Towards sensory Langmuir monolayers consisting of macrocyclic pentaaminoanthraquinone†
Elena
Ranyuk
a,
Elizaveta V.
Ermakova‡
b,
Laura
Bovigny§
a,
Michel
Meyer
*a,
Alla
Bessmertnykh-Lemeune
a,
Roger
Guilard
a,
Yoann
Rousselin
a,
Aslan Yu.
Tsivadze
b and
Vladimir V.
Arslanov
*b
aInstitut de Chimie Moléculaire de l'Université de Bourgogne (ICMUB), UMR CNRS 6302, 9 avenue A. Savary, 21078 Dijon Cedex, France. E-mail: michel.meyer@u-bourgogne.fr; Fax: +33 3 80 39 61 17; Tel: +33 3 80 39 37 16
bFrumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Leninsky Pr., 31 GSP-1, Moscow, 119071, Russia. E-mail: pcss_lab@mail.ru
First published on 29th October 2013
Abstract
A pentaazamacrocycle incorporating an intracyclic anthraquinone fragment (PENTAQ) was synthesized with the aim of forming metal-responsive Langmuir monolayers. PENTAQ allows a good discrimination by naked eye of copper ions in methanol–water solutions (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v). Spectrophotometric investigations of the protonation and Cu2+ binding properties of PENTAQ were undertaken in order to gain a deeper insight into the pH-dependent speciation as well as the color changing process. PENTAQ monolayers at air/water and air/methanol–water interfaces were prepared according to the Langmuir procedure. The structure of the monolayers can be tuned by varying the pH of the aqueous subphase, since it was found that the relative orientation at the interface of the aromatic lipophilic and polyazaalkyl hydrophilic fragments is critically dependent upon the protonation state of the immersed pentaazamacrocyclic chain. In situ fiber-optic UV-vis measurements revealed the formation of H-aggregates in the PENTAQ monolayer deposited on a water subphase at pH = 5.5. The ability of these films to detect copper(II) in the aqueous or methanol–water (20:80 v/v, pH = 9.8) subphase was explored by combining several experimental techniques, including pressure–area measurements (π–A isotherms), in situ UV-vis absorption spectrophotometry, and XPS spectroscopy.
:50 v/v). Spectrophotometric investigations of the protonation and Cu2+ binding properties of PENTAQ were undertaken in order to gain a deeper insight into the pH-dependent speciation as well as the color changing process. PENTAQ monolayers at air/water and air/methanol–water interfaces were prepared according to the Langmuir procedure. The structure of the monolayers can be tuned by varying the pH of the aqueous subphase, since it was found that the relative orientation at the interface of the aromatic lipophilic and polyazaalkyl hydrophilic fragments is critically dependent upon the protonation state of the immersed pentaazamacrocyclic chain. In situ fiber-optic UV-vis measurements revealed the formation of H-aggregates in the PENTAQ monolayer deposited on a water subphase at pH = 5.5. The ability of these films to detect copper(II) in the aqueous or methanol–water (20:80 v/v, pH = 9.8) subphase was explored by combining several experimental techniques, including pressure–area measurements (π–A isotherms), in situ UV-vis absorption spectrophotometry, and XPS spectroscopy.
Introduction
In recent years, there has been an upsurge of interest in the development of optical chemosensors specific for toxic metal ions because of the increasing contamination levels found in the environment and the resulting bother for the health of living organisms. In the past decades, major scientific efforts have focused on the design of novel molecular indicators sought as promising detection tools for on-line field monitoring.1–10 Although significant progress has been achieved, as testified by a number of reports describing efficient optical and fluorescent chemosensors for metal cations, further advances are still required to push the field towards operational analytical devices. While direct monitoring of toxic cations in environmental and biological aqueous samples is actively pursued, one of the major hurdles to overcome is the lack of hydrophilicity evidenced by most of the new generation chemosensors. Hence, synthetic efforts are more and more directed towards the development of water-soluble chromoionophores.11–13As an attractive alternative, the deposition at the air-solution interface of the molecular detectors as Langmuir monolayers and the post-formation of Langmuir–Blodgett films (LB films) can also be envisaged.14,15 This deposition technique is also valuable because it provides a high surface density of chemosensors due to the molecular organization at the air–liquid interface.16–18 Development of ultra-thin films for detection purposes is becoming an important direction of modern sensory science as it reflects nowadays trends of increasing the efficiency and miniaturization of information devices. Detectors of this type possess undoubted advantages, such as compactness, high speed performance, and availability of analytes. Moreover, a huge number of known chemosensors can be regarded as semi-amphiphilic compounds being constituted of a hydrophilic ionophore and a lipophilic signaling group. Hence, many of them should be suitable for preparing Langmuir monolayers on the surface of aqueous solutions, although the structural features of these molecules differ often significantly from those of classical amphiphiles designed for Langmuir monolayer formation. However, this field is still in its infancy as little is known about their behavior in the film. As a matter of fact, significant research efforts are mandatory in order to better understand the structural requirements and physico-chemical features that the molecular precursors should display for promoting an efficient optical detection at an air/liquid interface and enabling a successful transfer of these films onto solid supports.
Macrocyclic polyethers and to a lesser extent their aza or thia analogues have been widely used for monitoring alkaline, earth-alkaline, and transition metals in solutions.3,19–23 Thus, they are appealing candidates for obtaining nanostructured metal-responsive monolayers due to their high symmetry and tunable hydrophilic character. For example, amphiphilic crown-ethers, aza- and thiacrown derivatives are known to self-assemble at an air/water interface, while the characteristics of the resulting Langmuir monolayers are influenced by the presence of metal ions in the aqueous subphase.24–28 The high affinity exhibited by polyazamacrocycles for transition and main-group metal cations21,29–38 has been exploited for elaborating chromogenic probes working in aqueous or mixed water–organic solvent systems.20,22,39–60 However, reports describing the elaboration of Langmuir monolayers with these compounds are still relatively scarce. LB films consisting of co-polymers bearing cyclen derivatives have been investigated with the aim of detecting Cu2+ cations by refractive index changes.61 Other examples of macrocyclic ligands incorporated into Langmuir monolayers include N-substituted cyclen derivatives (1,4,7,10-tetraazacyclododecane) bearing n-alkyl chains,62–68 cyclam (1,4,8,11-tetraazacyclotetradecane) and hexacyclen (1,4,7,10,13,16-hexaazacyclooctadecane) functionalized with lipophilic substituents.69 For the latter compound, UV spectroscopic studies of LB films deposited on lipophilic quartz slides evidenced a linear correlation between the absorbance and the film thickness. However, to the best of our knowledge Langmuir monolayers of polyazamacrocyclic ligands incorporating a chromophore as signaling unit have not been considered so far for detecting heavy metals by absorption spectrophotometry.
Spurred by our previous work on water-soluble open-chain aminoanthraquinone-based specific chemosensors enabling the quantification of heavy metals such as Cu2+, Pb2+, and Hg2+ in aqueous media,12,13 we have designed a hemi-amphiphilic pentaazamacrocyclic receptor, denoted hereafter PENTAQ (Fig. 1), by connecting the 1 and 8 positions of anthraquinone with a linear pentaamine. A preliminary account of the sensing properties of PENTAQ in a methanol–water mixture has been recently reported.11 Thanks to an appropriate symmetry and a favorable balance between the hydrophilic chelating chain and the hydrophobic signaling motif, PENTAQ turned out to be well-suited for forming stable Langmuir monolayers. We disclose herein its coordination properties both in the bulk and at the air/water and air/methanol–water interfaces.
 | ||
| Fig. 1 Structural formula of PENTAQ. | ||
Results and discussion
PENTAQ synthesis
PENTAQ was readily synthesized following the Buchwald–Hartwig amination reaction (Scheme 1). Equimolar amounts of commercially available 1,8-dichloroanthraquinone and tetraethylenepentamine pentahydrochloride were reacted in the presence of the Pd(dba)2/BINAP catalytic system (8 mmol/16 mmol) and a large excess (10 equiv.) of Cs2CO3 in refluxing dioxane. The targeted compound was isolated in 41% yield after purification by column chromatography on silica gel. The red-violet PENTAQ was characterized using conventional methods, including MALDI-TOF-MS, 1H and 13C NMR spectroscopy, FTIR and UV-vis spectrophotometry. The strong absorption feature in the visible region is assigned to an intramolecular charge transfer (ICT) band centered at 562 nm in a methanol–water (80:20 v/v) mixture.
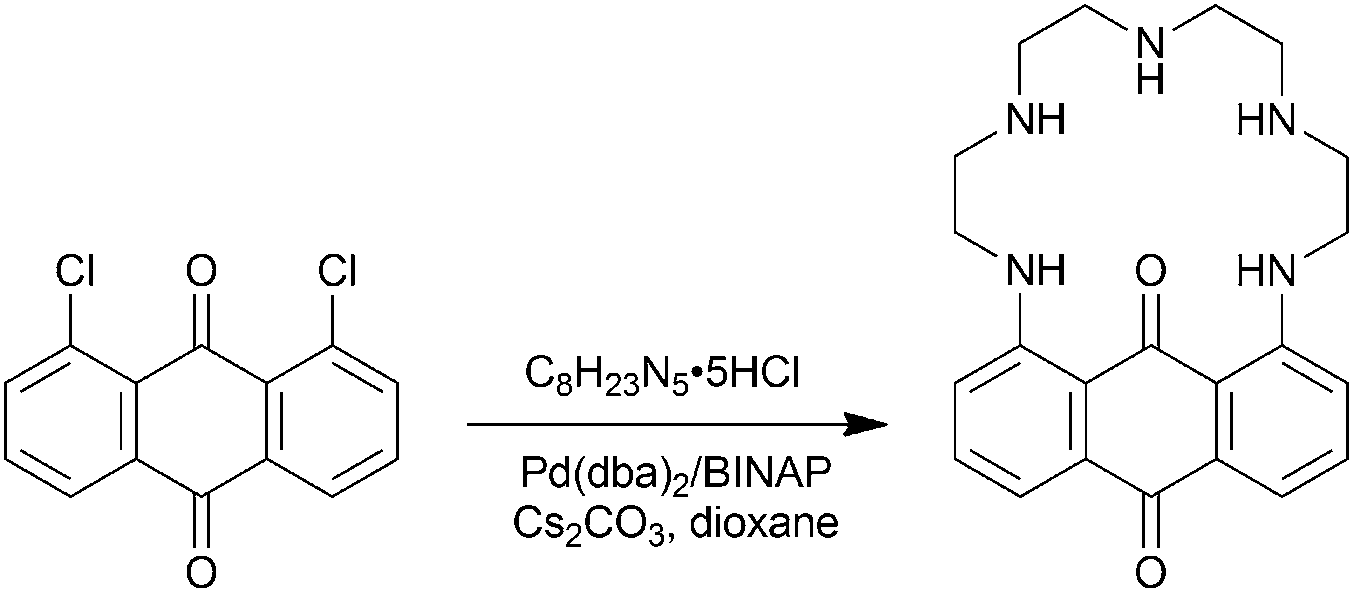 | ||
| Scheme 1 Synthesis of PENTAQ. | ||
X-ray crystallography
OLEX2 views70 of [(PENTAQ)H]CF3SO3·H2O and [(PENTAQ)H2](CF3SO3)2 showing the atom numbering and the intra- and intermolecular hydrogen bonds with the cocrystallized water and/or triflate anions are displayed in Fig. 2. In both structures, the protonated nitrogen atoms could be easily localized by considering the carbon–nitrogen bond lengths (Table 1) and the residual charge densities in the Fourier difference maps which could be assigned to the corresponding hydrogen atoms. In [(PENTAQ)H]+, the proton is located at the central amino group N3, the macrocycle adopting a nearly planar conformation as evidenced by the C14⋯C21⋯N3 angle of 172.4(2)°. In the [(PENTAQ)H2]2+ cation, the ammonium nitrogen atoms are N2 and N4. The aliphatic part of the macrocycle is significantly bent with respect to the aromatic ring as the C14⋯C21⋯N3 angle reaches 130.11(8)°. The conformational change undergone by PENTAQ upon addition of the second proton increases the curvature of the anthraquinone moiety, as reflected by the wider opening of the dihedral angle between benzene rings [9.8(1)° for [(PENTAQ)H]+vs. 17.8(1)° for [(PENTAQ)H2]2+]. In both structures, the carbonyl oxygen atom O1 is significantly bent out of the anthraquinone mean plane defined by atoms C9–C22, O1, and O2. The largest rms deviation is again observed for the dication [−0.431(2) vs. 0.246(3) Å, respectively], reflecting an unexpectedly high flexibility of the aromatic moiety. Intramolecular hydrogen bonds between the oxygen atom O1 and the proton borne by both adjacent nitrogen atoms N1 and N5 account well for the observed trends (Table S1 and S2, ESI†), although intermolecular interactions are certainly influencing to some extent the layout adopted by the macrocycle. In the case of [(PENTAQ)H]+, each macrocycle is interconnected to a triflate ion through a water solvate molecule that is hydrogen-bonded to the ammonium site N3 and the sulfonate oxygen atoms O4 and O6. In contrast, two triflates are located one on each side of the macrocyclic cavity in the dicationic structure. Remarkably, all three oxygen atoms of each sulfonate group are involved in hydrogen bonds with either an amine or an ammonium proton, only one of them (O4) is interacting with a neighboring [(PENTAQ)H2]2+ unit. | ||
| Fig. 2 OLEX2 views70 of the X-ray crystal structures of [(PENTAQ)H]CF3SO3·H2O (a) and [(PENTAQ)H2](CF3SO3)2 (b). Thermal ellipsoids are drawn at 50% probability level. The minor disordered part of the triflate counter anion in [(PENTAQ)H]CF3SO3 is omitted for the sake of clarity. The red dashed lines represent the hydrogen bonds. | ||
| Bond | [(PENTAQ)H]CF3SO3·H2O | [(PENTAQ)H2](CF3SO3)2 |
|---|---|---|
| N2–C2 | 1.446(6) | 1.500(2) |
| N2–C3 | 1.456(6) | 1.490(3) |
| N3–C4 | 1.490(6) | 1.464(3) |
| N3–C5 | 1.463(8) | 1.466(3) |
| N4–C6 | 1.451(6) | 1.496(3) |
| N4–C7 | 1.401(7) | 1.491(3) |
Visual detection of metal cations
The ability of PENTAQ to detect the presence of various amounts of environmentally-relevant metal ions was evaluated visually and by UV-vis spectroscopy in methanol–water solutions (50:50 v/v) (Fig. 3).
 | ||
| Fig. 3 Cation-induced color changes of PENTAQ (L) in CH3OH–H2O 50:50 v/v. [L]tot = [M]tot = 80 μM. | ||
The addition of an equimolar amount of different metal ions such as Na+, K+, Mg2+, Ca2+, Ba2+, Zn2+, Pb2+, Cd2+, or Ni2+ to a 80 μM PENTAQ solution has no noticeable effect (Fig. 3). In contrast, 1 equiv. of Cu2+ produces a color change from violet to pale blue that translates into a 94 nm bathochromic shift of the absorption maximum due to the deprotonation of an aromatic amine upon copper(II) binding, as observed earlier for various linear 1,8-diaminoanthraquinones.12,71
Solution binding properties
:20 v/v) containing 0.1 M N(CH3)4NO3 as the background electrolyte was selected for solubility reasons. Unfortunately, non-reproducible data were obtained in CH3OH–H2O mixtures containing less than 50% by volume of methanol, while increasing the methanol content to 80% was still not sufficient to reach the millimolar concentration level required for potentiometric or NMR titrations. Hence, the acid–base properties of PENTAQ, expressed as apparent stepwise protonation constants (K01h = [LHhh+]/[LHh−1(h−1)+][H+]), were unraveled by performing UV-vis spectrophotometric titrations in a large p[H] range (p[H] = −log[H+]).
A first batch experiment was performed at high acidities ([H+]tot = 0.02–4 M, −0.6 ≤ p[H] ≤ 1.7) by adding small aliquots of concentrated nitric acid to a 47 μM PENTAQ solution (Fig. S1, ESI†). As the total ionic strength did not remain constant, no quantitative information was extracted from these data, which showed a slight hypsochromic shift of 6 nm associated with a strong hyperchromic effect when the calculated p[H] increased from −0.6 to 0.3, while no significant spectral change could be noticed between p[H] 0.3 and 1.7. This behavior is fully consistent with the very low proton affinity exhibited by primary or secondary 1-amino and 1,8-diaminoanthraquinones for which logK011 values of ca. 0.4–0.5 in water have been reported.72 Delocalization of the electron lone-pair borne by the aromatic nitrogen atom and intramolecular hydrogen-bonding account for this extremely low basicity.
In the 1.7–12.2 p[H] range, the broad absorption band observed under acidic conditions (λmax = 520 nm) progressively shifts upon base addition to lower energies in apparently two distinct stages, until reaching its final position at λmax = 559 nm (Fig. 4a). No further spectral changes could be observed above p[H] 8. However, a closer examination of the spectral data at ca. 387 or 700 nm revealed a more complex absorbance–p[H] profile that could only be accounted for by assuming the occurrence of at least 4 absorbing species corresponding to the protonation of the aliphatic amines of PENTAQ (Fig. S2, ESI†).
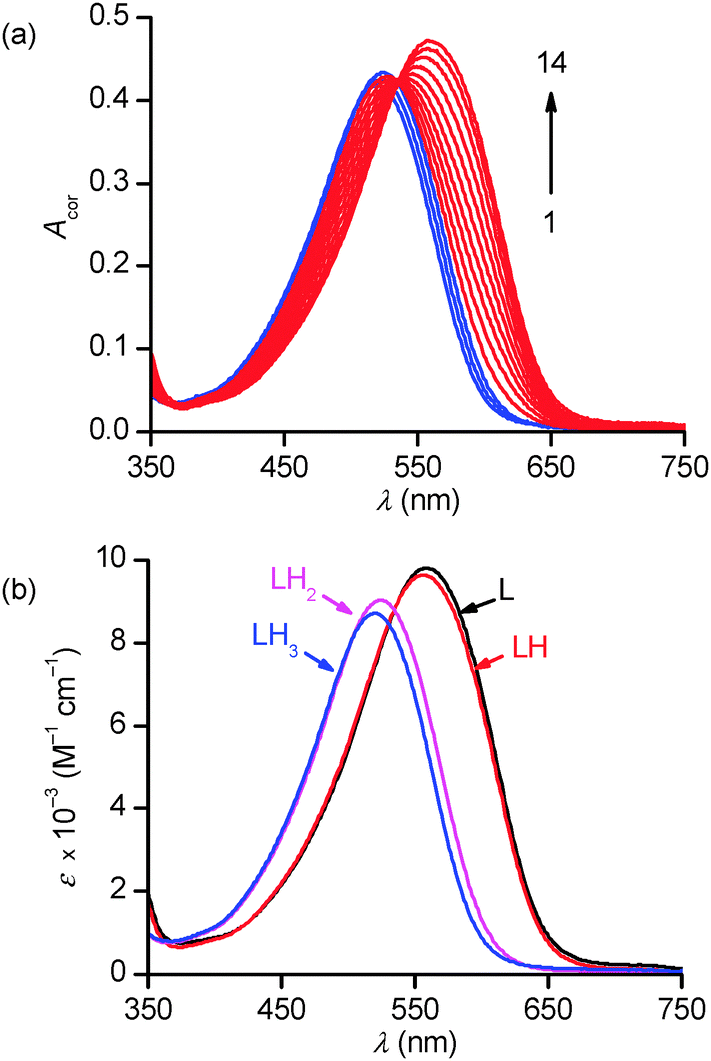 | ||
| Fig. 4 Spectrophotometric titration of PENTAQ (L) as a function of p[H] (a) and calculated electronic absorption spectra of L and its protonated forms (b) in CH3OH–H2O 80:20 v/v. [L]tot = 47 μM, I = 0.1 M N(CH3)4NO3, T = 298.2(2) K. Dilution-corrected spectra 1–14 (selected among 67): p[H] = 1.678, 2.386, 3.570, 4.627, 4.987, 5.227, 5.415, 5.608, 5.811, 6.050, 6.392, 6.972, 7.833, 11.922. | ||
Numerical data adjustment using the Specfit program73,74 with the model comprising the four L–LH33+ species was significantly better (r.s.d = 0.4–0.6 × 10−3) than with a 3-species (L–LH22+) model (r.s.d ∼ 1.0–1.2 × 10−3), in spite of the high similarities between the calculated spectra for L and LH+ on the one hand, and for those assigned to LH22+ and LH33+, on the other hand (Fig. 4b). The average values of the three refined stepwise protonation constants corresponding to 5 replicates are compiled in Table 2 together with literature values for other relevant ligands. The distribution diagram of PENTAQ and its protonated forms is shown in Fig. S3 (ESI†).
| PENTAQa | L1b | L2c | L3d | Cyclene | Cyclame | ||
|---|---|---|---|---|---|---|---|
|
a CH3OH–H2O 80:20 v/v, I = 0.1 M N(CH3)4NO3, T = 298.2(2) K.
b L1: 1-(2-dimethylaminoethylamino)anthraquinone, CH3OH–H2O 50:50 v/v, ref. 71.
c L2: 1,8-bis(2-dimethylaminoethylamino)anthraquinone, CH3OH–H2O 50:50 v/v, ref. 71.
d L3: N,N′-dimethylethylenediamine, H2O, I = 0.5 M KNO3, T = 303 K, ref. 75.
e H2O, I = 0.1 M, T = 298.2(2) K, ref. 76.
|
|||||||
| H+ | LogK011 |
7.2(1) | 7.96 | 7.91 | 10.17 | 10.65 | 11.4 |
| LogK012 |
5.51(3) | 5.09 | 7.30 | 9.64 | 10.28 | ||
| LogK013 |
2.5(1) | 1.4 | 1.6 | ||||
| LogK014 |
<0.5 | 0.7 | 2.1 | ||||
| Cu2+ | LogK110 |
8.60(4) | |||||
| LogK11–1 |
4.85(5) | ||||||
The protonation sequence for PENTAQ in solution can be established as follows (Scheme 2). In agreement with the X-ray crystal structure of [(PENTAQ)H]+ shown in Fig. 2a and the very similar absorption spectra calculated for the free-base and the monoprotonated form (Fig. 4b), the first added proton should mainly reside on the central nitrogen atom (N3) being the most remote from the anthraquinone fragment, affecting thereby only slightly the absorption properties of the chromophore. The second protonation produces both marked hypso- and hypochromic shifts of the absorption band, suggesting a molecular reorganization affecting nitrogen atoms quite close to the aromatic ring. Formation of a symmetric [(PENTAQ)H2]2+ species in which N2 and N4 are protonated while the middle amino group N3 is deprotonated, as observed in the crystal structure (Fig. 2b), would well account for the observed spectral changes and would be the most favorable arrangement for minimizing the electrostatic repulsion. Similar redistributions of protons in macrocyclic polyamines have been described elsewhere.77,78 In the third protonation step, the central N3 nitrogen atom is reprotonated, leaving the chromophore almost unaffected as evidenced by the close resemblance of the absorption spectra of [(PENTAQ)H2]2+ and [(PENTAQ)H3]3+. Finally, protonation occurs at the aromatic amines for acid concentrations higher than 0.5 M.
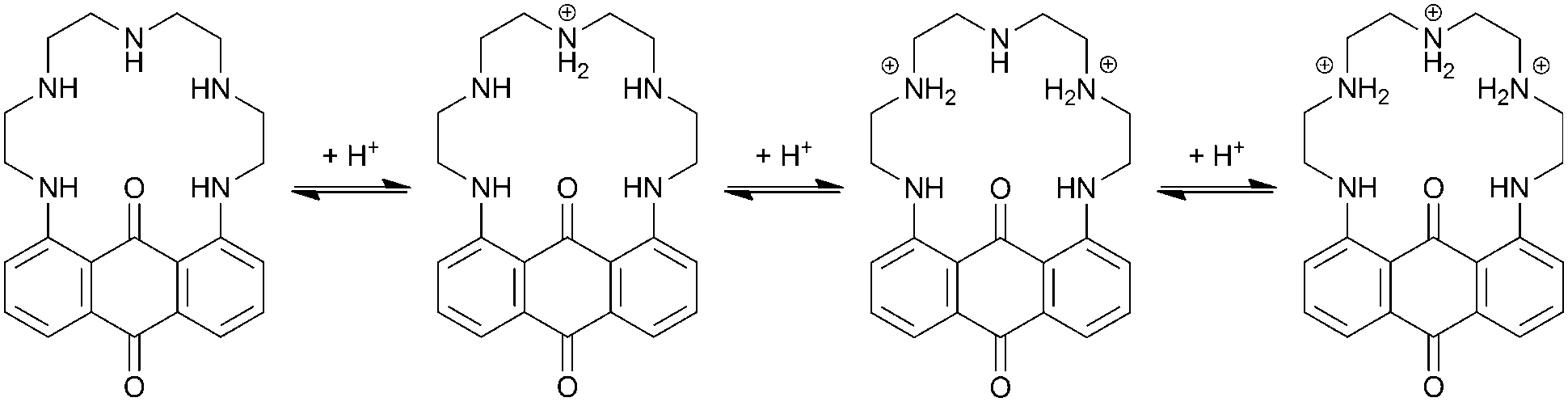 | ||
| Scheme 2 Proposed protonation sequence of the aliphatic chain of PENTAQ. The fourth protonation step occurs at the aromatic amino group (not shown). | ||
As can been seen in Table 2, the values found for the first and second protonation constants of PENTAQ are very close to those reported in the literature for related acyclic compounds, such as 1-(2-dimethylaminoethylamino)anthraquinone (L1) and 1,8-bis(2-dimethylaminoethylamino)anthraquinone (L2) in methanol–water 50:50 v/v,71 indicating that the steric strain induced by the ring closure does virtually not affect the basicity of the secondary aliphatic amines. However, these K011 and K012 values are significantly lower than those measured in water for N,N′-dimethylethylenediamine (DMED),75N-methyl- and N-ethyl-substituted diethylenetriamine,79 or for the saturated cyclic tetraamines cyclen and cyclam.76 Nevertheless, the ca. 3 log unit difference can to a large extent be explained by solvation effects and differences in solvent composition as it is well-known that the protonation constants of amines decrease by ca. 0.8 to 1.5 log units with respect to pure water when the methanol content is reaching 65–80%.80,81 The third protonation constant of PENTAQ (logK013 = 2.5) is three orders of magnitude lower than the second one due to intramolecular hydrogen bonding and electrostatic repulsion effects between neighboring secondary ammonium groups separated by an ethylene chain, as classically found for the cyclic tetraamines.
:20 v/v) have also been investigated by absorption spectrophotometry in the p[H] range 1.7–11.9. Under equimolar conditions (42.9 μM), fast binding kinetics was observed at p[H] values higher than 3, allowing the direct determination of the equilibrium constants by a stepwise addition of base (Fig. 5a). The spectral evolution upon raising the p[H] is compatible with the progressive deprotonation of the unbound ligand prevailing under acidic conditions (λmax = 520 nm) giving rise to bathochromic shifts with a concomitant splitting of the absorption band that spreads over the 400–850 nm range with a maximum centered at 658 nm. The almost 140 nm shift experienced between p[H] 1.7 and 8 (no significant changes occurring above) is responsible for a color change from pink to pale blue. Singular value decomposition by Specfit indicated the presence of five absorbing species. The best fit was achieved by a model assuming the formation of two complexes, [Cu(PENTAQ)]2+ [logK110 = 8.60(4)] and [Cu(PENTAQ)H–1]+ [logK11–1 = 4.85(5), eqn (1)] in equilibrium with the mono-, di-, and triprotonated ligand (Table 2). Unfortunately, direct comparison of our thermodynamic data with literature values is essentially meaningless, considering the differences in solvent composition.12,71,82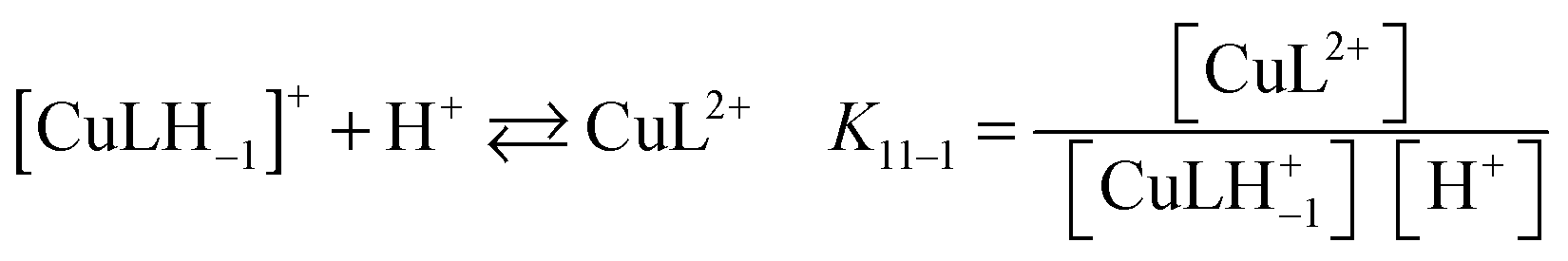 | (1) |
 | ||
| Fig. 5 Spectrophotometric titration of PENTAQ (L) in the presence of one equivalent of Cu2+ as a function of p[H] (a) and calculated electronic absorption spectra of CuL and CuLH–1 (b) in CH3OH–H2O 80:20 v/v. [L]tot = [Cu]tot = 42.9 μM, I = 0.1 M N(CH3)4NO3, T = 298.2(2) K. Dilution-corrected spectra 1–19 (selected among 95): p[H] = 1.691, 1.834, 2.096, 2.444, 3.266, 3.870, 4.011, 4.105, 4.218, 4.343, 4.477, 4.659, 4.897, 5.189, 5.522, 6.605, 8.175, 9.836, 11.584. | ||
The spectral changes accompanying the appearance of [Cu(PENTAQ)H−1]+ (Fig. 5b) are fully compatible with the copper-assisted deprotonation of the anthraquinolyl nitrogen atom and subsequent formation of a six-membered chelate cycle involving also the carbonyl oxygen atom. It can be also safely assumed that the adjacent ethylenediamine fragment N1–(CH2)2–N2 is involved in a second, five-membered chelate cycle.12,71,82
The distribution diagram shown in Fig. S4 (see ESI†) indicates that PENTAQ is an effective ligand for binding Cu2+ ions above p[H] 6 in the binary methanol–water mixture. Unfortunately, the rather limited water solubility hampers its use as a colorimetric copper(II) indicator for environmental or analytical purposes, but conversely PENTAQ is an appealing model compound for designing metal-responsive Langmuir-type films at an air/solution interface.
Langmuir monolayer formation
Surface pressure–area isotherms of PENTAQ monolayers on different subphases are shown in Fig. 6. The curve corresponding to a monolayer deposited on deionized water (pH ∼ 5.5) is significantly different from that formed on the surface of an alkaline solution (pH = 9.8, NaOH). On pure water, the limiting molecular area is rather low (28 Å per molecule), but this value increases to 37 Å2 per molecule when the subphase becomes basic, indicating that the pH of the aqueous phase significantly influences the structure of the monolayer as a consequence of the different protonation states of the molecule. | ||
| Fig. 6 Surface pressure–area isotherms of the PENTAQ monolayers deposited on deionized water at pH = 5.5 (1), a 0.1 mM Cu(ClO4)2 solution at pH = 9.8 (2), and an alkaline (NaOH) subphase at pH = 9.8 (3). T = 293(1) K. | ||
According to the solution studies performed in methanol–water 80:20 v/v, it can be reasonably anticipated that a film of PENTAQ deposited on the surface of pure water (pH ∼ 5) should be positively charged due to the partial protonation of the macrocycle (vide infra). In contrast, a solution with a bulk pH of 9.8 should be exclusively covered by non-protonated neutral PENTAQ molecules. Interestingly, Langmuir monolayers formed with a variety of amphiphilic compounds, including fatty acids and long-chain amines, are also pH dependent but an increase of the limiting molecular area is typically observed for charged molecules due to the electrostatic repulsion between the headgroups in the monolayers.83,84 Likewise, the surface pressure–area isotherms for N,N′′-di(n-cetyl)cyclen Langmuir monolayers follow the same trend upon acidifying the water subphase.63–68 This result was ascribed to both the electrostatic repulsions between positively charged macrocycles and to the increased rigidity of the tetraamine once protonated.62 The same behavior and rationale have been reported for cyclam and hexacyclen derivatives bearing long N-alkyl chains.69 Conversely, a significant decrease of the limiting molecular area has been observed for 1,4,7,10,13,16-hexakis((myristoyoxy)ethyl)-1,4,7,10,13,16-hexaazacyclooctadecane monolayers after lowering the pH of the aqueous subphase. In the latter case, a plausible explanation would be a structural modification of the monolayer originating from a departure of carboalkoxy groups from the surface.69
To explain the unexpected changes in the pressure–area isotherms recorded for PENTAQ, the electrostatic interactions have of course to be taken into consideration, but their influence is not limited to a simple intermolecular repulsion between molecules forming the monolayer. PENTAQ consists of two parts: a lipophilic anthraquinone motif floating on the air/water interface and a more hydrophilic pentaazaalkyl tether immersed in the aqueous bulk. Obviously, protonation of the secondary amines significantly increases the polarity of the chain, resulting in a better solvation by water molecules.85–87 While the aliphatic tether is plunging more deeply upon acidifying the solution, seaking to be remote from the less polar interfacial region, it can be reasonably assumed that the anthraquinone part takes also a different orientation at the interface, probably enforcing a less tilted position with respect to the horizontal plane (Fig. 7). Indeed, according to X-ray data of protonated forms described above, the PENTAQ molecule is flexible and can easily change its conformation depending on the degree of protonation and the formation of intermolecular hydrogen bonds.
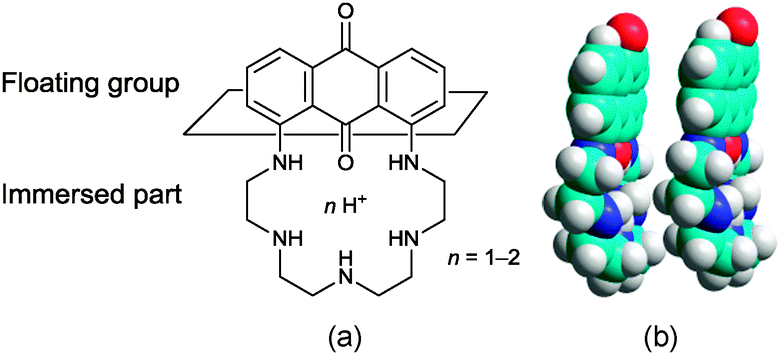 | ||
| Fig. 7 (a) Relative orientation of a protonated PENTAQ molecule in the monolayer at the air/water interface (pH = 5.5) and (b) structural model of a [(PENTAQ)H]+ cation pair corresponding to the closest approach distance based on sole steric considerations as calculated by HyperChem assuming the same macrocyclic conformation as found in the crystal structure of [(PENTAQ)H]CF3SO3·H2O. | ||
The observed limiting molecular area of 28 Å2 per molecule is in agreement with a projection area of 24 Å2 obtained for the orientation shown in Fig. 7. Closely packed PENTAQ monolayers are stabilized by face-to-face π–π stacking interactions.88 Moreover, this monolayer could be also stabilized by intermolecular hydrogen bonding between adjacent PENTAQ and water molecules. The low compressibility of the film is also in agreement with this structural proposal. Thus, protonated PENTAQ monolayers differ from those formed with fatty acids, amines and even alkylated polyazamacrocycles in that the positively charged groups are located rather far from the air/water interface as compared to other monolayers. Obviously, the electrostatic interactions impact in a different way the structure and the energy of these monolayers.
The increase of the limiting molecular area at higher pH values observed for PENTAQ monolayers can be ascribed to a different orientation of the floating anthraquinone fragment at the air/water interface. Indeed, the neutral free base is less polar compared to the protonated forms and therefore can be located closer to the interface. The electrostatic intermolecular interactions are therefore expected to be weaker in the film where the anthraquinone fragment can adopt a virtually horizontal layout at the air/water interface, thus forcing the pentaazamacrocycle to adopt a more folded conformation (Fig. 8). If two such units are brought together in order to maximize the overlap of the aromatic rings while avoiding a steric clash (Fig. 8), the calculated limiting area occupied by each molecule is 37 Å2, which again is in good agreement with the experimental results.
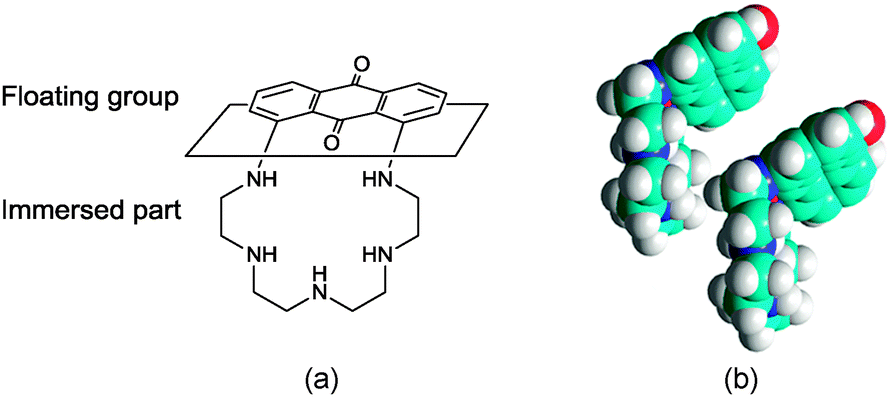 | ||
| Fig. 8 (a) Proposed relative orientation of the free-base PENTAQ molecule in the monolayer at the air/water interface (pH = 9.8) and (b) structural model of a folded PENTAQ pair corresponding to the closest approach distance based on sole steric considerations as calculated by HyperChem. The macrocyclic conformation found in the crystal structure of [(PENTAQ)H2](CF3SO3)2 was taken as a geometrical model for the folded free base (see the text for further explanations). | ||
To confirm these structural hypotheses, the different monolayers obtained at pH = 5.5 and 9.8 were further characterized by in situ UV-vis spectrophotometry (Fig. 9). On pure water, the intensity of the absorption band decreases with the molecular area occupied by each molecule on the surface. Moreover, the positions of three band maxima shown in Fig. 9a are similar and are blue-shifted by 25–30 nm compared to the solution spectrum of PENTAQ recorded in methanol–water (80:20 v/v). Such a shift can be ascribed to the formation of H-aggregates in the PENTAQ monolayer even for large monolayer areas (i.e. before the increase of the surface pressure). Such a situation is commonly encountered for Langmuir monolayers of amphiphilic aromatic derivatives adopting a vertical orientation with respect to the interface.88,89 If the absorption is normalized to the surface density of the monolayer, the intensity does not change below 40 Å2 per molecule, indicating the absence of structural modification during compression to higher surface pressures. These results further support the structural model for this monolayer shown in Fig. 7.
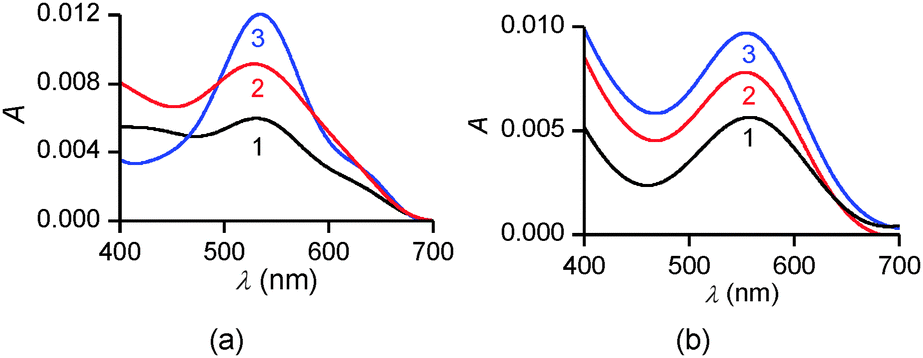 | ||
| Fig. 9 In situ UV-vis spectra of PENTAQ monolayers deposited on water subphases at pH = 5.5 (a) and 9.8 (b) for different areas: 100 (1a,b), 30 (2a,b), 20 (3a), and 25 Å2 per molecule (3b). | ||
In comparison, UV-vis absorption properties of PENTAQ monolayers on the alkaline subphase (Fig. 9b) differ from those of the monolayers at pH = 5.5. The band maximum is not shifted upon compression and appears at the same wavelength as in solution (λmax = 555 nm). This indicates the absence of self-assembly of PENTAQ molecules into H-aggregates which is prohibited by an unfavorable orientation of anthraquinone fragments on the surface. Such behavior is expected if the film packing leads to a tilted orientation of the anthraquinone moieties with respect to the interface (Fig. 8b). Moreover, our data confirm the structural stability of the monolayer until its collapse, as the molecular orientation at the interface doesn't change upon compression.
Copper(II) binding by Langmuir monolayers
At large molecular areas (60 Å2 per molecule), the UV-vis absorption properties (Fig. 10a) are similar to those found for a free-base PENTAQ solution in methanol–water (80:20 v/v) or the monolayer on an alkaline subphase (Fig. 9b). Nevertheless, the ICT band experiences a significant hypsochromic shift of 10 nm by compressing the monolayer. These data clearly rule out the formation of [Cu(PENTAQ)]2+ or [Cu(PENTAQ)H−1]+ complexes at the surfaces that should produce a strong bathochromic effect (Fig. 5). Conversely, the observed shift is compatible with the occurrence of free PENTAQ H-aggregates at the interface.
 | ||
| Fig. 10 In situ UV-vis spectra of a PENTAQ monolayer (a) deposited on an alkaline subphase (pH = 9.8) in the presence of Cu(ClO4)2 (0.1 mM) for different areas: 60 (1), 40 (2), 25 (3), and 20 Å2 per molecule (4); (b) collected from the surface and transferred to a quartz plate. | ||
Attempts to transfer by standard Langmuir–Blodgett procedure the monolayers onto quartz slides for subsequent spectroscopic characterization failed. Nevertheless, self-assembled films could be obtained in the following way. After compressing the monolayer at a surface pressure exceeding the collapse pressure, a quartz plate was introduced under this over-compressed film into the subphase. Then, the slide was pulled out while keeping a small angle between the solid substrate and the water surface.¶ Two air-dried probes prepared in this way have been characterized either directly after deposition (sample A) or after rinsing the plate for 30 s with deionized water (sample B). The absorption spectrum of sample A was similar to that of the Langmuir monolayer on an alkaline subphase, indicating the successful transfer of the film to the quartz slide (Fig. 10b).
XPS spectra of samples A and B evidenced two peaks at 285.1 and 399.9 eV, which are straightforwardly assigned to carbon (C 1s) and nitrogen (N 1s) atoms of uncharged secondary amines, respectively.91 In addition, sample A also showed two low-intensity features corresponding to binding energies of about 933 (Cu 2p3/2) and 952 eV (Cu 2p1/2), which support the occurrence of Cu+ and Cu2+ ions in the studied films (Fig. S5, ESI†).92,93 The presence of monovalent copper can be explained by X-ray-induced reduction of Cu2+ during sample irradiation. Nevertheless, the amount of copper ions in the film did not exceed 0.2%, while only trace amounts of copper could be detected after washing the film (sample B), suggesting most likely that copper was only physically adsorbed rather than being complexed by PENTAQ molecules. Contrary to all our expectations originating from the copper(II) binding affinity of cyclen-based monolayers64,65,94 and more generally of polyazamacrocycles in solution,21 self-assembled monolayers of PENTAQ do not ligate Cu2+ ions from an alkaline subphase.
In spite of it, spectrophotometric absorption data provided strong evidence for Cu2+ coordination by PENTAQ in the Langmuir monolayer floating above a methanol–water subphase (Fig. 11). Indeed, the spectrum exhibits two absorption bands with maxima located at 585 and 610 nm, both being bathochromically shifted with respect to the band seen in the absence of a metal salt (λmax = 555 nm). Most importantly, this spectral morphology corresponds to the signature of the [Cu(PENTAQ)]2+ complex in solution (Fig. 5b). In addition, the band intensity was shown to steadily increase upon compressing the monolayer, while the band shape remained unchanged. This is understandable because the number of molecules per unit area grows together with the applied pressure, an increase of the latter inducing no structural modification or aggregation in the monolayer.
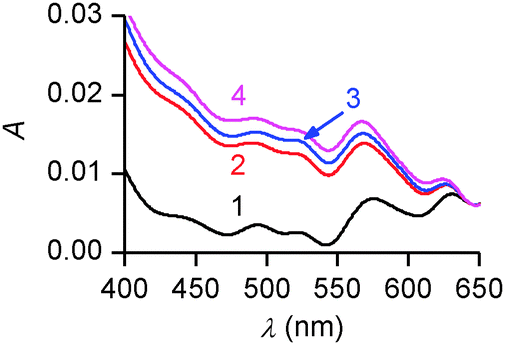 | ||
| Fig. 11
In situ UV-vis spectra of PENTAQ deposited on a methanol–water (20:80 v/v, pH* = 9.8) subphase in the presence of Cu(ClO4)2 (0.1 mM) for different monolayer areas: 140 (1), 125 (2), 100 (3), and 75 Å2 per molecule (4). Spectra have been normalized at 647 nm. | ||
Unfortunately, UV-vis and XPS studies evidenced no complex formation once the film was transferred according to the aforementioned procedure onto the surface of quartz plates. The UV-vis spectrum of the collected film was similar to that of the monolayer corresponding to the free ligand on an alkaline subphase. Likewise, the copper content found by XPS analysis did not exceed 1.6 atom% and, moreover, reached trace amounts after rinsing the sample with water. These data suggest a rather low stability of the copper complexes in the PENTAQ monolayer and their possible dissociation during the transfer onto the quartz slides. It turns out that only the free ligand is transferred onto the solid surface, whereas solvated copper ions remain in the liquid subphase.
The higher affinity of PENTAQ monolayers when methanol is added to the aqueous bulk solution might therefore be attributed to a higher local concentration of the organic solvent in the layer located just underneath the interface,95–97 as it is well known that metal–ligand complexes are stabilized under less-polar conditions. While the binding affinity becomes high enough for allowing the detection of submillimolar amounts of Cu2+ by PENTAQ spread over a methanol–water 20:80 v/v solution at pH* = 9.8, it is still too low for applying the LB film transfer technology.
Conclusions
Langmuir monolayer technology is a promising paradigm for the fast detection of toxic metal ions. As a first step towards that goal, a pentaazamacrocyclic chromogenic chemosensor incorporating an anthraquinone signaling unit (PENTAQ) has been designed. This ligand evidenced good discrimination ability for copper(II) with respect to divalent earth-alkaline, transition, and main group metal ions in methanol–water solutions, while exhibiting an immediate and reversible color change. Detailed spectrophotometric studies of the protonation and Cu2+ uptake highlighted the key role played by the copper-induced deprotonation of the aromatic amine in the optical response of the ligand. This original amphiphilic compound forms stable, albeit unusual, Langmuir monolayers on methanol–water subphases containing 80–100% of water. Most remarkably, the structure of the self-assembled layer turned out to be determined by the orientation of the bulky anthraquinone group which is influenced by the protonation state of the secondary polyamine chain immersed in the water subphase. As a consequence, the supramolecular film architecture was found to be highly pH dependent. As revealed by different analytical techniques, any polarity and solubility increase of PENTAQ resulting from the protonation of the aliphatic polyamine chain upon acidifying the aqueous phase enforces the tether to sink more deeply and, concomitantly, the lipophilic anthraquinone fragment to change its orientation by righting up with respect to the horizon. This pH-induced tuning of the monolayer structure is somehow reminiscent of the behaviour of the so-called schizophrenic micelles obtained from hydrophilic double block copolymers and should be taken into account in the future development of sensory Langmuir monolayers. Moreover, in situ fiber optic spectrophotometry ascertained the formation of PENTAQ H-aggregates on the water subphase at pH = 5.5.Optical detection of copper(II) could be ultimately achieved by PENTAQ monolayers spread over a 0.1 mM methanol–water solution (20:80 v/v, pH* = 9.8), thus validating our proof of concept. While we demonstrated thereby that amphiphilic anthraquinone-based chromogenic ligands possess the ability to self-assemble into stable Langmuir monolayers at air/solution interfaces, further fine-tuning of the molecular structure and binding affinity is required for improving the detection performances of the corresponding LB films. En route towards less pH-sensitive systems while maintaining excellent chromoionophoric properties with respect to copper and mercury, work involving open-chain aminoanthraquinone chemosensors bearing carbamoylmethylphosphoester groups is currently under progress.
Experimental
General consideration
Unless otherwise noted, all chemicals and starting materials were obtained commercially from Acros or Aldrich, and used without further purification. All metal salts used were perchlorates of general M(ClO4)n·xH2O formula. Caution! Although no problems were experienced, perchlorate salts are potentially explosive when combined with organic ligands and should be manipulated with care and used only in very small quantities. Dioxane was dried over metallic sodium and distilled before use. All reactions were performed in oven-dried glassware under a slight positive pressure of nitrogen. Analytical thin-layer chromatography (TLC) was carried out using Merck silica gel 60 plates (precoated sheets, 0.2 mm thick, with fluorescence indicator F254).NMR spectra were recorded on a 300 MHz Avance III NanoBay spectrometer from Bruker at the “Pôle Chimie Moléculaire”, the technological platform for chemical analysis and molecular synthesis (http://www.wpcm.fr) of the Institute of Molecular Chemistry of the University of Burgundy and Welience™. UV-vis spectra were collected either using a Cary 60 Probe or a Cary 5E (Agilent-Varian) spectrophotometer equipped with a double-jacketed cell holder connected to a RE 106 (Lauda) water circulator. The optical path length of the quartz cell (Hellma) was 1 cm. Unless otherwise noted (see the Solution thermodynamic measurements section), pH values are defined as −logaH3O+. Under such circumstances, the combined electrode connected to an Expert-pH (Econix, Russia) pH meter was calibrated using aqueous commercial buffers (pH = 4, 7, 10). According to this procedure, only apparent pH readings (denoted pH*) are reported for methanol–water 20:80 v/v mixtures, as no correction for the presence of the phase modifier was applied.
Synthesis of PENTAQ
PENTAQ was synthesized using a modified literature procedure.98 A 500 mL two-necked flask equipped with a magnetic stirrer and a back-flow condenser was charged with 1,8-dichloroanthraquinone (2.770 g, 10 mmol), tetraethylene-pentamine pentahydrochloride (3.715 g, 10 mmol), Pd(dba)2 (0.46 g, 8 mol%), BINAP (1.0 g, 16 mol%) and cesium carbonate (32.5 g, 100 mmol). The reaction vessel was evacuated and purged with N2 three times. Subsequently, 300 mL of dioxane was added and the mixture was stirred at reflux for 3 d. After cooling, the reaction mixture was filtered and the solvent evaporated under vacuum. The residue was subjected to column chromatography on silica gel using CH2Cl2–CH3OH (30%)/Et3N (1–5%) as eluent. PENTAQ was isolated as a violet solid in 41% yield (1.61 g). The spectral data of the isolated compound were in good agreement with the literature data.98Visual detection of metal ions
The chemosensing properties of PENTAQ in the presence of metal ions were evaluated visually and by UV-vis spectrophotometry by adding 1 equiv. of a 0.05–0.10 M aqueous solution of metal perchlorate to PENTAQ (c = 80 μM) dissolved in a binary methanol–water solvent mixture (50:50 v/v).
X-ray crystallography
Light violet prismatic crystals of [(PENTAQ)H]CF3SO3·H2O and red prismatic crystals of [(PENTAQ)H2](CF3SO3)2 were accidentally obtained by slow evaporation of an equimolar mixture of free ligand and copper(II) triflate in either chloroform or methanol containing traces of acid. All other attempts to grow single crystals of the copper complexes failed.![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 7.1884(2) Å, b = 11.2777(3) Å, c = 16.3837(6) Å, α = 107.004(2)°, β = 93.659(2)°, γ = 102.998(1)°, V = 1225.57(7) Å3, Z = 2, T = 115.0 K, μ(Mo-Kα1) = 0.206 mm−1, dcalc = 1.522 g cm−3, 9265 reflections measured (2.624 ≤ 2θ ≤ 54.964°), 5578 unique (Rint = 0.0413) which were used in all calculations. The final R1 was 0.0976 (I > 2σ(I)) and wR2 was 0.3203 (all data).
(no. 2), a = 9.2673(2) Å, b = 10.8959(3) Å, c = 15.1558(5) Å, α = 94.759(1)°, β = 91.357(1)°, γ = 105.731(1)°, V = 1466.27(7) Å3, Z = 2, T = 115(2) K, μ(Mo-Kα1) = 0.276 mm−1, dcalc = 1.571 g cm−3, 12162 reflections measured (5.734 ≤ 2θ ≤ 55.116°), 6709 unique (Rint = 0.0367) which were used in all calculations. The final R1 was 0.0443 (I > 2σ(I)) and wR2 was 0.1146 (all data).
(no. 2), a = 7.1884(2) Å, b = 11.2777(3) Å, c = 16.3837(6) Å, α = 107.004(2)°, β = 93.659(2)°, γ = 102.998(1)°, V = 1225.57(7) Å3, Z = 2, T = 115.0 K, μ(Mo-Kα1) = 0.206 mm−1, dcalc = 1.522 g cm−3, 9265 reflections measured (2.624 ≤ 2θ ≤ 54.964°), 5578 unique (Rint = 0.0413) which were used in all calculations. The final R1 was 0.0976 (I > 2σ(I)) and wR2 was 0.3203 (all data).
(no. 2), a = 9.2673(2) Å, b = 10.8959(3) Å, c = 15.1558(5) Å, α = 94.759(1)°, β = 91.357(1)°, γ = 105.731(1)°, V = 1466.27(7) Å3, Z = 2, T = 115(2) K, μ(Mo-Kα1) = 0.276 mm−1, dcalc = 1.571 g cm−3, 12162 reflections measured (5.734 ≤ 2θ ≤ 55.116°), 6709 unique (Rint = 0.0367) which were used in all calculations. The final R1 was 0.0443 (I > 2σ(I)) and wR2 was 0.1146 (all data).
Solution thermodynamic measurements
:20 v/v solvent mixture. The 0.1 M HNO3 solution was obtained by diluting a Merck concentrate (Titrisol®) in 500 mL of methanol. The 1 L volumetric flask was then filled up to the mark with water. The 0.1 M N(CH3)4OH solution was prepared likewise by dilution of a commercial solution (Aldrich, 25 wt% in water). Their exact titer was determined by standardizing them against oven-dried (120 °C for 2 h) Tris buffer (Aldrich-Sigma, 99.9%) and potassium hydrogen phthalate (Aldrich-Sigma, 99.99%), respectively. Equivalence points were calculated by the second-derivative method. The concentration of the standardized solutions corresponded to the average of at least five replicates and was known with a relative precision better than 0.15%. They were stored under purified argon using Ascarite II (Acros, 20–30 mesh) scrubbers in order to prevent absorption of carbon dioxide. The ∼0.01 M copper nitrate solutions were prepared in the solvent mixture and standardized by complexometric titration against 0.1000 M Na2H2EDTA (Titriplex III®) according to established procedures.17 Ligand stock solutions were prepared by careful weighing using a Precisa 262SMA-FR balance (precision: ±0.01 mg).
:20 v/v). Calibration data (1.7 ≤ p[H] ≤ 11.9) were processed according to the four-parameter extended Nernst equation [eqn (2)], which takes into account the standard potential (E0), the Nernst slope (S), and the correction terms accounting for the changes in the liquid junction potential in the acidic (Ja) and the alkaline (Jb) region.102 In addition, the base-concentration factor (γ) was also allowed to refine, whereas the ionic product of water was fixed (pKw = −14.09 was determined according to the method of Fischer and Byé).103| E = E0 + S log[H+] + Ja [H+] + JbKw [H+]−1 | (2) |
Visible absorption spectra were recorded in situ as a function of p[H] using a Cary 50 Probe (Varian) spectrophotometer equipped with an immersion probe of 1 cm path length made of SUPRASIL® 300 (Hellma, reference 661.202). The same titration cell and electrode calibration procedure as described above were used. Aliquots of base were added manually with the help of a Gilmont micropipette (2 μL resolution) to ca. 10−4 M ligand or complex solutions. Enough time was allowed after the addition of each base increment in order to reach the equilibrium. The potential-drift criterion was set at dE/dt < 0.1 mV min−1. The collection of absorption spectra was repeated with 2 min delays between two consecutive measurements until identical spectra were obtained. The entire multiwavelength datasets were decomposed into their principal components by factor analysis before adjusting the equilibrium constants and extinction coefficients by nonlinear least-squares analysis with the Specfit program.73,74 When processing metal/ligand titrations, the extinction coefficients for PENTAQ and [(PENTAQ)H]+ had to be fixed to the average values found for the ligand alone titrations (Fig. 5b) in order to reach convergence, while those for [(PENTAQ)H2]2+ and [(PENTAQ)H3]3+ could be adjusted as these two species prevail below p[H] 4. Comparison of the corresponding experimental and calculated spectra was used as a criterion for evaluating the refinement consistency and accuracy of the model. Distribution diagrams were computed with the Hyss program.104
Surface chemistry and spectroscopic characterizations
:20 v/v) mixture. The pH was adjusted to 9.8 using 0.1 M NaOH. The absence of Cu(OH)2 precipitate in the freshly prepared basic solutions was checked using light scattering techniques. When precipitation occurred, usually after a few hours, the stock solutions were discarded.
Acknowledgements
This work was carried out in the frame of the International Associated French-Russian Laboratory of Macrocycle Systems and Related Materials (LAMREM) of the Centre National de la Recherche Scientifique (CNRS) and the Russian Academy of Sciences (RAS). It was financially supported by the ARCUS Bourgogne-Russie project, the CNRS, the Conseil Régional de Bourgogne (program PARI IME SMT8), and the Russian Foundation for Basic Research (project 12-03-93105). L.B. thanks the Erasmus program for a travel grant. We thank also M. E. Espin and F. Dubare for preliminary solution thermodynamic studies, and Dr P. Richard for helpful discussions.Notes and references
- J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551–8588 CrossRef CAS PubMed.
- A. P. de Silva, J. Phys. Chem. Lett., 2011, 2, 2865–2871 CrossRef CAS.
- M. Formica, V. Fusi, L. Giorgi and M. Micheloni, Coord. Chem. Rev., 2012, 256, 170–192 CrossRef CAS PubMed.
- N. Kaur and S. Kumar, Tetrahedron, 2011, 67, 9233–9264 CrossRef CAS PubMed.
- I. Leray and B. Valeur, Eur. J. Inorg. Chem., 2009, 3525–3535 CrossRef CAS.
- L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59–83 CrossRef CAS.
- X. Qian, Y. Xiao, Y. Xu, X. Guo, J. Qian and W. Zhu, Chem. Commun., 2010, 46, 6418–6436 RSC.
- D. T. Quang and J. S. Kim, Chem. Rev., 2010, 110, 6280–6301 CrossRef CAS PubMed.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed.
- B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS.
- E. Ranyuk, C. Morkos-Douaihy, A. Bessmertnykh, F. Denat, A. Averin, I. Beletskaya and R. Guilard, Org. Lett., 2009, 11, 987–990 CrossRef CAS PubMed.
- E. Ranyuk, A. Uglov, M. Meyer, A. Bessmertnykh-Lemeune, F. Denat, A. Averin, I. Beletskaya and R. Guilard, Dalton Trans., 2011, 40, 10491–10502 RSC.
- E. Ermakova, J. Michalak, M. Meyer, V. Arslanov, A. Tsivadze, R. Guilard and A. Bessmertnykh-Lemeune, Org. Lett., 2013, 15, 662–665 CrossRef CAS PubMed.
- D. Prabhakaran, H. Nanjo and H. Matsunaga, Anal. Chim. Acta, 2007, 601, 108–117 CrossRef CAS PubMed.
- D. Prabhakaran, M. Yuehong, H. Nanjo and H. Matsunaga, Anal. Chem., 2007, 79, 4056–4065 CrossRef CAS PubMed.
- K. B. Blodgett and I. Langmuir, Phys. Rev., 1937, 51, 964–982 CrossRef CAS.
- Langmuir-Blodgett Films, ed. G. G. Roberts, Plenum Press, New York, 1990 Search PubMed.
- M. C. Petty, Langmuir Blodgett Films – An Introduction, Cambridge University Press, New York, 1996 Search PubMed.
- F. Fages, J.-P. Desvergne, H. Bouas-Laurent, J.-M. Lehn, Y. Barrans, P. Marsau, M. Meyer and A.-M. Albrecht-Gary, J. Org. Chem., 1994, 59, 5264–5271 CrossRef CAS.
- E. U. Akkaya, M. E. Huston and A. W. Czarnik, J. Am. Chem. Soc., 1990, 112, 3590–3593 CrossRef CAS.
- R. M. Izatt, K. Pawlak, J. S. Bradshaw and R. L. Bruening, Chem. Rev., 1995, 95, 2529–2586 CrossRef CAS.
- A. Beeby, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1996, 1565–1580 RSC.
- M. Kadarkaraisamy and A. G. Sykes, Polyhedron, 2007, 26, 1323–1330 CrossRef CAS PubMed.
- I. K. Lednev and M. C. Petty, Adv. Mater., 1996, 8, 615–630 CrossRef CAS.
- L. M. Goldenberg, J. F. Biernat and M. C. Petty, Langmuir, 1998, 14, 1236–1241 CrossRef CAS.
- P. Kele, J. Orbulescu, T. L. Calhoun, R. E. Gawley and R. M. Leblanc, Langmuir, 2002, 18, 8523–8526 CrossRef CAS.
- T. I. Sergeeva, S. P. Gromov, A. I. Vedernikov, M. S. Kapichnikova, M. V. Alfimov, V.-T. Lieu, D. Möbius, M. S. Tsarkova and S. Y. Zaitsev, Colloids Surf., A, 2005, 255, 201–209 CrossRef CAS PubMed.
- K. Wojciechowski, D. Grigoriev, R. Ferdani and G. W. Gokel, Langmuir, 2006, 22, 8409–8415 CrossRef CAS PubMed.
- R. D. Hancock and A. E. Martell, Chem. Rev., 1989, 89, 1875–1914 CrossRef CAS.
- R. D. Hancock, P. W. Wade, M. P. Ngwenya, A. S. De Sousa and K. V. Damu, Inorg. Chem., 1990, 29, 1968–1974 CrossRef CAS.
- S. F. Lincoln, Coord. Chem. Rev., 1997, 166, 255–289 CrossRef CAS.
- M. Meyer, V. Dahaoui-Gindrey, C. Lecomte and R. Guilard, Coord. Chem. Rev., 1998, 178–180, 1313–1405 CrossRef CAS.
- D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977–2010 CrossRef CAS PubMed.
- R. Delgado, V. Felix, L. M. P. Lima and D. W. Price, Dalton Trans., 2007, 2734–2745 RSC.
- J. Geduhn, T. Walenzyk and B. Koenig, Curr. Org. Synth., 2007, 4, 390–412 CrossRef CAS.
- P. Hermann, J. Kotek, V. Kubicek and I. Lukes, Dalton Trans., 2008, 3027–3047 RSC.
- C. Bazzicalupi, A. Bencini, A. Bianchi, A. Danesi, E. Faggi, C. Giorgi, S. Santarelli and B. Valtancoli, Coord. Chem. Rev., 2008, 252, 1052–1068 CrossRef CAS PubMed.
- S. Shinoda, Chem. Soc. Rev., 2013, 42, 1825–1835 RSC.
- E. Kimura, Tetrahedron, 1992, 48, 6175–6217 CrossRef CAS.
- D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1995, 1305–1314 RSC.
- T. Koike, T. Watanabe, S. Aoki, E. Kimura and M. Shiro, J. Am. Chem. Soc., 1996, 118, 12696–12703 CrossRef CAS.
- K. Kubo and T. Sakurai, Chem. Lett., 1996, 959–960 CrossRef CAS.
- J. Yoon, N. E. Ohler, D. H. Vance, W. D. Aumiller and A. W. Czarnik, Tetrahedron Lett., 1997, 38, 3845–3848 CrossRef CAS.
- P. Ghosh, P. K. Bharadwaj, J. Roy and S. Ghosh, J. Am. Chem. Soc., 1997, 119, 11903–11909 CrossRef CAS.
- E. Kimura and T. Koike, Chem. Soc. Rev., 1998, 27, 179–184 RSC.
- E. Kimura and S. Aoki, Biometals, 2001, 14, 191–204 CrossRef CAS.
- E. Kimura, S. Aoki, E. Kikuta and T. Koike, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3731–3736 CrossRef CAS PubMed.
- N. C. Lim, L. Yao, H. C. Freake and C. Brückner, Bioorg. Med. Chem. Lett., 2003, 13, 2251–2254 CrossRef CAS.
- R. Meallet-Renault, R. Pansu, S. Amigoni-Gerbier and C. Larpent, Chem. Commun., 2004, 2344–2345 RSC.
- S.-Y. Moon, N. J. Youn, S. M. Park and S.-K. Chang, J. Org. Chem., 2005, 70, 2394–2397 CrossRef CAS PubMed.
- N. J. Youn and S.-K. Chang, Tetrahedron Lett., 2005, 46, 125–129 CrossRef CAS PubMed.
- G. Bergamini, P. Ceroni, V. Balzani, L. Cornelissen, J. van Heyst, S.-K. Lee and F. Vogtle, J. Mater. Chem., 2005, 15, 2959–2964 RSC.
- S. H. Kim, J. S. Kim, S. M. Park and S.-K. Chang, Org. Lett., 2006, 8, 371–374 CrossRef CAS PubMed.
- S. M. Park, M. H. Kim, J.-I. Choe, K. T. No and S.-K. Chang, J. Org. Chem., 2007, 72, 3550–3553 CrossRef CAS PubMed.
- K.-C. Song, M. H. Kim, H. J. Kim and S.-K. Chang, Tetrahedron Lett., 2007, 48, 7464–7468 CrossRef CAS PubMed.
- H. Mu, R. Gong, Q. Ma, Y. Sun and E. Fu, Tetrahedron Lett., 2007, 48, 5525–5529 CrossRef CAS PubMed.
- Y. Shiraishi, S. Sumiya, Y. Kohno and T. Hirai, J. Org. Chem., 2008, 73, 8571–8574 CrossRef CAS PubMed.
- S. E. Plush, S. F. Lincoln and K. P. Wainwright, Inorg. Chim. Acta, 2009, 362, 3097–3103 CrossRef CAS PubMed.
- H. Kim, K.-S. Moon, S. Shim and J. Tae, Chem.–Asian J., 2011, 6, 1987–1991 CrossRef CAS PubMed.
- M. Li, H.-Y. Lu, R.-L. Liu, J.-D. Chen and C.-F. Chen, J. Org. Chem., 2012, 77, 3670–3673 CrossRef CAS PubMed.
- H. Knobloch, S. Woigk, L. Brehmer, R. Yusupov and A. Rakhnyanskaya, Thin Solid Films, 1995, 265, 89–91 CrossRef CAS.
- M. A. Kalinina, V. V. Arslanov, L. A. Tsar'kova and A. A. Rakhnyanskaya, Colloid J., 2000, 62, 545–549 CAS.
- M. A. Kalinina, V. V. Arslanov, L. A. Tsar'kova, V. D. Dolzhikova and A. A. Rakhnyanskaya, Colloid J., 2001, 63, 312–317 CrossRef CAS.
- M. A. Kalinina and V. V. Arslanov, Colloid J., 2002, 64, 49–55 CrossRef CAS.
- M. A. Kalinina, V. V. Arslanov and S. Z. Vatsadze, Colloid J., 2003, 65, 177–185 CrossRef CAS.
- M. A. Kalinina, O. A. Raitman, S. L. Selector, D. S. Turygin and V. V. Arslanov, IEEE Sens. J., 2006, 6, 450–457 CrossRef CAS.
- D. S. Turygin, M. Subat, O. A. Raitman, S. L. Selector, V. V. Arslanov, B. König and M. A. Kalinina, Langmuir, 2007, 23, 2517–2524 CrossRef CAS PubMed.
- M. A. Kalinina, V. V. Arslanov, D. S. Turygin and I. A. Gagina, Colloid J., 2009, 71, 627–633 CrossRef CAS.
- C. Mertesdorf, T. Plesnivy, H. Ringsdorf and P. A. Suci, Langmuir, 1992, 8, 2531–2537 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- N. Kaur and S. Kumar, Dalton Trans., 2006, 3766–3771 RSC.
- T. Shibusawa, T. Saito and T. Hamayose, Nippon Kagaku Kaishi, 1977, 264–271 CrossRef CAS.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 95–101 CrossRef CAS.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 251–264 Search PubMed.
- P. Paoletti, R. Barbucci, A. Vacca and A. Dei, J. Chem. Soc. A, 1971, 310–313 RSC.
- A. E. Martell, R. M. Smith and R. J. Motekaitis, NIST Critically Selected Stability Constants of Metal Complexes Database, NIST Standard Reference Database No. 46, Gaithersburg, MD, 2004 Search PubMed.
- M. Meyer, L. Frémond, A. Tabard, E. Espinosa, G. Y. Vollmer, R. Guilard and Y. Dory, New J. Chem., 2005, 29, 99–108 RSC.
- J.-C. Chambron and M. Meyer, Chem. Soc. Rev., 2009, 38, 1663–1673 RSC.
- J. W. Allison and R. J. Angelici, Inorg. Chem., 1971, 10, 2233–2238 CrossRef CAS.
- C. L. De Ligny, Recl. Trav. Chim. Pays-Bas, 1960, 79, 731 CrossRef CAS.
- D. B. Rorabacher, W. J. MacKellar, F. R. Shu and M. Bonavita, Anal. Chem., 1971, 43, 561 CrossRef CAS.
- K. Kaur and S. Kumar, Tetrahedron, 2010, 66, 6990–7000 CrossRef CAS PubMed.
- A. Datta, J. Kmetko, A. G. Richter, C. J. Yu, P. Dutta, K. S. Chung and J. M. Bai, Langmuir, 1999, 16, 1239–1242 CrossRef.
- G. Gabrielli, G. Caminati and M. Puggelli, Adv. Colloid Interface Sci., 2000, 87, 75–111 CrossRef CAS.
- G. L. Gaines, Insoluble monolayers at liquid-gas interfaces, Interscience Publishers, New York, 1966 Search PubMed.
- G. S. Patil, R. H. Matthews and D. G. Cornwell, J. Lipid Res., 1976, 17, 197–202 CAS.
- S. P. Walsh and J. B. Lando, Langmuir, 1994, 10, 252–256 CrossRef CAS.
- M. Kadarkaraisamy and A. G. Sykes, Inorg. Chem., 2006, 45, 779–786 CrossRef CAS PubMed.
- B. V. Shankar and A. Patnaik, Langmuir, 2006, 22, 4758–4765 CrossRef CAS PubMed.
- S. Selector, O. Fedorova, E. Lukovskaya, A. Anisimov, Y. Fedorov, N. Tarasova, O. Raitman, F. Fages and V. Arslanov, J. Phys. Chem. B, 2012, 116, 1482–1490 CrossRef CAS PubMed.
- R. A. Ando, G. M. do Nascimento, R. Landers and P. S. Santos, Spectrochim. Acta, Part A, 2008, 69, 319–326 CrossRef PubMed.
- G. Z. Xing, J. B. Yi, J. G. Tao, T. Liu, L. M. Wong, Z. Zhang, G. P. Li, S. J. Wang, J. Ding, T. C. Sum, C. H. A. Huan and T. Wu, Adv. Mater., 2008, 20, 3521–3527 CrossRef CAS.
- S.-W. Cao, Y.-J. Zhu, J. Wu, K.-W. Wang and Q.-L. Tang, Nanoscale Res. Lett., 2010, 5, 781–785 CrossRef CAS PubMed.
- M. A. Kalinina, V. V. Arslanov, D. S. Turygin, E. Y. Tereshchenko and S. I. Zheludeva, Russ. J. Phys. Chem. A, 2008, 82, 623–629 CAS.
- J. A. Morrone, K. E. Haslinger and M. E. Tuckerman, J. Phys. Chem. B, 2006, 110, 3712–3720 CrossRef CAS PubMed.
- L. B. Partay, P. Jedlovszky, A. Vincze and G. Horvai, J. Phys. Chem. B, 2008, 112, 5428–5438 CrossRef CAS PubMed.
- L. Dougan, S. P. Bates, R. Hargreaves, J. P. Fox, J. Crain, J. L. Finney, V. Reat and A. K. Soper, J. Chem. Phys., 2004, 121, 6456–6462 CrossRef CAS PubMed.
- I. P. Beletskaya, A. G. Bessmertnykh, A. D. Averin, F. Denat and R. Guilard, Eur. J. Org. Chem., 2005, 281–305 CrossRef CAS.
- Z. Otwinowski and W. Minor, in Methods Enzymol., ed. C. W. Carter, R. M. Sweet, J. N. Abelson and M. I. Simon, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
- A. Avdeef and J. J. Bucher, Anal. Chem., 1988, 50, 2137–2142 CrossRef.
- R. Fischer and J. Byé, Bull. Soc. Chim. Fr., 1964, 2920–2929 CAS.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Hydrogen bond distances and angles, spectrophotometric titration of PENTAQ in the high acidity range, absorbance–p[H] titration profiles at 700 and 387 nm of PENTAQ, species distribution diagrams for PENTAQ and Cu2+/PENTAQ systems as a function of p[H], XPS spectra of PENTAQ monolayers collected from the subphase surface. CCDC 945039 and 945040. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj01121f |
| ‡ On leave from Higher Chemical College, Russian Academy of Sciences, Moscow, Russia. |
| § Erasmus fellow on leave from the Ecole d'Ingénieurs et d'Architectes de Fribourg, boulevard de Pérolles 80, 1705 Fribourg, Switzerland. |
| ¶ Close structural analogy of films collected from the subphase by this way and genuine LB films is supported by the collapse mechanism of the monolayers. Indeed, compression above the 2D–3D transition point leads to bilayer structures which are chaotically spread over the surface. Nevertheless, the molecular organization inside the film has minor importance in the present case, as we are only interested in its composition probed by UV-vis and XPS techniques. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |