson, P. B. Hitchcock, C. H. L. Kennard, R. N. Lamb and C. L. Raston, J. Chem. Soc., Chem. Commun., 1990, 1394 Search PubMed.
- F. G. N. Cloke, G. R. Hanson, M. J. Henderson, P. B. Hitchcock and C. L. Raston, J. Chem. Soc., Chem. Commun., 1989, 1002 RSC.
- A. Hinchliffe, F. S. Mair, E. J. l. McInnes, R. G. Pritchard and J. E. Warren, J. Chem. Soc., Dalton Trans., 2008, 222 RSC.
- D. A. Atwood, Coord. Chem. Rev., 1998, 176, 407 CrossRef CAS.
- J. A. C. Clyburne, R. D. Culp, S. Kamepalli, A. Cowley and A. Decken, Inorg. Chem., 1996, 35, 6651 CrossRef CAS PubMed.
- H. M. Lee, J. Y. Zeng, C.-H. Hu and M.-T. Lee, Inorg. Chem., 2004, 43, 6822 CrossRef CAS PubMed.
- J. Keijsper, H. van der Poel, L. H. Polm, G. van Koten, K. Vrieze, P. F. A. B. Seignette, R. Varenhorst and C. Stam, Polyhedron, 1983, 2, 1111 CrossRef CAS.
- L. H. Staal, J. Keijsper, G. van Koten, K. Vrieze, J. A. Cras and W. P Bosman, Inorg. Chem., 1981, 20, 555 CrossRef CAS.
- A. Flores-Parra, P. Suárez-Moreno, S. A. Sánchez-Ruiz, M. Tlahuextl, J. Jaen-Gaspar, H. Tlahuext, R. Salas-Coronado, A. Cruz, H. Nöth and R. Contreras, Tetrahedron: Asymmetry, 1998, 9, 1661 CrossRef CAS.
- Y. Lu, Y. Liu, H. Li, X. Zhu, H. Liu and W. Zhu, J. Phys. Chem. A, 2012, 116, 2591 CrossRef CAS PubMed.
- The Importance of Pi-Interactions, in Crystal Engineering: Frontiers in Crystal Engineering, ed. E. R. T. Tiekink and J. Zukerman-Schpector, Wiley, 2012, ch. 8 Search PubMed.
- C. Estrella, A. Frontera, D. Quiñonero and P. M. Deyà, ChemPhysChem, 2011, 12, 2742 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98, Revision A.7, Gaussian, Inc., Pittsburgh PA, 1998 Search PubMed.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871 CrossRef CAS.
- S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2009, 131, 14176 CrossRef CAS PubMed.
- B. Bantu, G. M. Pawar, K. Wurst, U. Decker, A. M. Schmidt and M. R. Buchmeiser, Eur. J. Inorg. Chem., 2009, 1970 CrossRef CAS.
- M. Zettlitzer, H. T. Dieck, E. T. K. Haupt and L. Stamp, Chem. Ber., 1986, 119, 1868 CrossRef CAS.
- H. van der Poel, G. van Koten, M. Kokkes and C. H. Stam, Inorg. Chem., 1981, 20, 2941 CrossRef CAS.
- G. Bencivenni, T. Lanza, M. Minozzi, D. Nanni, P. Spagnolo and G. Zanardi, Org. Biomol. Chem., 2010, 8, 3444 CAS.
- J. H. Cotgreave, D. Colclough, G. Kociok-Köhn, G. Ruggiero, C. G. Frost and A. S. Weller, J. Chem. Soc., Dalton Trans., 2004, 1519 RSC.
- A. Higelin, S. Keller, C. Göhringer, C. Jones and I. Krossing, Angew. Chem., Int. Ed., 2013, 52, 4941 CrossRef CAS PubMed.
- M. L. Cole, D. E Hibbs, C. Jones, P. C. Junk and N. A. Smithies, Inorg. Chim. Acta, 2005, 358, 102 CrossRef CAS PubMed.
- S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2009, 131, 14176 CrossRef CAS PubMed.
- W. A. Herrmann and Ch. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162 CrossRef CAS.
- O. Kühl, Functionalised N-Heterocyclic Carbene Complexes, Wiley, 2010 Search PubMed.
- M. Shukla, N. Srivastava and S. Saha, Interactions and Transitions in Imidazolium Cations. Based Ionic Liquids. Ionic Liquids–Classes and Properties, ed. S. Handy, 2011, http://www.intechopen.com.books/ionic-liquids Search PubMed.
- T. Kano, K. Sasaki and K. Marouka, Org. Lett., 2005, 7, 1347 CrossRef CAS PubMed.
- Y. Suzuki, K. Muramatsu, K. Yamauchi, Y. Morie and M. Sato, Tetrahedron, 2006, 62, 302 CrossRef CAS PubMed.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, 3rd edn, 1988 Search PubMed.
- S. Roland, P. Mangeney and A. Alexakis, Synthesis, 1999, 228 CrossRef CAS PubMed.
- G. Alvaro, F. Grepioni and D. Savoia, J. Org. Chem., 1997, 62, 4180 CrossRef CAS.
- T. Dieck and H. Dietrich, Chem. Ber., 1984, 117, 694 CrossRef.
- G. M. Sheldrick, SHELX 97-2 Users Manual, University of Göttingen, Germany, 1977 Search PubMed.
- P. W Betteridge, J. R. Carruthers, R. I Cooper, K. Prout and D. J. J. Watkin, Appl. Crystallogr., 2003, 36, 1487 CrossRef.
- M. Camalli, M. C. Burla, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, G. Polidori and R. J. Spagna, Appl. Crystallogr., 2003, 36, 1103 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray diffraction analysis of (R)-1-chloro-butan-2-amine hydrochloride (S1). Macromolecular helix arrangement in the crystal of compound 2 (S2). Theoretical conformational analysis of 1,4-dialkyl-1,4-diazabutadienes 1–4 (S3). Anagostic interactions H⋯Al found in the X-ray diffraction analysis of compound 10 (S4). Cell packing representation of compound 14 with [I3]− as the anion. (S5) Optimized conformers of compounds: 5, 17, 18, 20–22 (S6). Representation of the electrostatic potentials of the indium compounds 16–18 and of the ligand 3 coordinated to one and two InCl3 molecules (S7). CCDC 960655–960662. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj01226c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |