Bioelectrocatalytic reduction of O2 at a supramolecularly associated laccase electrode†
Manuel
Antuch
,
Darío G.
Abradelo
and
Roberto
Cao
*
Laboratorio de Bioinorgánica, Universidad de La Habana, Zapata y G, Vedado, La Habana, 10400 Cuba. E-mail: caov@fq.uh.cu; Fax: +53 7 8733502; Tel: +53 7 8792145
First published on 22nd November 2013
Abstract
In order to improve the bioelectrocatalytic reduction of dioxygen, a method for the supramolecular immobilization of Trametes versicolor laccase is reported. The immobilization support consisted of a self-assembled monolayer (SAM) of mercaptoundecanoic acid tyrosine amide on gold electrodes. To mediate the electron transfer between the enzyme and the electrode surface, ferrocenehexanethiol was introduced into the SAM as a second component. The resulting system was studied by cyclic voltammetry. Reduction of dioxygen started at 0.80 V, with a reduction peak at 0.52 V and a decrease in its intensity was observed when the solution was deaerated. The achieved current density for dioxygen reduction (23 μA cm−2, open to air) suggests a high surface density of active enzyme prone to bioelectrocatalytic activity. This system offers new insights into the supramolecular design of biofuel cells.
Introduction
The search for renewable low energy sources for biomedical devices is very active. Efforts have been made to develop enzymatic biofuel cells, which take advantage of the natural efficiency of oxidoreductase enzymes to oxidize or reduce easily accessible substrates.1–6 Several enzymes have been employed in biofuel cells. In particular, dioxygen reducing enzymes, such as laccases, have attracted great attention due to their ability to decrease the activation barrier of dioxygen reduction.6,7Laccases are copper containing oxidoreductase enzymes that couple the oxidation of a wide family of aromatic substrates to the concomitant reduction of dioxygen to water.6–8 In general, they contain four copper atoms as the prosthetic group. The oxidation of the substrate takes place at the type 1 (T1) copper, from which the electrons are transferred to a trinuclear cluster containing type 2 (T2) and type 3 (T3) copper atoms, where the reduction of dioxygen occurs.7,9 Laccases have been investigated from structural,10 spectroscopic11 and electrochemical12 points of view. It has been found that the T1, T2 and T3 copper sites have standard potentials within the range of 0.69–1.00 V, 0.36 V, and 0.57–1.00 V (vs. Ag/AgCl), respectively.3,9 Thus, the onset of the dioxygen reduction reaction is always near the potential of the T1 site.13–19
Several approaches have been used for the immobilization of laccases such as entrapment, encapsulation, self-immobilization, covalent binding or adsorption.7,20 Immobilization processes should guarantee the appropriate orientation of the electron-receptor T1 site towards the surface of the electrode in order to achieve an adequate electrochemical communication with it.21 As the T1 site of laccase is rich in aromatic aminoacids,14,19 it may be assumed that surfaces modified with tyrosine (Tyr) should be able to achieve an adequate orientation of the enzyme when a supramolecular procedure is used.17,22 We have recently reported the immobilization of laccase on gold nanoparticles modified with a SAM of a thiolated derivative of Tyr, solely by means of multivalent supramolecular interactions.23
On the other hand, the presence of additional redox species is frequently necessary to mediate the electron transfer process between the redox enzyme and the supporting electrode.6 Many species have been used for this purpose, such as carbon nanotubes,24 certain phenolic dyes or osmium complexes.25 Redox mediation of immobilized laccase with ferrocene has been less studied, even when it offers a stable electrochemical signal.6,26
Herein we describe the reduction of dioxygen at gold electrodes modified layer-by-layer with a mixed SAM of mercaptoundecanoic acid tyrosine amide (MUATyr) and ferrocenehexanethiol (FcH) (Fig. 1). Laccase from Trametes versicolor, Lac (EC 1.10.3.2), was supramolecularly immobilized on the mixed SAM. In such a system, the terminal Tyr moieties should have directed the orientation of the T1 site of laccase towards the electrode surface while the ferrocene derivative promoted the dioxygen reduction.
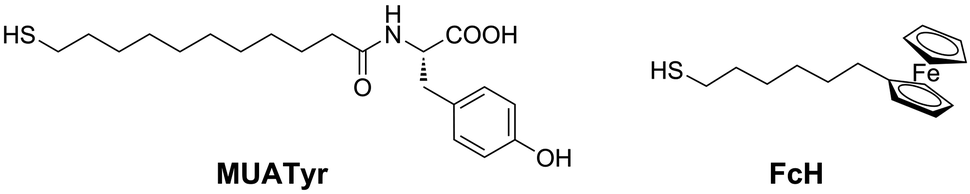 | ||
| Fig. 1 Components of the mixed SAM. | ||
Experimental section
Materials
All reagents and solvents employed were commercially available high-grade purity materials (Aldrich Chemicals) and used as supplied without further purification.Spectroscopy
1H and 13C NMR spectra were recorded on a Variant Gemini 400 spectrometer. UV-Vis spectra were recorded on an Ultrospec III (Pharmacia-LKB). The X-ray photoelectron spectra (XPS) were obtained using a VG Escalab 250 iXL ESCA instrument (VG Scientific). The measurements were carried out using monochromatic Al-Kα radiation (1486.92 eV). Photoelectrons were collected from a takeoff angle of 90° relative to the sample surface. The measurement was obtained in a constant analyzer energy mode (CAE) with 100 eV pass energy for survey spectra and 20 eV pass energy for high-resolution spectra. The binding scale was referenced by setting Au4f7/2 BE at 84.0 eV. Mass spectra were obtained using a ToF-SIMS spectrometer of Ion-TOF GmbH Germany. The sample was bombarded with a pulsed gallium ion beam (25 keV) at 45° incidence. The secondary ions generated were extracted with a 10 kV voltage, and their time-of-flight from the sample to the detector was measured using a reflectron mass spectrometer.Synthesis of MUATyr
A mixture of 11-mercaptoundecanoic acid (100 mg, 0.458 mmol) and N-hydroxysuccinimide (52.7 mg, 0.458 mmol) was stirred at −5 °C in THF (60 mL) for 15 min. A solution of dicyclohexylcarbodiimide (94.3 mg, 0.458 mmol) in THF (5 mL) was added dropwise for 45 min. The mixture was stirred for another 24 h and filtered. An additional solution of Tyr (83 mg, 0.458 mmol) in ethanol was prepared by the addition of NaOH (0.5 M) dropwise. Both solutions were mixed and stirred for 24 h. The solvent was evaporated under vacuum; the white product was washed several times with a hydroalcoholic solution and dried in air. Yield 160.1 mg (86.6%). 1H NMR (400 MHz, D2O, 25 °C): δ = 2.50–0.90, (w, 20H), 3.12 (s, Ha), 2.40 (s, Hb), 4.54 (s, H), 6.77 (dd, 4H); 13C NMR (125 MHz, DMSO-d6, 25 °C) δ = 30–23 (10C), 34, 39, 41, 49, 116, 125, 130, 153, 168, 170.Preparation of the SAMs
Gold sheets were cleaned consecutively with NaBH4 and a dilute nitric acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solution for 3 h and then thoroughly washed with doubly distilled water prior to the immobilization.27,28 The gold electrode was prepared by annealing the tip of a gold wire (1 mm diameter, 5 cm long, 99.99%), which had previously been cleaned overnight with nitric acid (68%). The formed gold bead was electrochemically pretreated in HClO4 (0.1 M) at 2.3 V until gold(III)oxide formation (red color) was observed in order to guarantee the formation of a Au{111} surface. The electrode was then cleaned with HCl (0.1 M) and thoroughly rinsed with doubly distilled water (3 μS). The SAM of MUATyr was obtained by immersion of a gold sheet in a 10−3 M methanolic solution for 24 h. The SAM of FcH was obtained by immersion of gold electrodes in a 10−2 M ethanolic solution for 20 h. The mixed SAM of MUATyr and FcH was obtained by 20 h immersion of gold electrodes into an equimolar ethanolic solution of both components (total thiol concentration 10−2 M). The SAMs obtained were thoroughly rinsed with methanol, ethanol and double distilled water to remove residual reactants.
:1 v/v) solution for 3 h and then thoroughly washed with doubly distilled water prior to the immobilization.27,28 The gold electrode was prepared by annealing the tip of a gold wire (1 mm diameter, 5 cm long, 99.99%), which had previously been cleaned overnight with nitric acid (68%). The formed gold bead was electrochemically pretreated in HClO4 (0.1 M) at 2.3 V until gold(III)oxide formation (red color) was observed in order to guarantee the formation of a Au{111} surface. The electrode was then cleaned with HCl (0.1 M) and thoroughly rinsed with doubly distilled water (3 μS). The SAM of MUATyr was obtained by immersion of a gold sheet in a 10−3 M methanolic solution for 24 h. The SAM of FcH was obtained by immersion of gold electrodes in a 10−2 M ethanolic solution for 20 h. The mixed SAM of MUATyr and FcH was obtained by 20 h immersion of gold electrodes into an equimolar ethanolic solution of both components (total thiol concentration 10−2 M). The SAMs obtained were thoroughly rinsed with methanol, ethanol and double distilled water to remove residual reactants.
Immobilization of laccase
Laccase was immobilized onto the SAM of MUATyr by incubation in a 1 mg mL−1 laccase buffered solution (pH 5) for 2 h. The enzymatic activity was determined in a buffered aqueous solution (pH 5) containing ABTS (5 mM, 100 μL). Absorbance values were acquired every 15 s at 420 nm.28,29 For the electrochemical measurements, laccase was immobilized in the same fashion onto the mixed SAM of FcH and MUATyr.Cyclic voltammetry
All glassware was cleaned using “piranha” solution followed by thoroughly rinsing with double distilled water. Cyclic voltammetry was performed using a commercial EG&G Model 384B Polarographic Analyzer in a three electrode cell configuration. A gold bead, Pt, and Ag/AgCl (KCl saturated) were used as the working, counter, and reference electrodes, respectively. Na2SO4 (0.1 M) was employed as the supporting electrolyte for all measurements, while KOH (0.5 M) was used for all the reductive desorption experiments. Purging of dissolved O2 was achieved by N2 bubbling via an external source.Results and discussion
Spectroscopic characterization of the SAM of MUATyr and laccase supramolecularly immobilized on it
The formation of a SAM of MUATyr on gold sheets was confirmed by ToF-SIMS (Fig. 2a). The spectrum showed peaks corresponding to Au-MUATyr+ (591 u.m.a.) and MUATyr+ (394 u.m.a.) fragments. Such a SAM served as a host for the supramolecular immobilization of the enzyme, which took place spontaneously within 2 h of immersion. Direct evidence for the immobilization process of laccase onto the SAM of MUATyr (Au-MUATyr-Lac) was observed in the XPS spectrum in the N 1s region (Fig. 2b) as the number of nitrogen atoms increased from 3.3 to 7.9% (semiquantitative evaluation), caused by the presence of the amino acid residues of laccase.30 The enzymatic activity of the SAM of MUATyr-Lac was determined by the oxidation kinetics of ABTS and normalized against the area of the gold sheets (5.19 × 10−6 U cm−2).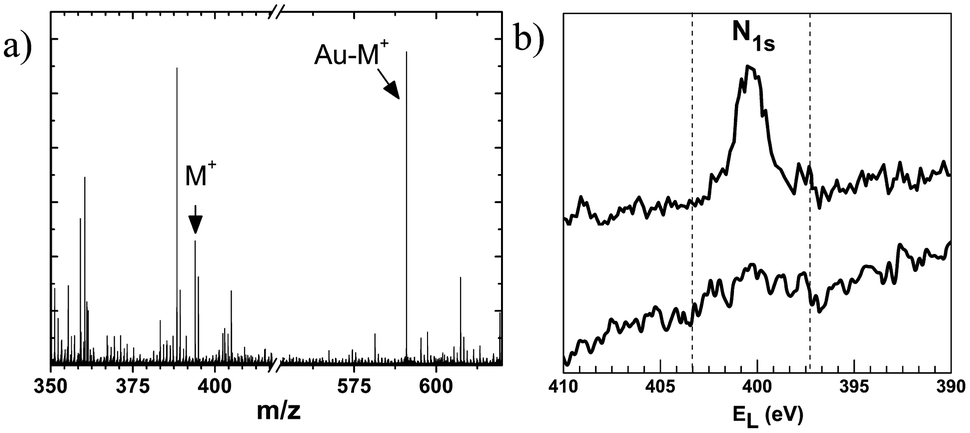 | ||
| Fig. 2 (a) ToF-SIMS of the SAM of Au-MUATyr. Arrows indicate the fragments at 394 (MUATyr+) and 591 u.m.a. (Au-MUATyr+). (b) XPS spectra of the SAM of MUATyr on gold sheets with (up) and without (down) laccase. | ||
Electrochemical behaviour of the mixed SAM of FcH and MUATyr
The ferrocene moiety of FcH (Fig. 1) was chosen to enhance the electron transfer process between the electrode and the immobilized enzyme as its equilibrium formal potential is lower than that reported for the T1 site of all laccases.22,31The electrochemical behaviour of the mixed SAM of MUATyr and FcH was studied by cyclic voltammetry (CV). The chemically reversible redox waves of FcH and the irreversible tyrosine oxidation signal were observed at 0.42 V and 0.84 V, respectively (Fig. 3a, dashed). The anodic shift of about 100 mV in the ferrocene signal is in agreement with previous reports of SAM containing embedded ferrocene derivatives.32 The electrochemical confirmation of the formation of this SAM was assessed by the linear dependence of the intensity of the anodic and cathodic waves on the applied scan rate (Fig. S1, ESI†).33 In the reductive desorption experiment, peaks corresponding to FcH at −1.0 V (Γ = 9.5 × 10−11 mol cm−2) and MUATyr at −1.2 V (Γ = 8.5 × 10−11 mol cm−2) confirmed the presence of the mixed monolayer (Fig. S2, ESI†), with a ratio very similar (1:1.18) to the equimolar proportion used in the formation of the mixed SAM.
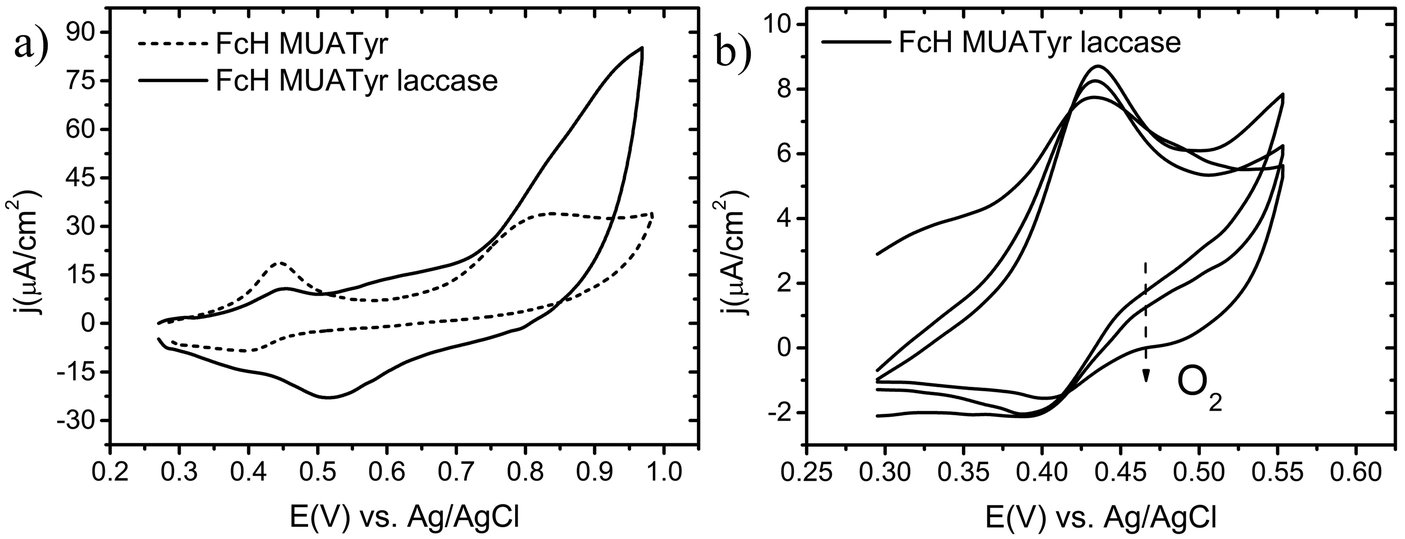 | ||
| Fig. 3 Cyclic voltammograms of (a) the mixed SAM of FcH and MUATyr (dashed) and the mixed SAM of FcH, MUATyr and immobilized laccase as a second monolayer (solid) where the oxygen reduction is observed as a broad signal with its maximum at 0.52 V. (b) Increase in oxygen reduction as it diffuses to the modified electrode containing laccase. All scans were performed in the anodic direction; scan rate = 100 mV s−1. | ||
Additional information on the formed mixed SAM regarding the interaction of terminal ferrocene species with MUATyr was obtained. The obtained width at the half maximum of the oxidation and reduction waves (ΔEfwhm) of 80 mV for FcH suggested an almost ideal (90.6/n mV) electrochemical behaviour for this species.34 In this mixed SAM, FcH presented a reversible (i.e. fast) electron transfer (Ea − Ec = 40 mV) with a small wave separation independently of the scan rate (Fig. S1, ESI†).
In contrast to the latter, SAM of pure FcH exhibited broad redox waves (ΔEfwhm = 280 mV, Fig. S1 and S2, ESI†) which indicated strong interactions between the terminal ferrocene moieties. Moreover, the SAM of FcH presented high peak separation (Ea − Ec = 440 mV at 100 mV s−1) with a simultaneous dependence on the scan rate, indicating quasireversible (i.e. slow) electron transfer.
The difference found in the ΔEfwhm values of the pure and mixed SAM of FcH suggests that FcH and MUATyr are distributed homogeneously on the gold surface.35 The increase in the rate of electron transfer of the ferrocene moieties, due to the presence of MUATyr, was also desirable since FcH was included in the SAM to promote a fast electron transfer with the enzyme.
Biocatalytic reduction of dioxygen
The electrochemical properties of electrodes modified with FcH, MUATyr and supramolecularly associated laccase were studied. The electrochemical cell was initially deaerated with a flow of nitrogen and the potential was swept in the anodic direction up to 0.55 V in order to avoid the irreversible oxidation of the Tyr moiety.36 Under these conditions it was possible to detect only the chemically reversible response of FcH. After additional scans with the electrochemical system open to air, the cathodic current increased and a second wave at 0.52 V was recorded, corresponding to the presence of inflowing dioxygen (Fig. 3b).37,38After each deaeration of the cell, the same tendency was observed, confirming the dependence of this new electrochemical signal on the concentration of dioxygen. Moreover, when the potential was swept up to 1.00 V, not only the signals of FcH and MUATyr were detected, but also that of the reduction of dioxygen (Fig. 3a, solid). It has to be highlighted that the current density of the dioxygen reduction reaction (j = 23 μA cm−2, air saturated) was even more intense than that of FcH, suggesting a high surface density of catalytic sites (active laccase) with the appropriate orientation of the enzyme (Fig. 4). The same electrochemical determination, but with a SAM that only contained MUATyr (without FcH), yielded only a slight increase in the cathodic current upon the diffusion of dioxygen, while the cathodic wave was not detected (Fig. S3, ESI†). This way the mediator role of FcH was confirmed.
 | ||
| Fig. 4 Scheme representing the appropriate orientation of the T1 site of laccase towards the electrode and a possible electron pathway for dioxygen reduction. | ||
Both the onset of dioxygen reduction at 0.80 V, within the range reported for laccaseT1 site potential (0.69–1.00 V), and the maximum reduction current reached at 0.52 V are in agreement with previous reports and support the assignment of the latter electrochemical signal to dioxygen reduction.15,18 These results confirmed the capability of this bioelectrode to effectively immobilize laccase and to promote the reduction of molecular dioxygen at low potentials.
Conclusions
Laccase was supramolecularly immobilized as a second monolayer on gold surfaces previously modified with a mixed SAM of MUATyr and FcH. In the studied system the terminal Tyr moieties on the electrode served to associate to the T1 site through π–π interactions in order to properly orient it towards the surface. When a multivalent supramolecular association of an enzyme is accomplished, as is our case, the conformation should be externally stabilized.22 Under such conditions, an efficient ET process between the enzyme and the gold electrode enabled the reduction of dioxygen with a high current density. Operational variables (pH, ionic strength) and lifetime are under investigation for future biofuel cell applications.Notes and references
- A. Ramanavicius, A. Kausaitea and A. Ramanavicienea, Biosens. Bioelectron., 2008, 24, 761–766 CrossRef CAS PubMed.
- P. Atanassov, C. Apblett, S. Banta, S. Brozik, S. C. Barton, M. Cooney, B. Y. Liaw, S. Mukerjee and S. D. Minteer, Interfaces, 2007, 16, 28–31 CAS.
- S. C. Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867–4886 CrossRef CAS.
- F. Barriére, P. Kavanagh and D. Leech, Electrochim. Acta, 2006, 51, 5187–5192 CrossRef PubMed.
- I. Ivanov, T. Vidakovič-Koch and K. Sundmacher, Energies, 2010, 3, 803–846 CrossRef CAS.
- D. Leech, P. Kavanagh and W. Schuhmann, Electrochim. Acta, 2012, 84, 223–234 CrossRef CAS PubMed.
- M. Fernández-Fernández, M. Sanromán and D. Moldes, Biotechnol. Adv., 2013, 131, 405–412 Search PubMed.
- I. Bento, L. O. Martins, G. G. Lopes, M. A. Carrondo and P. F. Lindley, Dalton Trans., 2005, 3507–3513 RSC.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2605 CrossRef CAS PubMed.
- K. Piontek, M. Antorini and T. Choinowski, J. Biol. Chem., 2002, 277, 37663–37669 CrossRef CAS PubMed.
- R. P. Bonomo, B. M. G. Castronovo and A. M. Santoro, Dalton Trans., 2004, 104–112 RSC.
- C. Fernández-Sánchez, T. Tzanov, G. M. Gübitz and A. Cavaco-Paulo, Bioelectrochemistry, 2002, 58, 149–156 CrossRef.
- K. Karnicka, K. Miecznikowski, B. Kowalewska, M. Skunik, M. Opallo, J. Rogalski, W. Schuhmann and P. J. Kulesza, Anal. Chem., 2008, 80, 7643–7648 CrossRef CAS PubMed.
- M. S. Thorum, C. A. Anderson, J. J. Hatch, A. S. Campbell, N. M. Marshall, S. C. Zimmerman, Y. Lu and A. A. Gewirth, J. Phys. Chem. Lett., 2010, 1, 2251–2254 CrossRef CAS PubMed.
- X. Wu, Y. Hu, J. Jin, N. Zhou, P. Wu, H. Zhang and C. Cai, Anal. Chem., 2010, 82, 3588–3596 CrossRef CAS PubMed.
- S. C. Barton, H.-H. Kim, G. Binyamin, Y. Zhang and A. Heller, J. Phys. Chem. B, 2001, 105, 11917–11921 CrossRef CAS.
- M. Pita, C. Gutierrez-Sanchez, D. Olea, M. Velez, C. Garcia-Diego, S. Shleev, V. M. Fernandez and A. L. D. Lacey, J. Phys. Chem. C, 2011, 115, 13420–13428 CAS.
- N. S. Parimi, Y. Umasankar, P. Atanassov and R. P. Ramasamy, ACS Catal., 2012, 2, 38–44 CrossRef CAS.
- X. Wang, R.-M. Latonen, P. Sjöberg-Eerola, J.-E. Eriksson, J. Bobacka, H. Boer and M. Bergelin, J. Phys. Chem. C, 2011, 115, 5919–5929 CAS.
- T. Kuwahara, T. Asano, M. Kondo and M. Shimomura, Bioelectrochemistry, 2013, 91, 28–31 CrossRef CAS PubMed.
- B. Willner, E. Katz and I. Willner, Curr. Opin. Biotechnol., 2006, 17, 589–596 CrossRef CAS PubMed.
- R. Villalonga, R. Cao and A. Fragoso, Chem. Rev., 2007, 107, 3088–3116 CrossRef CAS PubMed.
- D. G. Abradelo, R. Cao and S. Schlecht, RSC Adv., 2013, 3, 21461–21465 RSC.
- Y. Liu and S. Dong, Electroanalysis, 2008, 20, 827–832 CrossRef CAS.
- P. Scodeller, R. Carballo, R. Szamocki, L. Levin, F. Forchiassin and E. J. Calvo, J. Am. Chem. Soc., 2010, 132, 11132–11140 CrossRef CAS PubMed.
- A. Heller and B. Feldman, Chem. Rev., 2008, 108, 2482–2505 CrossRef CAS PubMed.
- C. Nogues and M. Wanunu, Surf. Sci., 2004, 573, L383–L389 CrossRef CAS PubMed.
- M. C. Rodríguez-Argüelles, R. Villalonga, C. Serra, R. Cao, M. Sanromán and M. A. Longo, J. Colloid Interface Sci., 2010, 348, 96–100 CrossRef PubMed.
- M. L. Niku-Paavola, E. Karhunen, A. Kantelinen, L. Viikari, T. Lundell and A. J. Hatakka, Biotechnol., 1990, 13, 211–221 CAS.
- C. Gruian, E. Vanea, S. Simon and V. Simon, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824, 873–881 CrossRef CAS PubMed.
- T. Auletta, F. C. J. M. van Veggel and D. N. Reinhoudt, Langmuir, 2002, 18, 1288–1293 CrossRef CAS.
- A. Hosseini, J. P. Collman, A. Devadoss, G. Y. Williams, C. J. Barile and T. A. Eberspacher, Langmuir, 2010, 26, 17674–17678 CrossRef CAS PubMed.
- R. Voicu, T. H. Ellis, H. Ju and D. Leech, Langmuir, 1999, 15, 8170–8177 CrossRef CAS.
- J. J. Gooding and S. Ciampi, Chem. Soc. Rev., 2011, 40, 2704–2718 RSC.
- A. L. Eckermann, D. J. Feld, J. A. Shaw and T. J. Meade, Coord. Chem. Rev., 2010, 254, 1769–1802 CrossRef CAS PubMed.
- T. A. Enache and A. M. Oliveira-Brett, J. Electroanal. Chem., 2011, 655, 9–16 CrossRef CAS PubMed.
- X. Wang, M. Falk, R. Ortiz, H. Matsumura, J. Bobacka, R. Ludwig, M. Bergelin, L. Gorton and S. Shleev, Biosens. Bioelectron., 2012, 31, 219–225 CrossRef CAS PubMed.
- U. Salaj-Kosla, S. Pöller, W. Schuhmann, S. Shleev and E. Magner, Bioelectrochemistry, 2013, 91, 15–20 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further electrochemical considerations. See DOI: 10.1039/c3nj01143g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |