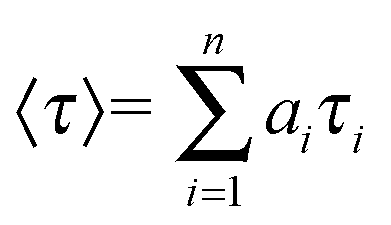DOI:
10.1039/C3NJ00814B
(Paper)
New J. Chem., 2014,
38, 376-385
Photophysical study of a π-stacked β-sheet nanofibril forming peptide bolaamphiphile hydrogel†
Received
(in Montpellier, France)
23rd July 2013
, Accepted 16th October 2013
First published on 17th October 2013
Abstract
We describe the state of molecular self-assembly of a peptide based bolaamphiphile molecule using spectroscopic and microscopic techniques. The tryptophan and phenylalanine containing peptide bolaamphiphile forms a hydrogel upon sonication under physiological conditions. Sonication helps to reorient the peptide molecules by providing the required energy for the self-assembly process. The disassembly and self-assembly processes are influenced by various stimuli, including heating–cooling and shaking–rest methods. The extensive hydrogen bonding and π–π stacking interactions are responsible for the self-assembly process, which is confirmed by FT-IR, temperature dependent NMR and fluorescence spectroscopy studies. FT-IR and powder X-ray diffraction studies reveal that the gelator molecules self-assemble into an antiparallel β-sheet type structure. The TEM image of the hydrogel shows a well-defined amyloid-like nanofibrillar structure. The amyloid-like behaviour of the fibril forming peptide bolaamphiphile hydrogel is confirmed by ThT and Congo red binding studies. The effect of concentration, time and temperature on the self-assembly mechanism of the peptide bolaamphiphile hydrogel is investigated by time resolved fluorescence spectroscopy.
1. Introduction
A wide range of scientific investigations have established a growing interest in peptide-based hydrogels,1–4 which form through the self-assembly5–10 of small peptide molecules. In many cases, it is necessary to use external stimuli such as enzymes,11–14 light,15,16 heat,17,18 sonication19,20 or change in pH,21,22 which have a drastic effect on the self-assembly mechanism. These supramolecular nanostructured hydrogels have been used in drug delivery systems,23–25 tissue engineering26–29 and various fields of nanotechnology.30–32 The self-assembly of molecules starts from a nucleation point and finally ends with higher ordered supramolecular nanostructured materials. This process is dynamic in nature,33 strongly depends on the surrounding environment and is dominated by non-covalent interactions. Therefore, it is essential to investigate the detailed mechanism of the self-assembly process to understand the fundamental non-covalent interactions at the supramolecular level.
Peptide or protein mis-folding and anomalous aggregation into amyloid fibrils are responsible for several neurodegenerative diseases like Alzheimer's disease, Prion disease, Huntington's disease and Parkinson's disease.34–36 The peptides and proteins aggregate into a β-sheet structure, which is common to all amyloid fibrils. Another example of amyloid disease is type II diabetes, where misfolded islet amyloid polypeptides (IAPP) form amyloid fibrils in the pancreas.37 Amyloid fibrils are large, highly ordered polypeptide aggregates with high mechanical and chemical stability, which have several applications in nanotechnology.38–44
Bolaamphiphiles are another interesting class of amphiphilic molecules, which can self-assemble to form well-defined nanostructures. Generally, two hydrophilic end groups are covalently bonded with a hydrophobic spacer in a bolaamphiphile molecule. Recently, several groups have explored the self-assembly process of bolaamphiphiles.45–48 Amino acid and sugar based bolaamphiphiles have also been reported, which can be used for gene delivery.49 Matsui and co-workers have reported peptide bolaamphiphiles which self-assemble to form nanotubes.50L-Glutamic acid-based bolaamphiphiles also self-assemble and form a hollow nanotubular structure.51,52
Currently, we are focused on stimuli responsive peptide bolaamphiphile self-assembly, which can tune the nanostructure at a supramolecular level. We have designed peptide bolaamphiphile molecules with two different hydrophobic dipeptides, which are covalently connected by a succinic acid moiety. The balance between the hydrophobic and hydrophilic groups can lead the peptide bolaamphiphile to self-assemble in aqueous media. There are hydrogen-bonding sites in the two dipeptides, along with two carboxylic acid groups. The hydrophobic aromatic side chains can also take part in the self-assembly process through π–π stacking interactions. These molecules are ideal for designing self-assembling soft materials through extensive hydrogen bonding and π–π stacking interactions.
The self-assembly process of peptides and proteins has been studied using infra-red spectroscopy, fluorescence, powder X-ray diffraction, rheological studies and various microscopic techniques. The secondary structure of peptides and proteins is analyzed by CD and FT-IR spectroscopy.53–55 Fluorescence spectroscopy provides direct information about π–π stacking interactions, which play a role during the self-assembly process of peptides/proteins containing aromatic residues.56,57 MacPhee et al. reported peptide self-assembly using a time correlated single photon counting (TCSPC) technique. In this case, the peptide is modified with a fluorophore group (Fmoc), which is capped with an N-terminus.58 Early aggregation in the self-assembly of prion peptides is characterized by nonlinear and ultrafast time-resolved fluorescence spectroscopy.59 As a consequence, the study of the self-assembly process of a peptide bolaamphiphile hydrogel by time-resolved fluorescence spectroscopy is rare. Our objective is to use time-resolved fluorescence spectroscopy and other spectroscopic and microscopic techniques to establish the self-assembly process of a peptide bolaamphiphile based hydrogel.
In the present study, we have used a tryptophan and phenylalanine rich peptide bolaamphiphile 1 which self-assembles into a β-sheet structure and forms a self-supporting hydrogel. The hydrogel was characterized by FT-IR, temperature dependent NMR, TEM, powder X-ray diffraction and rheological experiments. The photophysical behavior of the self-assembled peptide was explored by means of steady state and time resolved fluorescence spectroscopy.
2. Results and discussion
2.1. Hydrogel formation
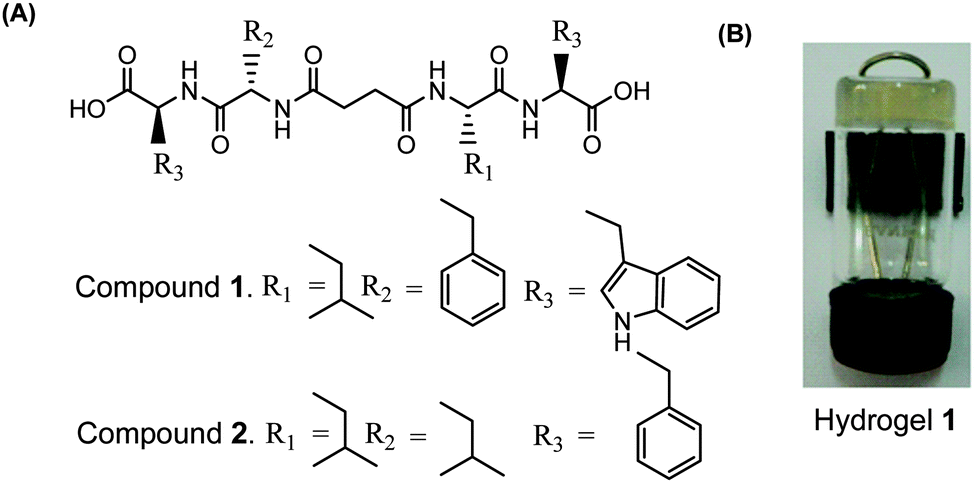
Two peptide bolaamphiphiles containing a centrally located succinic acid moiety attached to dipeptides have been synthesized for the self-assembly study (Fig. 1A). The hydrogelation behaviour of both peptide bolaamphiphiles has been investigated upon sonication under physiological conditions. Peptide bolaamphiphile 1 (HO-Trp-Phe-Suc-Leu-Trp-OH) is capable of forming a transparent hydrogel in phosphate buffer (pH 8, 10 mmol) under physiological conditions (Fig. 1B), but the peptide bolaamphiphile 2 (HO-Phe-Leu-Suc-Leu-Phe-OH) is unable to form a gel under the same conditions. The self-assembly process of peptide bolaamphiphile 1 was studied using various spectroscopic and microscopic techniques along with time-resolved fluorescence spectroscopy.
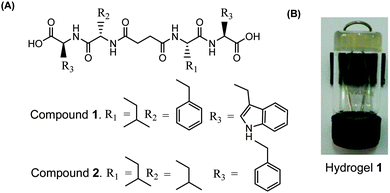 |
| | Fig. 1 (A) Structures of peptide bolaamphiphiles 1–2 and (B) a photograph of the self-supporting hydrogel formed from peptide bolaamphiphile 1. | |
2.2. Effect of stimuli
The self-assembly behaviour of the peptide bolaamphiphile hydrogel 1 is influenced by external stimuli. The peptide bolaamphiphile 1 was dispersed in phosphate buffer and sonicated for 30 minutes to form a self-supporting hydrogel. Sonication helps to dissolve the peptide bolaamphiphile molecules in aqueous solution and provides sufficient energy to reorient the molecules into a self-assembled soft material through extensive hydrogen bonding and π–π stacking interactions.60 The peptide bolaamphiphile 1 forms a thermoreversible and mechano-sensitive hydrogel (Fig. 2A). The hydrogel turns to solution on heating, and turns back into a gel again at room temperature. The gel turns to solution on shaking, but it turns back into a gel on rest. Thus, heating and mechanical shaking disassemble the peptide bolaamphiphile molecules by rupturing the non-covalent interactions. The peptide molecules again self-assemble and regain their gelation property on cooling and resting (Fig. 2B).61
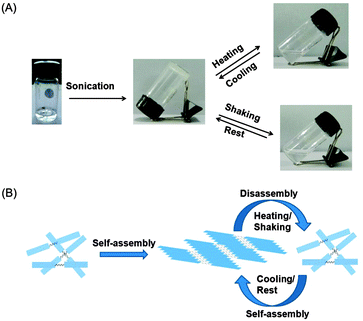 |
| | Fig. 2 (A) Stimuli responsive assembly–disassembly of peptide bolaamphiphile 1 and (B) a scheme of the assembly–disassembly of 1. | |
2.3. Rheological and FT-IR studies
The dynamic mechanical behaviour of hydrogel 1 was investigated by a rheological study. Fig. 3A shows that the storage modulus (G′) exceeds that of the loss modulus (G′′) over the oscillating frequency. In the non-destructive frequency sweep mode, the storage modulus dominates the loss modulus, exhibiting the characteristics of a typical solid-like material.62,63 The gel is moderately stiff, with G′ > 105 Pa at low frequency. The value of the storage modulus G′ exceeds that of the loss modulus G′′ by a factor of 8 in the higher frequency region, which indicates the formation of a strong and rigid hydrogel. The FT-IR study of the solid compound and its corresponding hydrogel was performed to investigate the secondary structural arrangement of the peptide bolaamphiphile hydrogel (Fig. 3B). A N–H band at around 3298 cm−1 was observed for solid compound 1, which indicates hydrogen bonding interactions in the solid state. In the gel state, the peak was shifted to 3040 cm−1, which indicates a greater extent of hydrogen bonding interactions. The peak at 2939 cm−1 is ascribed to the CH3 stretching band of the leucine moiety in the gel state. The peak of the carboxylic acid group appeared at 1718 cm−1 in the solid state, and shifted to 1710 cm−1 in the gel state. The shift of the corresponding acid peak suggests that a large amount of hydrogen bonded carboxylic acid groups are involved in the hydrogelation process. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band at 1736 cm−1 can be assigned to non-hydrogen bonded carboxylic acid groups.64
O stretching band at 1736 cm−1 can be assigned to non-hydrogen bonded carboxylic acid groups.64
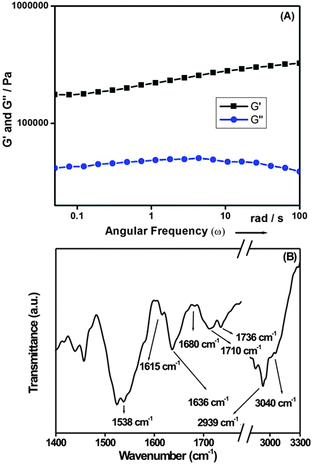 |
| | Fig. 3 (A) Dynamic rheology (frequency sweep) of the hydrogel of peptide bolaamphiphile 1 (C = 10 mmol L−1). The storage modulus G′ is higher than the loss modulus G′′. G′ > 105 Pa at low frequency, and the storage modulus is higher than the loss modulus by a factor of 6–10, indicating the excellent solid-like properties of the gel materials. (B) FT-IR spectrum of the hydrogel of peptide bolaamphiphile 1, indicating an anti-parallel β-sheet structure in the gel phase. | |
The amide I band at 1636 cm−1 along with a peak at 1615 cm−1 indicates the formation of a hydrogen bonded supramolecular β-sheet structure in the gel state. Another band at 1538 cm−1 is assigned to the amide II band, which is another characteristic feature of the β-sheet arrangement. Furthermore, a weak intensity peak at 1680 cm−1 suggests that the peptides are arranged in an antiparallel manner to form a higher order supramolecular β-sheet structure.55
2.4. Steady state fluorescence study
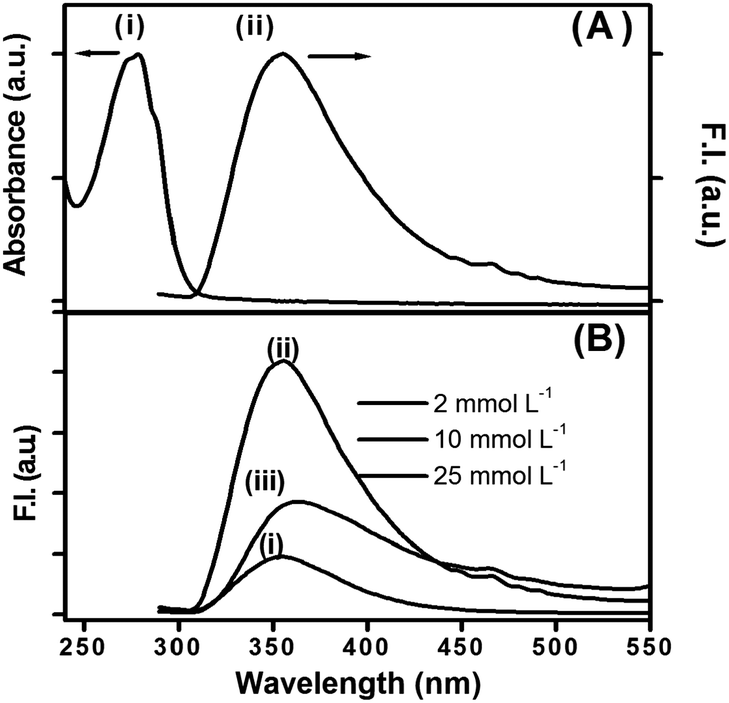
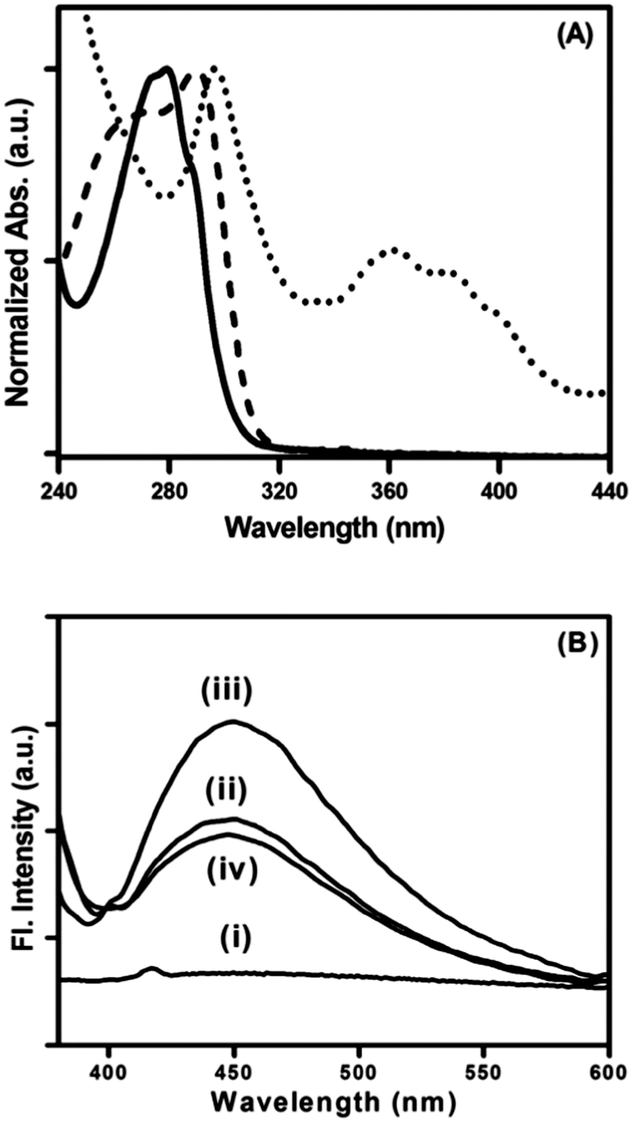
Peptide bolaamphiphile hydrogel 1 shows an absorption band at 280 nm (Fig. 4A (i)). Excitation at 280 nm results a in clear emission band centered at 355 nm (Fig. 4A (ii)). The emission spectrum is quite broad and shows a weak shoulder at around 460 nm. Fig. 4B shows the effect of peptide bolaamphiphile concentration on the fluorescence behaviour. The fluorescence intensity increases with the increase in peptide bolaamphiphile concentration from 2 mmol L−1 (solution state) to 10 mmol L−1 (gel state) with no significant peak shift. However, further increase in peptide bolaamphiphile concentration to 25 mmol L−1 results in a decrease in fluorescence intensity with a peak shift of ∼9 nm. Interestingly, the weak shoulder at 460 nm is observed only at and above 10 mmol L−1 concentration. To determine the origin of this weak shoulder at 460 nm, we recorded the excitation spectrum65 with the emission wavelength fixed at 460 nm. The excitation spectra of the peptide bolaamphiphile hydrogel recorded at emission wavelengths of 355 nm and 460 nm differ significantly (Fig. 5A). The excitation spectrum for the 355 nm emission band shows a peak at 290 nm with a shoulder at 266 nm, which is similar to that of the absorption spectrum with a 10 nm red shift. On the other hand, the excitation spectrum for 460 nm shows a maximum peak at 296 nm and a new red shifted broad band at around 365 nm. Excitation at 365 nm results a large Stokes shifted emission band centered at 450 nm at a concentration of 10 mmol L−1 (Fig. 5B (ii)). The fluorescence intensity of this band is strongly dependent on the peptide bolaamphiphile concentration (Fig. 5B). The emission at 450 nm was observed for peptide bolaamphiphile 1 at and above 10 mmol L−1 concentration. No such emission was observed for peptide bolaamphiphiles in solution phase at a concentration of 2 mmol L−1 (Fig. 5B). Interestingly, the fluorescence intensity at 450 nm increases with the increase in peptide bolaamphiphile concentration up to 20 mmol L−1. Subsequent increase in concentration to 25 mmol L−1 results in a decrease in fluorescence intensity. This decrease in fluorescence intensity is accompanied by a 9 nm red shift in the fluorescence spectrum.
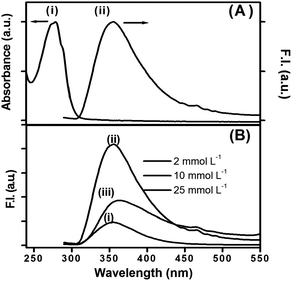 |
| | Fig. 4 (A) Absorption and emission spectra of peptide bolaamphiphile hydrogel 1 at 10 mmol L−1 (λex = 280 nm). (B) Emission spectra of peptide bolaamphiphile 1 at different concentrations: (i) 2 mmol L−1, (ii) 10 mmol L−1 and (iii) 25 mmol L−1. | |
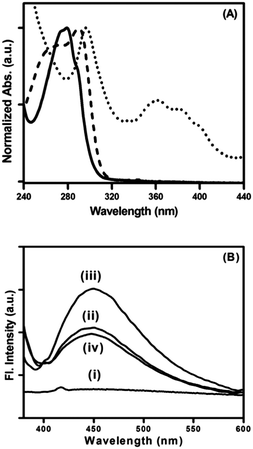 |
| | Fig. 5 (A) Excitation spectra of peptide bolaamphiphile hydrogel 1 at 350 nm (dashed line) and 450 nm (dotted line) overlapped with the absorption spectrum (solid line). (B) Fluorescence spectra of peptide bolaamphiphile 1 in solution (i) and as a hydrogel at (ii) 10 mmol L−1, (iii) 20 mmol L−1 and (iv) 25 mmol L−1 concentration with 365 nm excitation. | |
The initial increase and subsequent decrease in fluorescence intensity at 355 nm signifies the formation of some kind of stable ordered arrangement of peptide bolaamphiphiles below 25 mmol L−1.
At 2 mmol L−1 concentration, peptide bolaamphiphiles in the solution state show very low fluorescence due to non-radiative decay processes, mainly intramolecular torsional motion of the aromatic rings. In the gel state at 10 mmol L−1 concentration, all non-radiative torsional motion of the aromatic rings freezes, resulting in enhanced fluorescence. The observed fluorescence quenching at 25 mmol L−1 concentration may be due to either destabilization of the ordered arrangement, or self-quenching (concentration quenching) due to the very close proximity of peptide bolaamphiphiles. However, the peptide bolaamphiphile remains in the gel form even at 25 mmol L−1 concentration and shows a 9 nm red shift in the fluorescence spectrum. This indicates that at and above 25 mmol L−1 concentration the peptide bolaamphiphiles are very closely packed, which leads to self-quenching due to non-radiative energy loss.66,67 The different excitation spectra at two different emission wavelengths (365 nm and 450 nm) clearly signifies that the molecular origin of the two emission bands is different. This kind of different excitation spectra at different wavelengths often signifies ground state heterogeneity.68 The absence of a similar band in the solution phase at 2 mmol L−1 concentration indicates that the species responsible for the characteristic emission is only formed with the formation of the gel state. Earlier, MacPhee and coworkers have observed a similar kind of large Stokes shifted emission (∼440 nm) along with the normal emission (∼302 nm) from nanoscale fibrillar aggregates.58 Based on their results they assigned the red shifted emission peak to excimer formation. However, our results clearly signify that peptide bolaamphiphiles in the gel state form higher order aggregates in the ground state through π-stacking interactions between aromatic moieties.69 These aggregates absorb near 365 nm and show characteristic emission around 450 nm. At higher concentrations these aggregates are self-quenched due to close proximity of the peptide bolaamphiphiles. To further establish this proposed model, we performed excited state decay measurements.
2.5. Time resolved self-assembly study
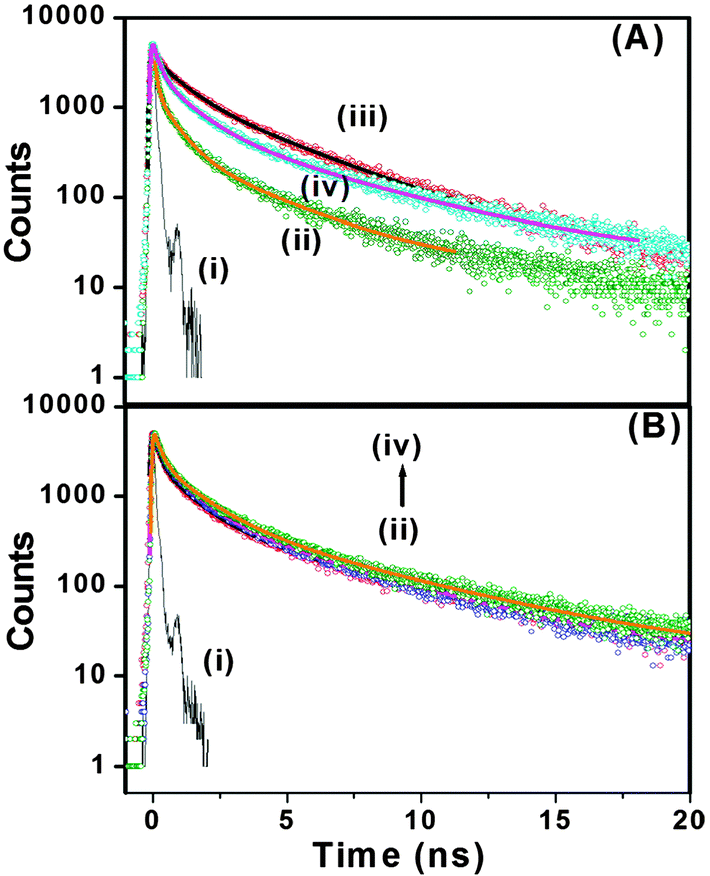
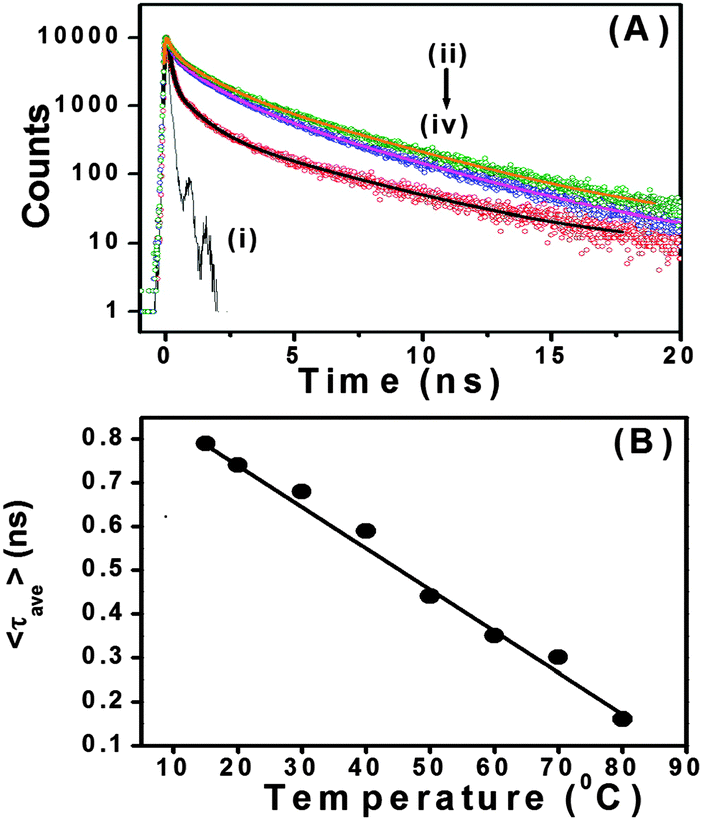
With our current time correlated single photon counting (TCSPC) setup, we measured decay traces of the peptide bolaamphiphiles with an excitation at 375 nm (Fig. 6A). Here, it is important to remember that only the aggregated peptide bolaamphiphiles show characteristic emission at 460 nm with 375 nm excitation. All the traces have been fitted with a three exponential function. The average lifetime of peptide bolaamphiphiles in the liquid state at 2 mmol L−1 concentration is 0.32 ns, with lifetime components of 0.68 ns (16%), 1.05 ns (80%) and 3.15 ns (0.04%). However, in the gel state at a concentration of 10 mmol L−1, the average lifetime increases to 0.94 ns with lifetime components of 0.24 ns (62%), 1.19 ns (28%) and 4.76 ns (10%), which is very similar to the increases in fluorescence intensity observed in the steady state experiment. Further increase in peptide bolaamphiphile concentration up to 20 mmol L−1 results in a gradual increase in lifetime. However, at 25 mmol L−1 concentration the average lifetime decreased to 0.75 ns with lifetime components of 0.08 ns (66%), 0.96 ns (20%) and 3.60 ns (14%) (ESI,† Table S1). The very short lifetime at 2 mmol L−1 concentration (solution state) of the peptide bolaamphiphiles indicates the involvement of non-radiative decay processes, mainly intramolecular torsional motion of the aromatic rings of free peptide bolaamphiphiles. However, in the gel state, the molecules self-assemble to form a rigid framework of β-sheet structures through π-stacking and hydrogen bonding interactions, which results in increased lifetime.70,71 In this β-sheet structure, the intramolecular torsional motion of aromatic rings is hindered, and the excited state non-radiative decay process is somewhat hampered. Further increase in lifetime with increasing peptide bolaamphiphile concentration indicates a more compact ordered arrangement, in which the aggregates are further stabilized. However, at 25 mmol L−1 concentration the lifetime decreases, similar to that observed in our steady state fluorescence data. Interestingly, at 25 mmol L−1 concentration, a new lifetime component with a very short decay (0.08 ns) time has been observed. This major (66%) decay component having a lifetime of 0.08 ns was neither observed in the solution state nor in the gel states with lower concentration. These results clearly support the idea that at and above 25 mmol L−1 concentration, peptide bolaamphiphiles are packed very close to each other which results in self-quenching due to non-radiative energy loss.72Fig. 6B shows the decay traces of the peptide bolaamphiphiles at a concentration of 10 mmol L−1 with varying time at room temperature. The average lifetime of the gel after 8 h is 0.87 ns, while that after 4 days is 1.12 ns. The change in lifetime is very minor but noticeable. This nominal increase in lifetime (ESI,† Table S2) with time indicates the progressive arrangement of peptide bolaamphiphiles into the ordered β-sheet structure. The effect of temperature on the gelation dynamics has been investigated by recording their decay traces at different temperatures (Fig. 7A). The temperature of the hydrogel at a concentration of 10 mmol L−1 was varied from 15 °C to 80 °C. At 15 °C, the hydrogel shows an average lifetime of 1.30 ns, which gradually decreases upon increasing the temperature (Fig. 7B), and at 80 °C it shows a value of 0.31 ns. Interestingly, the average lifetime at 80 °C matches exactly with that of the solution state of the peptide bolaamphiphiles with 2 mmol L−1 concentration. These results (ESI,† Table S3) strongly suggest that the aggregated peptide bolaamphiphile slowly dissociates into monomeric units with the increase in temperature. At 80 °C, which is just above the gel melting temperature of the hydrogel, a phase transition occurs and all the peptide bolaamphiphile molecules disassembled into the monomeric units.
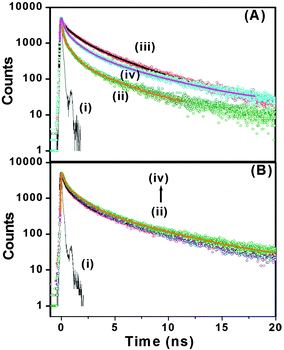 |
| | Fig. 6 (A) Decay traces of peptide bolaamphiphile 1 at (ii) 2 mmol L−1, (iii) 10 mmol L−1, and (iv) 25 mmol L−1. (B) Decay traces of peptide bolaamphiphile 1 hydrogel (10 mmol L−1) at (ii) 8 h, (iii) 1 day and (iv) 4 days. Curve (i) represents the IRF. | |
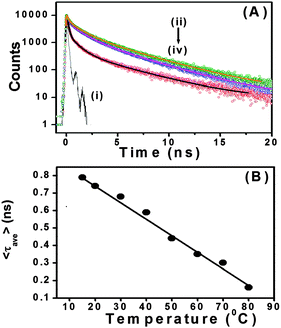 |
| | Fig. 7 (A) Decay traces of peptide bolaamphiphile 1 hydrogel (10 mmol L−1) at (ii) 15 °C, (iii) 40 °C and (iv) 80 °C temperature; (i) represents the IRF. (B) Changes in the average lifetime of peptide bolaamphiphile hydrogel 1 with temperature. | |
2.6. Temperature dependent 1H NMR study
The 1H NMR technique was used to confirm the π–π stacking interactions between the aromatic residues of peptide bolaamphiphile 1 during the self-assembly process. The hydrogel (10 mmol L−1) was prepared in sodium phosphate buffer in D2O, and the temperature dependent 1H NMR study was performed (Fig. 8). The study mainly focused on the aromatic region of the peptide bolaamphiphile molecule to confirm the π–π stacking interactions. In the gel state, the NMR spectrum appeared as a broad signal at room temperature (25 °C). The broad signal is a characteristic feature of self-assembled molecules. Fig. 8 shows that the aromatic protons underwent sharpening with the increase in temperature. At the gel melting point at 75 °C, the broad signals disappeared, and relatively sharp peaks with multiplicity appeared. At this stage, the disassembly of the peptide molecules occurred via disruption of π–π stacking interactions, and the hydrogel turned into a solution. The temperature dependent NMR study revealed that the π–π stacking interactions play a vital role in the self-assembly process of peptide bolaamphiphile 1 to form a hydrogel.73
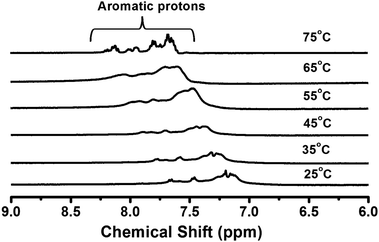 |
| | Fig. 8 Temperature dependent 1H NMR spectra showing the π–π stacking interactions during the self-assembly process. | |
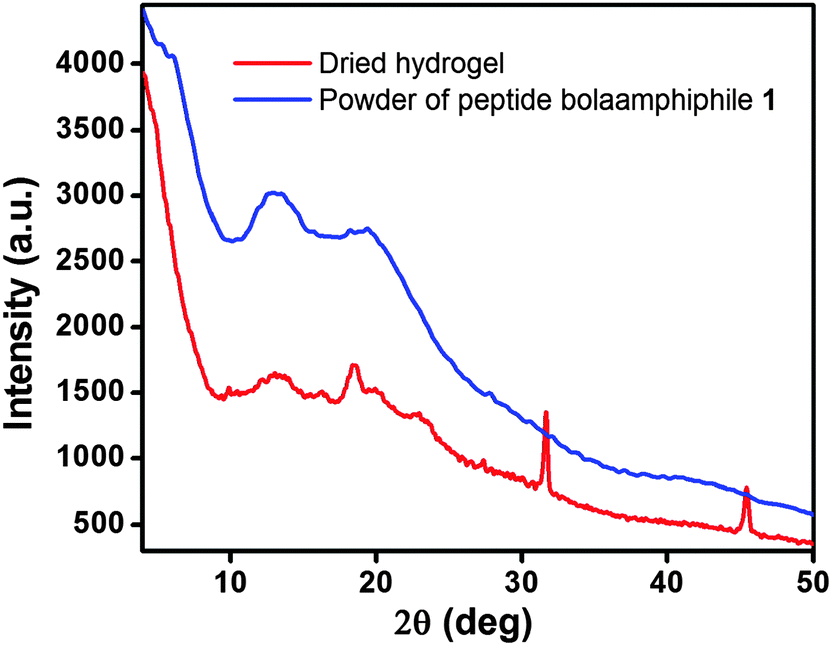
2.7. X-ray diffraction study
In order to obtain advanced structural information, the dried hydrogel of compound 1 was characterized by wide angle X-ray scattering (WAXS). The scattering patterns show a series of characteristic diffraction peaks (Fig. 9), from which we are able to correlate the different features of the self-assembly process. For the dried hydrogel of compound 1, the peak at 2θ = 10.20° corresponds to a d-spacing of 8.67 Å, which is expected to be the distance between two β sheets (inter-sheet distance). A characteristic peak at 2θ = 18.57° corresponding to a d-spacing of 4.77 Å, indicates the distance between two peptide molecules within the β-sheet structure.74 Another two characteristic reflection peaks are observed at 3.86 Å (2θ = 23.00°) and 2.81 Å (2θ = 31.75°). The 3.86 Å peak reveals the π–π stacking interactions between two aromatic groups, and the peak at 2.81 Å suggests the hydrogen bonding interactions between the CO and N–H of the peptide bolaamphiphiles.75
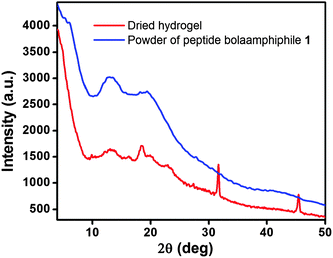 |
| | Fig. 9 Powder X-ray diffraction of solid powder of peptide bolaamphiphile 1 and its dried hydrogel. | |
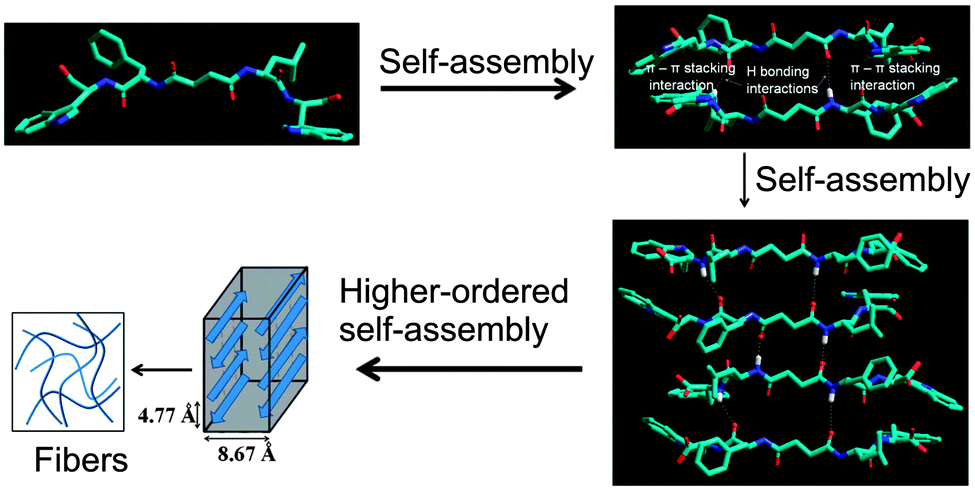
We propose a molecular model76 of the self-assembly mechanism for the supramolecular hydrogel of peptide bolaamphiphile 1. The model shows that the peptide bolaamphiphile molecules self-assembled through hydrogen bonding interactions and π–π stacking interactions between the tryptophan residues and form an anti-parallel β-sheet structure (Fig. 10). These findings are well correlated with the results of the FT-IR, fluorescence spectroscopy and X-ray diffraction studies.
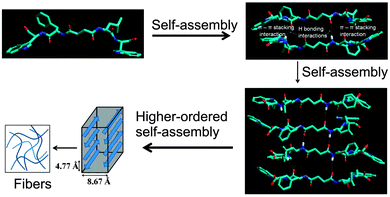 |
| | Fig. 10 A model structure was created for peptide bolaamphiphile 1, which is arranged in an anti-parallel β-sheet motif. Aromatic tryptophan moieties are stacked by π-stacking interactions, and peptide chains are associated with hydrogen bonding interactions, which results in the formation of nanostructured fibrils. | |
2.8. Transmission electron microscopy study
The nanostructural morphology77,78 of hydrogel 1 at the supramolecular level was characterized using transmission electron microscopy (TEM). The TEM image shows that the peptide bolaamphiphile molecules self-assembled into amyloid-like nanofibrillar networks which are responsible for formation of the supramolecular hydrogel. The average diameter of the nanofibers is 17 nm and each fiber is several micrometers in length (Fig. 11). In the TEM image, there are some circular domains besides the fibrillar structures. This indicates that the peptide bolaamphiphiles self-assemble to form spherical nanostructures.
 |
| | Fig. 11 Transmission electron microscopic image of peptide bolaamphiphile hydrogel 1 showing that the peptide bolaamphiphile molecules self-assembled into a nanofibrillar structure. | |
2.9. Characterisation of amyloid-like fibrils
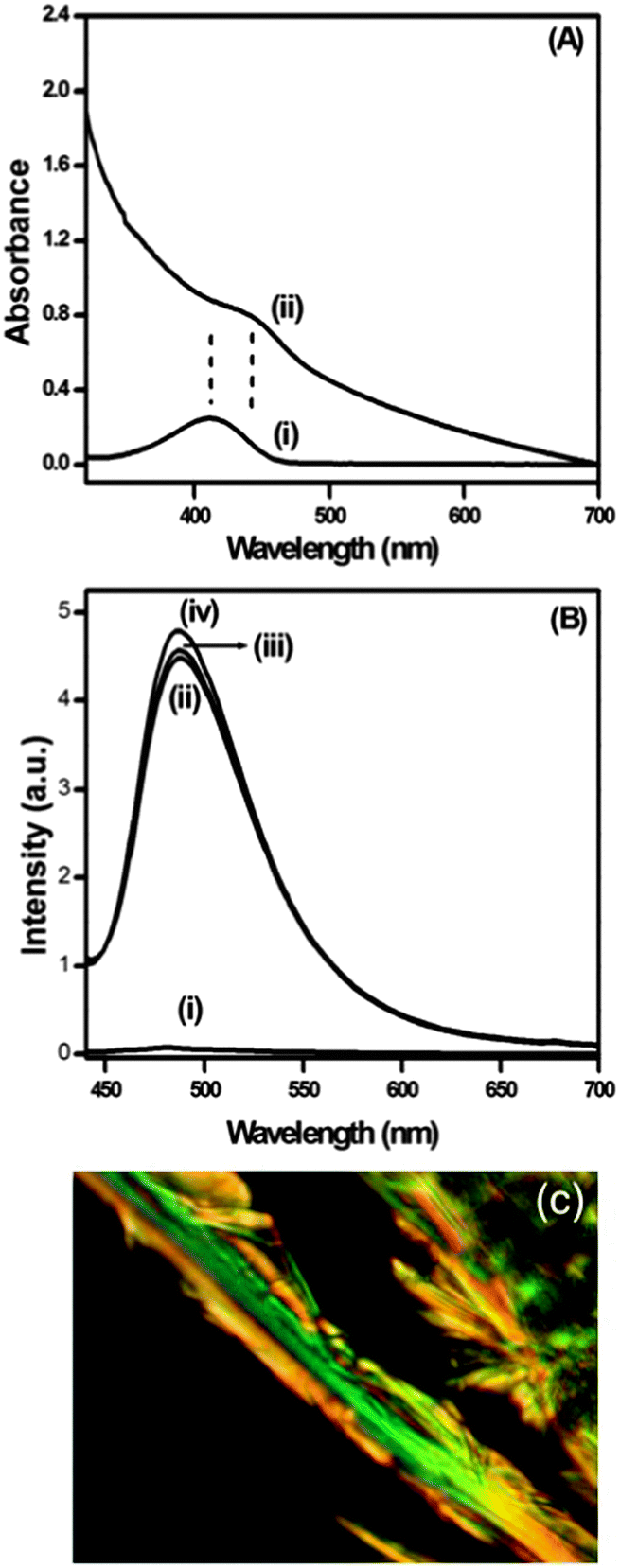
Thioflavin T (ThT) is a positively charged dye, which is widely used to monitor and detect the formation of amyloid-like fibrils.79 ThT shows a very low fluorescence quantum yield in low viscosity media like water because of its non-radiative bond twisting process around the central C–C bond in the excited state. Here, the binding of ThT (10 μM) with the peptide bolaamphiphile 1 gel was monitored by means of spectral changes in UV-Vis absorption and fluorescence spectroscopy. The absorption spectrum of ThT in phosphate buffer shows a peak at 412 nm. However, the peak shifted to 442 nm in the presence of peptide bolaamphiphile hydrogel 1 with increase in absorbance (Fig. 12A). The 30 nm red shift of the absorption peak clearly suggests that ThT was strongly bound with the β-sheet type structures resulting from peptide bolaamphiphile 1 in the gel state.80Fig. 12B shows the fluorescence spectra of ThT in the absence and presence of peptide bolaamphiphile hydrogel 1. ThT shows very weak emission at 480 nm in phosphate buffer on excitation at a wavelength of 412 nm. A significant increase in fluorescence intensity has been observed for ThT in peptide hydrogel 1 with a peak shift of 8 nm. We observed a sixty-fold enhancement in fluorescence intensity for the hydrogel containing ThT compared to ThT alone in phosphate buffer. Earlier, Banerjee et al. reported a similar kind of ThT binding assay of an amyloid fibril forming peptide using absorption and fluorescence spectroscopy.81 The significant enhancement in the fluorescence intensity with 8 nm red shift strongly indicates the binding of ThT with the amyloid-like fibril forming82 peptide bolaamphiphile hydrogel 1. Nanofibrils resulting from the hydrogel of compound 1 were stained with a physiological dye (Congo red) and viewed through a cross polarizer under a polarized microscope (Fig. 12C). Fibrils of peptide hydrogel 1 bind with Congo red and exhibit a characteristic green-gold birefringence under the cross polarizer, which indicates amyloid-like behaviour.83 Thus, peptide 1 forms amyloid type fibrils in the hydrogel state, as is evident from the typical green-gold birefringence after staining with Congo red and the enhancement of fluorescence upon binding with thioflavin T.
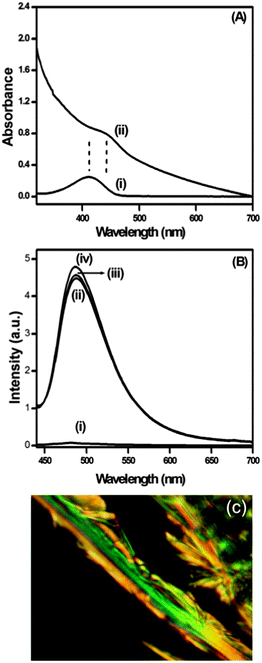 |
| | Fig. 12 (A) Binding of ThT into the amyloid fibrils of hydrogel 1 monitored by spectral changes in absorption spectra (i) in phosphate buffer, and (ii) in peptide bolaamphiphile 1 at 7 h. (B) Fluorescence spectra of ThT (i) in phosphate buffer, (ii) in peptide bolaamphiphile 1 at 7 h, (iii) in peptide bolaamphiphile 1 at 1 day, and (iv) in peptide bolaamphiphile 1 at 2 days. (C) Optical microscopic image of peptide 1 fibrils stained with Congo red dye showing a green-gold birefringence, which is a characteristic feature of amyloid fibrils. | |
3. Conclusion
In this report, the self-assembly mechanism and photophysics of amyloid-like fibril forming peptide bolaamphiphile 1 have been investigated. Hydrogen bonding interactions and π–π stacking interactions are the driving force for the self-assembly process, which results in the formation of a self-supporting hydrogel where the peptide bolaamphiphile molecules adopt an anti-parallel β-sheet structure. The self-assembly mechanism is influenced by temperature and change in concentration, which was studied by time dependent 1H NMR and time-resolved fluorescence spectroscopy. Peptide bolaamphiphile hydrogel 1 turns to solution with increase in temperature, which correlates with the results of temperature dependent TCSPC studies. The average life time of the peptide bolaamphiphiles decreases linearly with increase in temperature. With increase in concentration, the peptide bolaamphiphile molecules are organized in a more compact fashion and form a β-sheet structure. The average life time of the peptide bolaamphiphiles increases with increase in concentration up to a certain limit, which was studied by concentration dependent TCSPC measurement. The peptide bolaamphiphile molecules self-assemble into a well-defined nanostructured morphology at the supramolecular level upon sonication.
4. Experimental section
4.1. Synthesis of peptide bolaamphiphiles
Peptide bolaamphiphiles 1 and 2 were synthesized by conventional solution phase methodology. The C-terminus of the amino acids was protected as methyl ester. Couplings were mediated by dicyclohexylcarbodiimide and 1-hydroxybenzotriazole (DCC-HOBt). The final compounds were purified and fully characterized by FT-IR, 1H NMR, 13C NMR and mass spectral studies.
4.2. NMR spectroscopy
All NMR characterizations were carried out on a Bruker AV 400 MHz spectrometer at 300 K. Compound concentrations were in the range 1–10 mmol in (CD3)2SO and CDCl3. The temperature dependent NMR study was done in a phosphate buffer of D2O.
4.3. Mass spectrometry
Mass spectra were recorded on a Waters HPLCMS system (Column Symmetry C18, 7 mm) by negative mode electrospray ionisation and Bruker micrOTOF-Q II by positive mode electrospray ionisation.
4.4. Polarimetry study
Specific rotations of the synthesized compounds were measured on an Autopol V automatic polarimeter (Rudolph Research Analytical). The cell (length = 100 mm, capacity = 2 mL) was used for this study at 20 °C.
4.5. Rheology
Oscillating rheology was used to quantify the final mechanical properties of the peptide bolaamphiphile hydrogels. In each case, 2 mL of peptide bolaamphiphile hydrogel (10 mmol L−1) was prepared. The experiment was done on a Paar Physica Modular Compact Rheometer (MCR 301, Austria). A 25 mm cone plate with 1° angle configuration was used and the temperature was set to be constant at 25 °C. Storage (G′) and loss (G′′) moduli were measured at 0.1% strain with a true gap of 0.05 mm. To determine the exact strain for the frequency sweep experiment, the linear viscoelastic (LVE) regime was performed at a constant frequency of 10 rad s−1.
4.6. FT-IR spectroscopy
All reported FT-IR spectra were taken using a Bruker (Tensor 27) FT-IR spectrophotometer. The solid-state measurements were performed using the KBr pellet technique with a scan range between 400 and 4000 cm−1 over 64 scans at a resolution of 4 cm−1 and an interval of 1 cm−1. The gel sample was prepared in D2O, placed between crystal Zn–Se windows and scanned between 900 to 4000 cm−1 over 64 scans at a resolution of 4 cm−1 and an interval of 1 cm−1.
4.7. UV-Vis spectroscopy
UV-Vis absorption spectra of the hydrogel were recorded using a Varian Cary100 Bio UV-Vis spectrophotometer.
4.8. Fluorescence spectroscopy
Fluorescence and excitation spectra of the gels were recorded on a Horiba Scientific Fluoromax-4 spectrophotometer with a 1 cm path length quartz cell at room temperature. The slit width for the excitation and emission was set at 5 nm with a 1 nm data pitch. Excitations of samples were performed at 280 nm and 365 nm, and the data range was between 290 to 575 and 375 nm to 625 nm, respectively.
4.9. Time correlated single photon counting (TCSPC)
A 2 mL of gel sample was prepared in a quartz cuvette (1 cm × 1 cm) and time correlated single photon counting (TCSPC) studies were performed on a Horiba Yovin (model: Fluorocube-01-NL) instrument. Samples were excited at 376 nm using a picosecond diode laser (model: Pico Brite-375L). The signals were collected at magic angle (54.70°) polarization using a photomultiplier tube (TBX-07C) as a detector, which had a dark count of less than 20 cps. The instrument response function (IRF, FWHM ∼ 140 ps) was recorded using a very dilute scattering solution. Data analysis was performed using IBH DAS (version 6, HORIBA Scientific, Edison, NJ) decay analysis software.
The amplitude-weighted lifetime was estimated by
where
τi is the fluorescence lifetime of various fluorescent species and
ai are the normalized pre-exponential factors. The goodness of the fit was judged by the reduced chi-square (
χ2) value.
4.10. Wide angle X-ray diffraction
The XRD measurements were carried out using a Bruker D8 Advance X-ray diffractometer. The X-rays were produced using a sealed tube, and the wavelength of the X-rays was 0.154 nm (Cu Kα). The X-rays were detected using a fast counting detector based on silicon strip technology (Bruker LynxEye detector).
4.11. Molecular modeling
Models were constructed using the Hyper-Chem program. The amber force field was used to minimize the energy of the structures formed in this program.
4.12. Transmission electron microscopy
High resolution transmission electron microscopy images were taken using a Philips electron microscope (CM 200), operated at an accelerating voltage of 200 kV. The hydrogel (15 mmol L−1) was diluted to 0.75 mmol L−1 with double distilled water and characterized using a transmission electron microscope. Dilute solutions of the hydrogel were dried on carbon-coated copper grids (300 mesh) by slow evaporation in air, and then allowed to dry separately in a vacuum at room temperature.
4.13. Congo red binding study
An alkaline saturated Congo red solution was prepared. The peptide fibrils were stained with the alkaline Congo red solution (70% methanol + 30% 0.08 M NaOH solution in double distilled water) for 15 min, and then the excess stain (Congo red) was removed by rinsing the stained fibrils with 70% methanol + 30% double distilled water solution several times. The stained fibrils were dried at room temperature and then visualized at 100× magnification. The birefringence was observed between crossed polarizers.
Acknowledgements
AKD is thankful to the DST (Grant no. SR/FT/CS-67/2010) and CSIR (Grant no. 02(0056)/12/EMR-II), New Delhi, India for financial support. IM is indebted to CSIR, New Delhi, India for his fellowship for this work. We thank the UGC-DAE Consortium for Scientific Research, Indore for the use of powder XRD.
Notes and references
- X. Li, X. Du, Y. Gao, J. Shi, Y. Kuang and B. Xu, Soft Matter, 2012, 8, 7402–7407 RSC.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- S. Roy and A. Banerjee, RSC Adv., 2012, 2, 2105–2111 RSC.
- C. Ou, J. Zhang, X. Zhang, Z. Yang and M. Chen, Chem. Commun., 2013, 49, 1853–1855 RSC.
- G. Colombo, P. Soto and E. Gazit, Trends Biotechnol., 2007, 25, 211–218 CrossRef CAS PubMed.
- C.-A. Palma, M. Cecchini and P. Samori, Chem. Soc. Rev., 2012, 41, 3713–3730 RSC.
- S. E. Paramonov, H.-W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291–7298 CrossRef CAS PubMed.
- A. R. Hirst, B. Escuder, J.-F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- S. Scanlon and A. Aggeli, Nano Today, 2008, 3, 22–30 CrossRef CAS.
- A. L. Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295–4306 RSC.
- D. B. Rasale, I. Maity and A. K. Das, RSC Adv., 2012, 2, 9791–9794 RSC.
- A. K. Das, R. Collins and R. V. Ulijn, Small, 2008, 4, 279–287 CrossRef CAS PubMed.
- Y. Gao, F. Zhao, Q. Wang, Y. Zhang and B. Xu, Chem. Soc. Rev., 2010, 39, 3425–3433 RSC.
- J.-B. Guilbaud, E. Vey, S. Boothroyd, A. M. Smith, R. V. Ulijn, A. Saiani and A. F. Miller, Langmuir, 2010, 26, 11297–11303 CrossRef CAS PubMed.
- J. H. Collier, B.-H. Hu, J. W. Ruberti, J. Zhang, P. Shum, D. H. Thompson and P. B. Messersmith, J. Am. Chem. Soc., 2001, 123, 9463–9464 CrossRef CAS.
- L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025–17029 CrossRef CAS PubMed.
- R. P. Nagarkar, R. A. Hule, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2008, 130, 4466–4474 CrossRef CAS PubMed.
- D. W. Lowik, E. Leunissen, M. Van den Heuvel, M. Hansen and J. C. van Hest, Chem. Soc. Rev., 2010, 39, 3394–3412 RSC.
- I. Maity, D. B. Rasale and A. K. Das, Soft Matter, 2012, 8, 5301–5308 RSC.
- J. Wu, Q. Tian, H. Hu, Q. Xia, Y. Zou, F. Li, T. Yi and C. Huang, Chem. Commun., 2009, 4100–4102 RSC.
- I. Maity, D. B. Rasale and A. K. Das, RSC Adv., 2013, 3, 6395–6400 RSC.
- L. Chen, S. Revel, K. Morris and D. J. Adams, Chem. Commun., 2010, 46, 4267–4269 RSC.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- J. Kopecek and J. Yang, Angew. Chem., Int. Ed., 2012, 51, 7396–7417 CrossRef CAS PubMed.
- J. L. Frandsen and H. Ghandehari, Chem. Soc. Rev., 2012, 41, 2696–2706 RSC.
- G. A. A. Saracino, D. Cigognini, D. Silva, A. Caprini and F. Gelain, Chem. Soc. Rev., 2013, 42, 225–262 RSC.
- C. A. DeForest and K. S. Anseth, Angew. Chem., Int. Ed., 2012, 51, 1816–1819 CrossRef CAS PubMed.
- C. E. Semino, J. Kasahara, Y. Hayashi and S. Zhang, Tissue Eng., 2004, 10, 643–655 CrossRef CAS PubMed.
- R. N. Shah, N. A. Shah, M. M. D. R. Lim, C. Hsieh, G. Nuber and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3293–3298 CrossRef CAS PubMed.
- B. Adhikari and A. Banerjee, Chem.–Eur. J., 2010, 16, 13698–13705 CrossRef CAS PubMed.
- K. H. Smith, E. Tejeda-Montes, M. Poch and A. Mata, Chem. Soc. Rev., 2011, 40, 4563–4577 RSC.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
-
B. Alberts, D. Bray, J. Lewis, M. Raff, K. Roberts and J. D. Watson, in Molecular biology of the cell, Garland Publishing, New York, 3rd edn, 1994 Search PubMed.
- S. L. Bernstein, T. Wyttenbach, A. Baumketner, J.-E. Shea, G. Bitan, D. B. Teplow and M. T. Bowers, J. Am. Chem. Soc., 2005, 127, 2075–2084 CrossRef CAS PubMed.
- M. E. MacDonald and J. F. Gusella, Curr. Opin. Neurobiol., 1996, 6, 638–643 CrossRef CAS.
- M. Goedert, M. G. Spillntini and S. W. Davies, Curr. Opin. Neurobiol., 1998, 8, 619–632 CrossRef CAS.
- Y. Porat, S. Kolusheva, R. Jelinek and E. Gazit, Biochemistry, 2003, 42, 10971–10977 CrossRef CAS PubMed.
- S. K. Maji, D. Schubert, C. Rivier, S. Lee, J. E. Rivier and R. Riek, PLoS Biol., 2008, 6, e17 Search PubMed.
- I. Cherny and E. Gazit, Angew. Chem., Int. Ed., 2008, 47, 4062–4069 CrossRef CAS PubMed.
- S. Prigent, A. Ballesta, F. Charles, N. Lenuzza, P. Gabriel, L. M. Tine, H. Rezaei and M. Doumic, PLoS One, 2012, 7, e43273 CAS.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- S. Mankar, A. Anoop, S. Sen and S. K. Maji, Nano Rev., 2011, 2, 6032, DOI:10.3402/nano.v2i0.6032.
- K. K. M. Sweers, M. L. Bennink and V. Subramaniam, J. Phys.: Condens. Matter, 2012, 24, 243101 CrossRef CAS PubMed.
- C. Li and R. Mezzenga, Nanoscale, 2013, 5, 6207–6218 RSC.
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117–9149 RSC.
- C. Tomasini and N. Castellucci, Chem. Soc. Rev., 2013, 42, 156–172 RSC.
- S. Ray, A. K. Das and A. Banerjee, Chem. Mater., 2007, 19, 1633–1639 CrossRef CAS.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1443 CrossRef CAS PubMed.
- S. Denoyelle, A. Polidori, M. Brunelle, P. Y. Vuillaume, S. Laurent, Y. ElAzhary and B. Pucci, New J. Chem., 2006, 30, 629–646 RSC.
- Z. Zhao, I. A. Banerjee and H. Matsui, J. Am. Chem. Soc., 2005, 127, 8930–8931 CrossRef CAS PubMed.
- Q. Jin, L. Zhang, H. Cao, T. Wang, X. Zhu, J. Jiang and M. Liu, Langmuir, 2011, 27, 13847–13853 CrossRef CAS PubMed.
- C. Zhan, P. Gao and M. Liu, Chem. Commun., 2005, 462–464 RSC.
- J. T. Pelton and L. R. McLean, Anal. Biochem., 2000, 277, 167–176 CrossRef CAS PubMed.
- A. Aggeli, M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. B. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS PubMed.
- P. Zhu, X. Yan, Y. Su, Y. Yang and J. Li, Chem.–Eur. J., 2010, 16, 3176–3183 CrossRef CAS PubMed.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. van Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS PubMed.
- M. Ma, Y. Kuang, Y. Gao, Y. Zhang, P. Gao and B. Xu, J. Am. Chem. Soc., 2010, 132, 2719–2728 CrossRef CAS PubMed.
- K. J. Channon, G. L. Devlin, S. W. Magennis, C. E. Finlayson, A. K. Tickler, C. Silva and C. E. MacPhee, J. Am. Chem. Soc., 2008, 130, 5487–5491 CrossRef CAS PubMed.
- Y. Wang and T. Goodson III, J. Phys. Chem. B, 2007, 111, 327–330 CrossRef CAS PubMed.
- P. Duan and M. Liu, Langmuir, 2009, 25, 8706–8713 CrossRef CAS.
- Y. Li, T. Wang and M. Liu, Tetrahedron, 2007, 63, 7468–7473 CrossRef CAS PubMed.
- F. Song and L. M. Zhang, J. Phys. Chem. B, 2008, 112, 13749–13755 CrossRef CAS PubMed.
- G. Pont, L. Chen, D. G. Spiller and D. J. Adams, Soft Matter, 2012, 8, 7797–7802 RSC.
- M. Kogiso, T. Hanada, K. Yase and T. Shimizu, Chem. Commun., 1998, 1791–1792 RSC.
- A. R. Leon, A. O. Olatunde, J. R. Morrow and C. Achim, Inorg. Chem., 2012, 51, 12597–12599 CrossRef PubMed.
- X. Shi, D. Duft and J. H. Parks, J. Phys. Chem. B, 2008, 112, 12801–12815 CrossRef CAS PubMed.
- S. K. Mandal, T. Kar, D. Das and P. K. Das, Chem. Commun., 2012, 48, 1814–1816 RSC.
- I. Yamazaki, N. Tamai and T. Yamazaki, J. Phys. Chem., 1987, 91, 3572–3577 CrossRef CAS.
- A. K. Das, A. R. Hirst and R. V. Ulijn, Faraday Discuss., 2009, 143, 293–303 RSC.
- O. F. A. Larsen, I. H. M. Stokkum, A. Pandit, R. Grondelle and H. Amerongen, J. Phys. Chem. B, 2003, 107, 3080–3085 CrossRef CAS.
- J. Xu and J. R. Knutson, J. Phys. Chem. B, 2009, 113, 12084–12089 CrossRef CAS PubMed.
- A. Imhof, M. Megens, J. J. Engelberts, D. T. N. de Lang, R. Sprik and W. L. Vos, J. Phys. Chem. B, 1999, 103, 1408–1415 CrossRef CAS.
- Q. Jin, L. Zhang and M. Liu, Chem.–Eur. J., 2013, 19, 9234–9241 CrossRef CAS PubMed.
- T. R. Jahn, O. S. Makin, K. L. Morris, K. E. Marshall, P. Tian, P. Sikorski and L. C. Serpell, J. Mol. Biol., 2010, 395, 717–727 CrossRef CAS PubMed.
- M. Ikeda, T. Tanida, T. Yoshii and I. Hamachi, Adv. Mater., 2011, 23, 2819–2822 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC.
- D. B. Rasale, I. Maity, M. Konda and A. K. Das, Chem. Commun., 2013, 49, 4815–4817 RSC.
- N. Debeljuh, C. J. Barrow and N. Byrne, Phys. Chem. Chem. Phys., 2011, 13, 16534–16536 RSC.
- V. Pradines, A. J Stroia and P. Faller, New J. Chem., 2008, 32, 1189–1194 RSC.
- S. Ray, A. K. Das, M. G. B. Drew and A. Banerjee, Chem. Commun., 2006, 4230–4232 RSC.
- Z. Gong, L. Huang, Y. Yang, X. Chen and Z. Shao, Chem. Commun., 2009, 7506–7508 RSC.
- A. K. Das, M. G. B. Drew, D. Haldar and A. Banerjee, Org. Biomol. Chem., 2005, 3, 3502–3507 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: fluorescence decay and synthesis of peptide bolaamphiphiles. See DOI: 10.1039/c3nj00814b |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.