(4-Ferrocenylphenyl)propargyl ether derived carbohydrate triazoles: influence of a hydrophobic linker on the electrochemical and cytotoxic properties†
Sadanala Bhavya
Deepthi
a,
Rajiv
Trivedi
*a,
Lingamallu
Giribabu
a,
Pombala
Sujitha
b,
C.
Ganesh Kumar
b and
B.
Sridhar
c
aInorganic and Physical Chemistry Division, CSIR-Indian Institute of Chemical Technology (IICT), Hyderabad, Andhra Pradesh, India 500 007. E-mail: trivedi@iict.res.in; rtrajiv401@gmail.com; Fax: +91-40-2716 0921; Tel: +91-40-27191667
bChemical Biology Laboratory, Medicinal Chemistry and Pharmacology Division, CSIR-IICT, Hyderabad, Andhra Pradesh, India 500 007
cCentre for X-ray crystallography, CSIR-IICT, Hyderabad, Andhra Pradesh, India 500 007
First published on 2nd October 2013
Abstract
In this article we describe a detailed study of the influence of a hydrophobic linker on the properties of a series of ferrocene–carbohydrate conjugates. The alkyne, (4-ferrocenylphenyl)propargyl ether, was prepared from the reaction of propargyl bromide and 4-ferrocenylphenol. The structure of the alkyne was determined using a single crystal X-ray diffraction study. A benzyl triazole (3a) was synthesised for comparison purposes in the electrochemical and cytotoxic study. Herein, we report the synthesis and characterization of a series of carbohydrate triazole conjugates (3b–h) by using the Huisgen 1,3-dipolar cycloaddition reaction of a series of carbohydrate azides and (4-ferrocenylphenyl)propargyl ether. UV-visible spectroscopy and electrochemical studies were performed on the alkyne and triazole compounds. The data generated from the cyclic voltammetric studies was used to calculate the diffusion coefficients (Df) of the ferrocene–carbohydrate conjugates in DMSO and DMSO–buffer solutions. Slightly higher diffusion coefficient values were obtained for the carbohydrate triazole conjugates (3b–h) in the buffer solution, perhaps due to the hydrophobic nature of the linker and the absence of a hydrogen-bonding substituent adjacent to the ferrocene core. Furthermore, the triazole conjugate (3f) derived from ribofuranose exhibited significant cytotoxicity against a hormone dependent breast cancer cell line (MCF-7) with an IC50 value lower than 10 μM.
Introduction
Over the past decade, ferrocifen,1,2 ferroquine3 and their analogues4 have made a tremendous impact on the development of bioorganometallic compounds of ferrocene,5–8 particularly for their role in the exploration of anticancer and antimalarial drugs. During the course of such studies, it has been observed that the formation of a ferrocenium ion via reversible one electron oxidation is often cited as one of the factors that determines the biological activity.9,10 Although the actual mechanism by which ferrocenium salts exhibit cytotoxicity is unknown, Osella and co-workers have reported that under physiological conditions ferrocenium salts produce hydroxyl radicals (HO˙) leading to the oxidative damage of DNA.11The spacer or linker that connects the ferrocene to the organic scaffold also plays a significant role, contributing towards the bioactivity of the molecule as a whole.12 Several biologically important moieties such as amides, esters, triazoles, pyrazoles, imidazoles etc., have been reported to form bridges between ferrocene and organic or biomolecular fragments.13 Few of these linkers have been capable of stabilizing their organometallic conjugates in organic or buffer solutions via strong hydrogen bonds.14,15 Among these linkers, triazoles have certain advantages such as their facile synthesis via the Huisgen 1,3-dipolar cycloaddition reaction16 and their ability to act as peptide bioisosteres.17 Metzler-Nolte and co-workers have reported the synthesis and biological study of triazole containing ferrocene conjugates of peptides and PNA oligomers.18–20 However, there have been instances, as reported by Kenny and co-workers, where the ferrocene and biomolecules are connected via benzoyl or naphthyl moieties to form aryl ferrocene conjugates.21–23 Presumably, the conjugated aromatic linker lowers the redox potential of the ferrocene conjugate, thereby assisting oxidation to the ferrocenium species. Extensive research has been devoted to developing aryl ferrocenyl amino acid and peptide conjugates that can interact with other biomolecules via hydrogen bonding.21–23 In contrast, reports on the study of aryl ferrocene conjugates of other biomolecules such as carbohydrates are scarce.
Besides their crucial role in biology, carbohydrates possess a unique set of chemical and structural features that make them particularly attractive as molecular scaffolds. Ferrocene–carbohydrate conjugates have been at the forefront for use as electrochemical probes24–26 and as biologically active compounds.27–31 Most biologically active ferrocene–carbohydrate conjugates have been prepared from sugars such as glucose and glucosamine and they have demonstrated significant antimalarial activity.32–35
From our recent findings, it was observed that the linker connecting the ferrocene and carbohydrate scaffold played a significant role in the cytotoxic activity exhibited by the conjugates.36–38 Promising cytotoxicity was observed using amide-triazole linkers, which was to some extent due to their hydrogen bonding properties.38 In order to investigate the importance of the presence of hydrogen bonding groups in ferrocene–carbohydrate conjugates, herein we describe the synthesis of new ferrocene–carbohydrate conjugates, in which the ferrocene and carbohydrate scaffolds are connected through phenoxymethyl triazole linkers. This work also provides a detailed cyclic voltammetric study of the ferrocene–carbohydrate conjugates in polar and buffer solutions under physiological pH. The anticancer activity of the compounds was also studied using different cancer cell lines.
Experimental section
Optical rotations were determined using a JASCO J-120 polarimeter. THF was distilled over sodium pieces under an argon atmosphere. 1H NMR spectra were recorded in CDCl3 solutions using a 300 MHz (Avance 300) instrument. Chemical shifts for the protons were reported using tetramethylsilane (TMS) as an internal reference. 13C NMR spectra were recorded at 75.5 MHz on an Avance 300 instrument and the carbon shifts are referenced to the 13C signal of CDCl3 at 77.0 ppm. Coupling constants (J) are expressed in Hz. Infrared spectra were obtained with a Thermo Nicolet Nexus 670 spectrometer using KBr discs. Melting points were determined with a Toshniwal melting point apparatus and are uncorrected. The UV-visible spectra were recorded with a Varian Cary 500 spectrophotometer over the range 250–550 nm using 1 cm path length cuvettes. Cyclic voltammetry (CV) was performed with a conventional three-electrode configuration consisting of glassy carbon as a working electrode, platinum as an auxiliary electrode and a saturated calomel electrode (SCE) as a reference electrode. Cyclic voltammograms were recorded in presence of 0.1 M tetrabutylammonium perchlorate (TBAP) as the supporting electrolyte at a scan rate of 100 mV s−1, using a CHI620 model electrochemical analyzer at room temperature. The carbohydrate azides and 4-ferrocenylphenol were prepared according to the literature procedures.38,39Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) solvent mixture. Yield: 0.28 g (91%); mp: 98 °C; IR (KBr, νmax/cm−1) = 3448, 3269, 2926, 2116, 1523, 1223, 1020; 1H NMR (300 MHz, CDCl3, Me4Si): δ (ppm) = 2.53 (t, 1H, J = 2.3 Hz, CH), 4.03 (s, 5H, C5H5), 4.27 (t, 2H, J = 1.7 Hz, C5H4), 4.57 (t, 2H, J = 1.7 Hz, C5H4), 4.69 (d, 2H, J = 2.3 Hz, OCH2), 6.91 (d, 2H, J = 8.7 Hz, Ar), 7.41 (d, 2H, J = 8.7 Hz, Ar); 13C NMR (75.5 MHz, CDCl3): δ (ppm) = 50.88, 66.17, 68.55, 69.46, 75.44, 78.70, 85.16, 114.83, 127.12, 132.31, 155.98; ESI-MS (in CH3OH): m/z = 316 [M]+; anal. calc. for C19H16FeO (316.05): C 72.18, H 5.10; found: C 71.81, H 5.01; UV-Vis in CH2Cl2: λmax [ε (dm3 mol−1 cm−1)] = 283 (1390), 442 (480) nm.
:1 mixture of acetone and water (8 mL). To this solution, sodium ascorbate (0.05 g, 0.2 mmol) and CuSO4·5H2O (0.02 g, 0.09 mmol) were added. The heterogeneous mixture was stirred at room temperature until the starting materials disappeared. The reaction mixture was quenched with saturated NH4Cl solution and then extracted with CH2Cl2 (2 × 5 mL). The combined organic layers were dried over Na2SO4 and the solvent removed under vacuum affording a crude mass, which was purified by column chromatography on alumina.
:5) solvent mixture. Yield: 0.28 g (91%); mp: 98 °C; IR (KBr, νmax/cm−1) = 3448, 3269, 2926, 2116, 1523, 1223, 1020; 1H NMR (300 MHz, CDCl3, Me4Si): δ (ppm) = 2.53 (t, 1H, J = 2.3 Hz, CH), 4.03 (s, 5H, C5H5), 4.27 (t, 2H, J = 1.7 Hz, C5H4), 4.57 (t, 2H, J = 1.7 Hz, C5H4), 4.69 (d, 2H, J = 2.3 Hz, OCH2), 6.91 (d, 2H, J = 8.7 Hz, Ar), 7.41 (d, 2H, J = 8.7 Hz, Ar); 13C NMR (75.5 MHz, CDCl3): δ (ppm) = 50.88, 66.17, 68.55, 69.46, 75.44, 78.70, 85.16, 114.83, 127.12, 132.31, 155.98; ESI-MS (in CH3OH): m/z = 316 [M]+; anal. calc. for C19H16FeO (316.05): C 72.18, H 5.10; found: C 71.81, H 5.01; UV-Vis in CH2Cl2: λmax [ε (dm3 mol−1 cm−1)] = 283 (1390), 442 (480) nm.
:1 mixture of acetone and water (8 mL). To this solution, sodium ascorbate (0.05 g, 0.2 mmol) and CuSO4·5H2O (0.02 g, 0.09 mmol) were added. The heterogeneous mixture was stirred at room temperature until the starting materials disappeared. The reaction mixture was quenched with saturated NH4Cl solution and then extracted with CH2Cl2 (2 × 5 mL). The combined organic layers were dried over Na2SO4 and the solvent removed under vacuum affording a crude mass, which was purified by column chromatography on alumina.
X-ray crystallography
X-ray data for compound 1 was collected at room temperature using a Bruker Smart Apex CCD diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) with a ω-scan method.40 Preliminary lattice parameters and orientation matrices were obtained from four sets of frames. The unit cell dimensions were determined using 5624 reflections in the range of 2.43° < θ < 27.92°. Integration and scaling of the intensity data were accomplished using the SAINT program.40 The structure was solved using direct methods with SHELXS9741 and the refinements were carried out using a full-matrix least-squares technique with SHELXL97.41 Anisotropic displacement parameters were included for all of the non-hydrogen atoms. H atoms were positioned geometrically and treated as riding on their parent C atoms [C–H = 0.93–0.97 Å and Uiso (H) = 1.2 Ueq (c) for the H atoms].Crystal data for 1: C19H16FeO, M = 316.17, red plate, 0.11 × 0.06 × 0.04 mm3, monoclinic, space group P21/c, a = 17.913(2), b = 9.5455(12), c = 8.8695(11) Å, α = 90°, β = 99.613(2)°, γ = 90°, V = 1495.3(3) Å3, Z = 4, Dc = 1.404 g cm−3, F000 = 656, CCD area detector, Mo Kα radiation, λ = 0.71073 Å, T = 294(2) K, 2θmax = 50.0°, 12814 reflections collected, 2628 unique reflections (Rint = 0.0244). Final GoOF = 1.037, R1 = 0.0309, wR2 = 0.0808, R indices based on 2296 reflections with I > 2σ(I) (refinement on F2), 190 parameters, 0 restraints, μ = 1.003 mm−1.
In vitro cytotoxicity testing (MTT assay)
The cell lines used for testing the in vitro cytotoxicity included HeLa derived from human cervical cancer cells (ATCC No. CCL-2), MDA-MB-231 derived from human breast adenocarcinoma cells (ATCC No. HTB-26) and MCF7 derived from human breast adenocarcinoma cells (ATCC No HTB-22), which were all obtained from the American Type Culture Collection, Manassas, VA, USA. All tumour cell lines were maintained in a modified DMEM medium supplemented with 10% fetal bovine serum, along with 1% non-essential amino acids except L-glutamine, 0.2% sodium hydrogen carbonate, 1% sodium pyruvate and 1% antibiotic mixture (10000 units penicillin and 10 mg of streptomycin per mL). The cells were washed and resuspended in the above medium, then 100 μL of this suspension was seeded into a 96-well bottom plate. The cells were kept at 37 °C in a humidified incubator (Model 2406 Shellab CO2 incubator, Sheldon, Cornelius, OR) under a 5% CO2 atmosphere. After incubation for 24 h, the cells were treated for 2 days with the test compounds at concentrations ranging from 0.1–100 μM in DMSO (1% final concentration) and were assayed at the end of the second day. Each assay was performed with two internal controls: (1) an IC0 with the cells only, and (2) an IC100 with the media only. After incubation for 48 h, the cells were subjected to an MTT colorimetric assay (5 mg mL−1). The effects of the different test compounds on the viability of the tumour cell lines were measured at 540 nm using a multimode reader (Infinite® M200, Tecan, Switzerland). The IC50 values (50% inhibitory concentration) were calculated from the plotted absorbance data of the dose–response curves. The assay was performed using doxorubicin and cisplatin as either standards or positive controls and 1% DMSO as a vehicle control. In order to account for the toxicity of DMSO, the values obtained for the DMSO control were subtracted from those of the test compounds. The IC50 values (in μM) are expressed as the average of two independent experiments.
Results and discussion
Synthesis and characterization
The alkyne, (4-ferrocenylphenyl)propargyl ether (1), was synthesized by treating 4-ferrocenyl phenol39 with sodium hydride and propargyl bromide in dry THF. The triazole derivatives 3a–h were obtained from the Huisgen 1,3-dipolar cycloaddition of alkyne 1 with the corresponding azide (2a–h) in the presence of CuSO4·5H2O and sodium ascorbate in a 2:1 mixture of acetone–water at 25 °C for 12 h (Scheme 1).
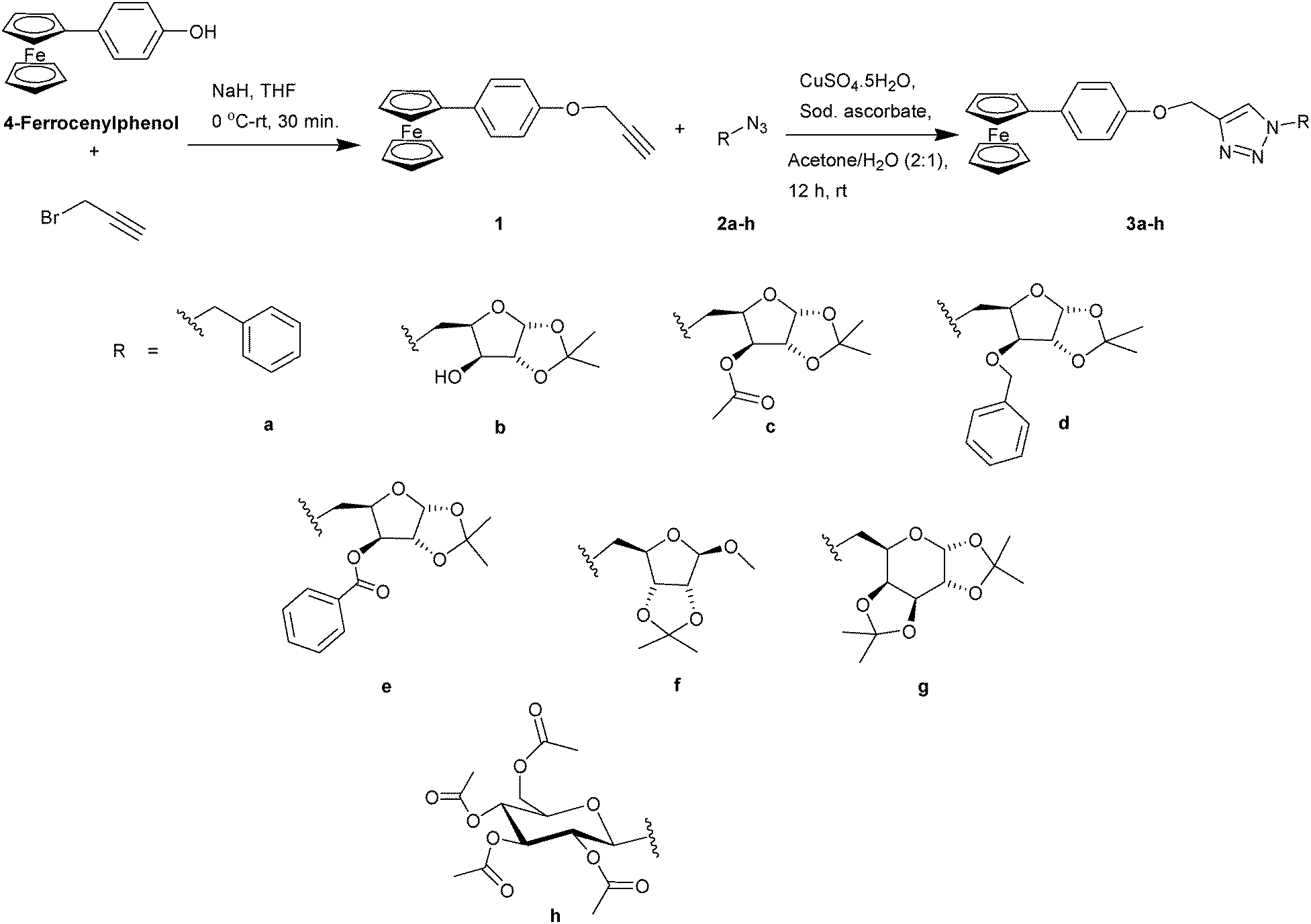 | ||
| Scheme 1 The synthesis of ferrocenyl triazoles using the Huisgen 1,3-dipolar cycloaddition of various azides and (4-ferrocenylphenyl)propargyl ether. | ||
In the 1H NMR spectrum of alkyne 1, the alkyne proton appeared as a triplet at 2.5 ppm. A sharp singlet was observed for the unsubstituted cyclopentadienyl (Cp) ring at 4.0 ppm, while the substituted cyclopentadienyl (Cp) ring protons appeared at 4.2 and 4.5 ppm. The –OCH2 protons appeared as a doublet at 4.6 ppm. The phenyl protons appeared as two doublets located at 6.9 ppm and 7.4 ppm. In the 13C NMR spectrum, the alkyne carbons were observed at 66.1 ppm and 68.5 ppm. The ferrocene and the aryl carbons appeared as reported in the literature.21–23 Compound 1 was further characterized by the presence of alkynyl C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C and C–H stretching bands at 2116 cm−1 and 3269 cm−1, respectively, in the IR spectrum.
C and C–H stretching bands at 2116 cm−1 and 3269 cm−1, respectively, in the IR spectrum.
In the 1H NMR spectra of the triazoles (3a–h), a characteristic singlet corresponding to the triazole proton was observed around 7.5–7.9 ppm. The protons of the unsubstituted cyclopentadienyl (Cp) ring appeared as a singlet at 4.0 ppm, while those for the substituted cyclopentadienyl (Cp) ring appeared around 4.2 ppm and 4.5 ppm. For the sugar triazoles (3b–f), the gem-dimethyl protons were observed as two singlets that appeared at 1.3 and 1.4 ppm, while for 3g, four singlets were observed in the region of 1.3–1.5 ppm. The anomeric proton of each sugar moiety was observed as a doublet between 5.6–6.2 ppm. In the case of compound 3f, a singlet was observed for the anomeric proton at 5.2 ppm.
In the 13C NMR spectra, for all of the triazoles, the triazole carbons were observed in the regions of 120 ppm and 140 ppm. In the case of the carbohydrate triazoles (3b–g), the gem-dimethyl group carbons appeared as singlets at around 24 ppm and 26 ppm. All of the triazoles exhibited characteristic C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching band in their IR spectra. With the exception of 3a and 3h, all of the triazoles were characterized using the [M + H]+ peak in their ESI+ mass spectra. For compound 3a, a [M]+ peak was observed, while for 3h a [M + Na]+ peak appeared in the mass spectrum.
C stretching band in their IR spectra. With the exception of 3a and 3h, all of the triazoles were characterized using the [M + H]+ peak in their ESI+ mass spectra. For compound 3a, a [M]+ peak was observed, while for 3h a [M + Na]+ peak appeared in the mass spectrum.
Using UV-visible spectroscopy, significant changes in the absorption spectra can be observed for the substituted ferrocene derivatives. For instance, in the case of p-aryl substituted ferrocenes, an increase in the intensity of the bands in the 300 nm region is observed due to the electronic interaction of ferrocene with the π-conjugated groups.42 Thus, it was expected that aryl ferrocene conjugates (3a–h) would also exhibit changes in the intensity of their bands.
The UV-visible spectra of alkyne 1 and the triazole compounds (3a–h) were recorded in dichloromethane over a wavelength range of 250–550 nm (see ESI†). All of the compounds exhibited two absorption maxima at around 310 and 450 nm corresponding to the Fe(a1g) → Cp(e2g) charge transfer and symmetry forbidden Fe(a1g) → Fe(e1g) transitions, respectively.43 It was observed that the bands corresponding to the Fe(a1g) → Fe(e1g) or d–d transitions were weak due to being symmetry forbidden transitions, whereas the intensity of the Fe(a1g) → Cp(e2g) or π → π* transition bands was considerably higher even at a concentration of 0.1 mM. Furthermore, these well resolved π → π* bands indicated that the ferrocene was conjugated with the aromatic moiety.43
Crystal structure of compound 1
The molecular structure of alkyne 1 was determined using X-ray diffraction studies (Fig. 1). Red crystals of 1 were obtained by the slow evaporation of a mixture of hexane–ethyl acetate (9:1). Compound 1 crystallized in the monoclinic space group P21/c. The crystallographically determined configuration and atom numbering scheme are shown in Fig. 1.
 | ||
| Fig. 1 A view of alkyne 1, showing the atom-labeling scheme. The displacement ellipsoids are drawn at the 30% probability level and H atoms are represented by circles with arbitrary radii. | ||
The pertinent bond angles and bond lengths of the compounds are given in Table 1. As can be seen from Fig. 1, the structure consists of a ferrocene unit and a propargyl ether group attached to the phenyl ring in a 1,4-configuation. While the ferrocene and aryl ring are almost co-planar, the propargyl group is aligned at an angle of 118.7° (C4–O1–C3) with respect to the ferrocenyl phenyl moiety. An angle of 178.1(4)° exists for the linear arrangement of the propargyl group (C1–C2–C3). The C1–C2 bond length was 1.149(4) Å, which indicates its triple bond character, and was found to be shorter than those reported by Wong and co-workers for their 4-ethynylphenylferrocene derivatives.43,44 The ferrocene and aryl rings are aligned in an almost planar fashion with a dihedral angle of 13.7(3)°. All other bond angles and bond lengths are consistent with standard literature values.44,45
| Bond distances (Å) | |||
| C15–Fe1 | 2.049(2) | C19–Fe1 | 2.034(2) |
| C1–C2 | 1.149(4) | C2–C3 | 1.452(4) |
| C3–O1 | 1.423(3) | C4–O1 | 1.373(3) |
| C7–C15 | 1.470(3) | C1–H1 | 0.930 |
| Bond angles (°) | |||
| C2–C1–H1 | 180.0 | C15–C7–C8 | 121.14(19) |
| C4–O1–C3 | 118.7(2) | C11–Fe1–C15 | 108.31(10) |
| C1–C2–C3 | 178.1(4) | C9–C4–C5 | 119.7(2) |
| O1–C3–C2 | 111.8(2) | ||
Electrochemical characterization
The redox behaviour of the ferrocene core is one of the factors responsible for the biological activity of the ferrocene conjugates. Hillard and coworkers reported proton coupled electron transfer in a series of ferrocifen-type compounds by using electrochemistry, while the redox behaviour of ferroquine has been reported by Biot and coworkers.46,47 Hence, electrochemical characterisation of the compounds was performed by means of cyclic voltammetry (CV) in 0.2 mM dichloromethane solution, using a three-electrode cell consisting of a glassy carbon working electrode, a platinum wire auxiliary electrode and a saturated calomel electrode as the reference electrode at a scan rate of 100 mV s−1 (Fig. 2a and b). All of the triazole compounds (3a–h) and the alkyne exhibited a quasi-reversible oxidation wave with high peak separation potentials of over 100 mV.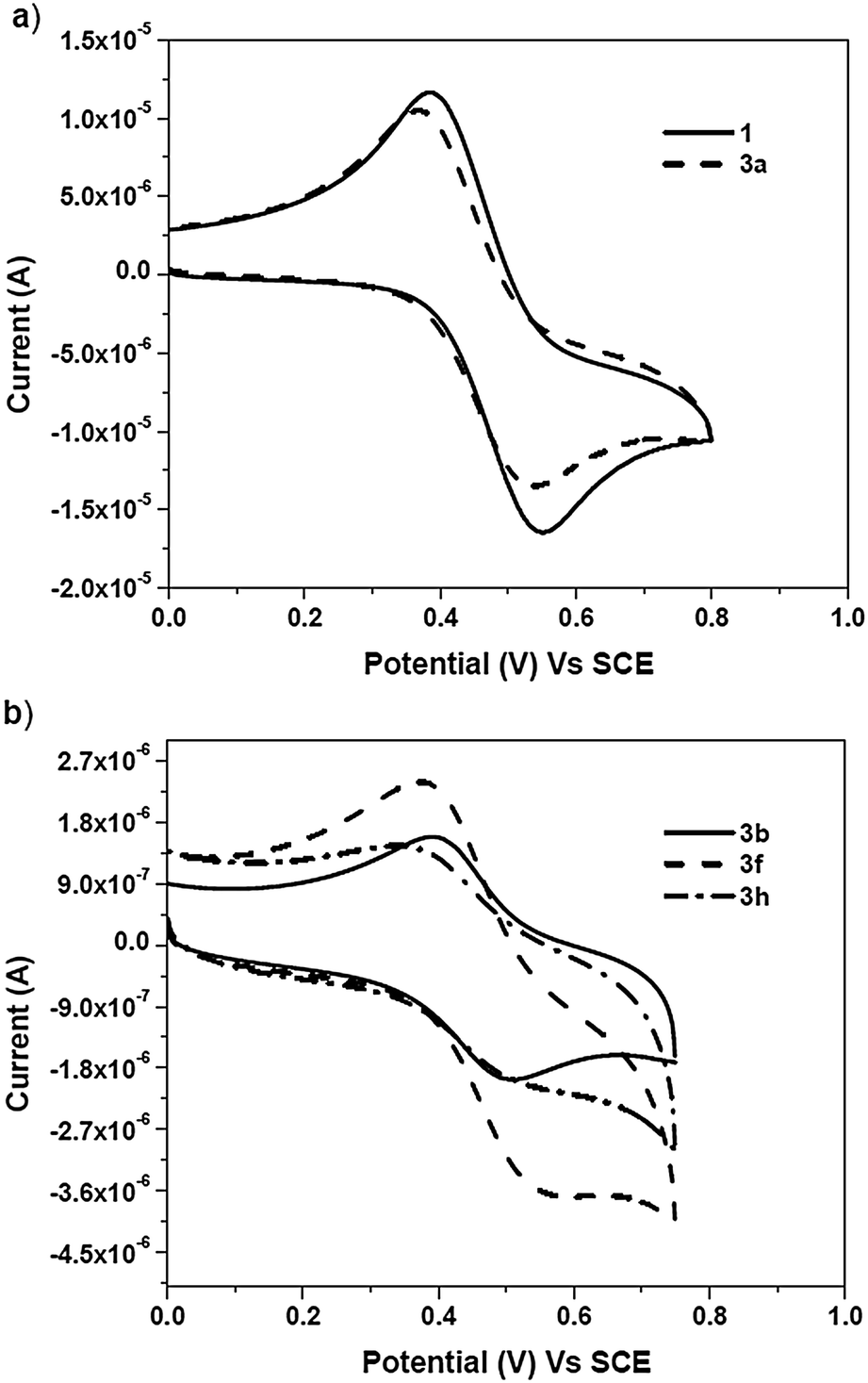 | ||
| Fig. 2 Cyclic voltammograms of (a) ferrocenyl alkyne 1 and benzyl triazole (3a) and (b) the ferrocene–carbohydrate triazoles (3b, 3f and 3h) in 0.2 mM dichloromethane at 25 °C. | ||
In dichloromethane solution, an E1/2 (vs. SCE) value of 433 mV was recorded for alkyne 1, whereas an E1/2 (vs. SCE) value of 452 mV was obtained for benzyl triazole 3a. In the case of the carbohydrate triazoles (3b–h), the E1/2 (vs. SCE) values obtained were in the range of 437–460 mV and depended on the type of sugar moiety. These results indicated that, in dichloromethane, the substituent on the triazole ring had a slight or no effect on the E1/2 values of the ferrocene conjugates (3a–h).
Cyclic voltammetry experiments for the ferrocene–carbohydrate conjugates (3b–h) were also conducted in DMSO and DMSO–buffer solutions (phosphate buffer pH = 7.4). It was observed that, in 0.2 mM DMSO solution, the compounds exhibited a reversible oxidation wave with potentials ranging from 414–426 mV (vs. SCE). However, solvent-induced oxidation of the ferrocene conjugates was not observed in DMSO. The absence of any oxidation indicates that these conjugates are stabilized in DMSO48via aromatic conjugation, which thereby prevents the expulsion of a Cp ring caused by the attack of a nucleophile at the Fe(III) centre.
Upon addition of a 0.1 M phosphate buffer solution (phosphate buffer with pH = 7.4) to each 0.2 mM DMSO solution of compounds 3b–h, a reversible oxidation wave corresponding to a ferrocenium ion was observed with E1/2 (vs. SCE) values ranging from 340–350 mV. The reversible nature of the oxidation wave for compounds 3b, 3f and 3h indicated the stability of these ferrocene conjugates in the buffer solution.36–38
From the CV experiments, it was observed that ferrocene–carbohydrate conjugates (3b–h) exhibited different electrochemical behaviour in dichloromethane and DMSO solutions. Furthermore, it was also observed, for all of the solvent systems, that the different carbohydrate scaffolds had no significant effect of on the E1/2 values of the ferrocene–carbohydrate conjugates (3b–h). This type of electrochemical behaviour indicated that the carbohydrate scaffolds were electrically neutral.49
Diffusion coefficient (Df) measurements for the ferrocene–carbohydrate conjugates
According to reports in the literature, the diffusion coefficient (Df) of a ferrocene–carbohydrate conjugate should depend on the geometry of the carbohydrate scaffold as well as its interaction with the solvent system, but should be independent of the molecular weight of the compound.25 However, in our recent work on ferrocene–carbohydrate conjugates with amide-triazole linkers, it was observed that the geometry and the spatial arrangement of the carbohydrate moiety had no significant effect on the diffusion coefficient values in buffer solutions.38 This behaviour suggested that the ferrocene–carbohydrate amide triazole ligands were stable in buffer solution. In contrast, in the present study, ferrocene–carbohydrate triazole conjugates (3b–h) contain a conjugated hydrophobic aromatic system, which is assumed to exhibited different behaviour in both polar and buffer solutions. Hence, the diffusion coefficients (Df) were calculated according to literature procedures,50 in order to investigate the extent of the stabilization of the compounds by solvent molecules, based on the reversible redox behaviour of the compounds in DMSO and DMSO–buffer solutions, under physiological conditions (pH = 7.4). In order to calculate the diffusion coefficient, the corresponding anodic peak currents (ia) were obtained by recording cyclic voltammograms at various scan rates ranging from 20–100 mV (see ESI†).The straight line obtained due to the diffusion controlled electrochemical process showed that the current was directly proportional to the scanning rate ν1/2, thereby indicating a reversible electrochemical reaction.49
The diffusion coefficients (Df) of (3b–h) were obtained using the Randles–Ŝevĉik relationship:50
| ia = 0.4463 × n3/2F3/2A(RT)1/2Df1/2Cν1/2 |
500 C mol–1, the above equation can be written as:| ia = 2.69 × 105 × ADf1/2Cν1/2 |
It was observed from the calculations that the compounds exhibited variable diffusion coefficients with an increase in the molecular weight (Table 2). In 0.2 mM DMSO solution, the Df values obtained for the ferrocenyl xylofuranose conjugates (3b, 3c and 3e), ranged between 4.25–5.25 × 10−8 m2 s–1. However, in the case of 3-O-benzyl xylofuranose derivative 3d, a higher Df value of 6.07 × 10−8 m2 s–1 was obtained. This observation could perhaps be explained on the basis of the hydrogen bonding ability of the protecting group present in the sugar moiety. The hydroxyl group and the ester functional group can form hydrogen bonds with the solvent molecules, yielding a lower diffusion coefficient value due to slower diffusion. In the case of the benzyl protected sugar 3d, since there is a lower possibility of hydrogen bonds forming between the sugar scaffold and the solvent molecules, a faster diffusion occurs resulting in a higher diffusion coefficient value. This demonstrates the effect that protecting groups on the carbohydrate moieties have on the diffusion coefficient. A Df value of 3.89 × 10−8 m2 s–1 was obtained for the triazole derived from ribose sugar (3f). Among the hexose sugars, the ferrocenyl galactose triazole (3g) exhibited a lower Df value compared to the ferrocene glucose triazole (3h). From this study, it was observed that, in DMSO solutions, the compounds exhibited different diffusion coefficient values depending on the geometry of the sugar scaffold and the protecting group present on the sugar moiety.
| Compound | Molecular weight (mg mmol−1) | Carbohydrate scaffold | D f in DMSO (×10−8 m2 s−1) | D f in DMSO–buffer (×10−8 m2 s−1) |
|---|---|---|---|---|
| 3b | 531 |

|
4.25 | 3.94 |
| 3c | 573 |
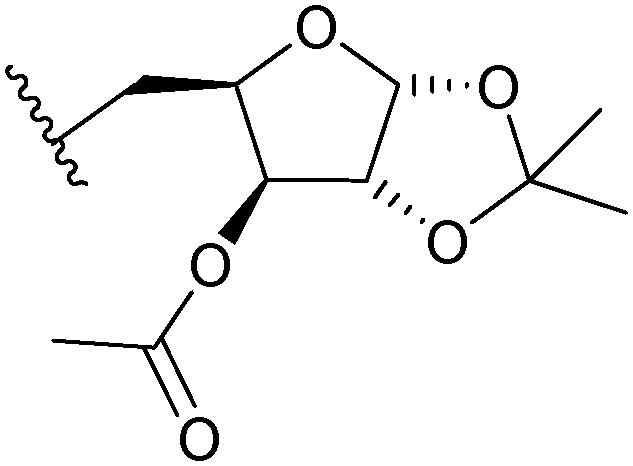
|
4.93 | 5.31 |
| 3d | 621 |
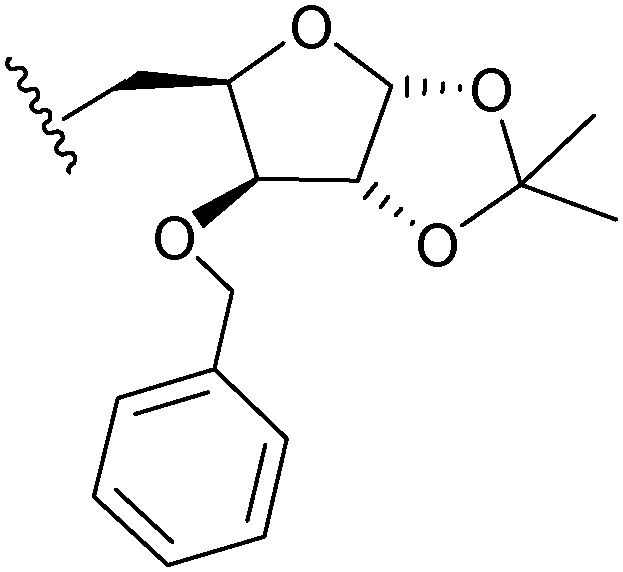
|
6.08 | 4.57 |
| 3e | 635 |
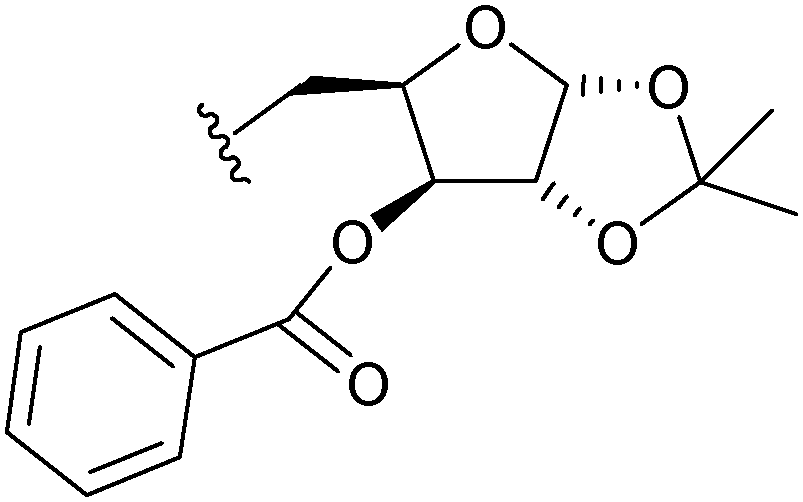
|
5.27 | 3.95 |
| 3f | 545 |
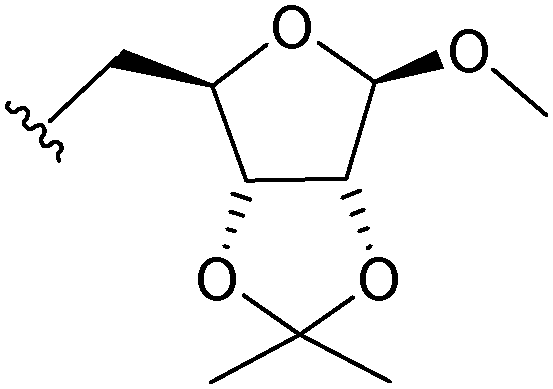
|
4.63 | 3.89 |
| 3g | 601 |
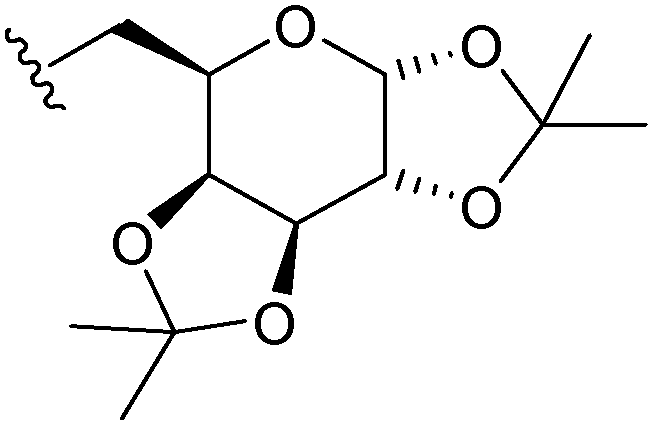
|
2.43 | 3.53 |
| 3h | 747 |
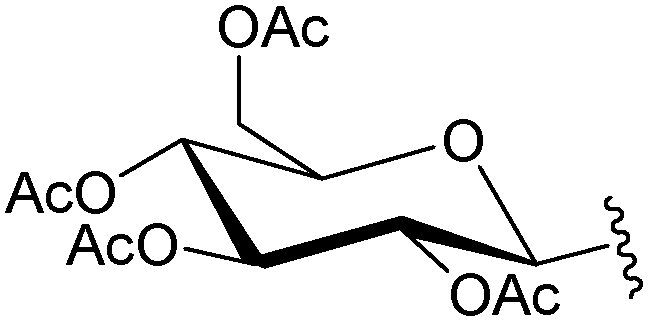
|
4.37 | 4.69 |
In the buffer solution, under physiological pH, the ferrocene–carbohydrate moieties were found to possess lower Df values due to the extensive stabilization of the compounds by solvent molecules. This was previously observed in cases of ferrocene–carbohydrate conjugates containing hydrogen bonding functionalities adjacent to the ferrocene scaffold.25,38 However, in the present study, a hydrophobic phenyl group attached to the ferrocene scaffold might also induce a stabilization effect of the compounds in buffer solutions. In order to observe this effect, diffusion coefficients were calculated in DMSO–buffer solution, under physiological pH. In the case of xylose (3b, 3d and 3e) and ribose derivatives (3f) a slight decrease in the Df values were obtained compared to those observed in DMSO solution. Among xylofuranose derivatives, a higher diffusion coefficient was obtained for compound 3c. Similarly, higher Df values were also obtained for the galactose and glucose triazoles in buffer solution when compared to DMSO solution. From these results, it can be concluded that the diffusion coefficient values in DMSO–buffer solutions are dependent on the hydrophobic and hydrophilic nature of the different moieties present in the ferrocene–carbohydrate conjugates (3b–h).
Biological activity
Recently, ferrocene–carbohydrate conjugates have demonstrated their usefulness by having various biological activities. Of particular interest in these conjugates, is the presence of different types of linker such as amides, triazoles, alkyl chains, esters, amide-triazoles etc.27–38 In some of these conjugates, hydrogen bonding units are present adjacent to the ferrocene core as well as in the carbohydrate scaffold. Only a few of these ferrocene–carbohydrate conjugates, such as ferrocene–iminosugar conjugates and amide-triazole linked ferrocene–carbohydrate conjugates have exhibited moderate to good cytotoxic activities. The presence of hydrogen bonding components in these conjugates might be one possible explanation for such behaviour. In order to determine the importance of the hydrogen bonding groups, we have synthesised ferrocene–carbohydrate conjugates with phenoxymethyl triazole linkers and have examined their cytotoxic activity.The cytotoxicity of ferrocene triazoles 3a–h was assessed on the basis of the measurement of in vitro cell growth in 96 well plates using the cell-mediated reduction of a tetrazolium salt to form water insoluble formazan crystals, carried out according to the literature procedure.51 These compounds were tested for cytotoxicity against different test cell lines up to a concentration of 100 μM. 4-Ferrocenylphenol was also tested on these cell lines for comparison purposes. The concentration of the compounds at which 50% of the cell growth was inhibited (IC50) was calculated and shown in Table 3.
| Compounda | IC50b (μM) | ||
|---|---|---|---|
| HeLa | MDA-MB-231 | MCF-7 | |
| a Compounds 3b–e and 3g demonstrated IC50 values > 100 μM against all of the tested cell lines. b The values are the mean of two independent experiments. c Positive control. | |||
| 4-Ferrocenylphenol | 22.91 (±0.81) | >100 | >100 |
| 1 | 33.11 (±0.59) | >100 | >100 |
| 3a | 41.71 (±0.46) | >100 | >100 |
| 3f | 61.79 (±0.15) | 41.18 (±0.78) | 9.0 (±0.03) |
| 3h | 36.71 (±0.97) | 19.07 (±0.08) | 19.91 (±0.03) |
| Doxorubicinc | 0.51 (±0.02) | 0.91 (±0.06) | 1.07 (±0.1) |
| Cisplatinc | 0.23 (±0.10) | 0.25 (±0.09) | 0.48 (±0.16) |
It was observed that 4-ferrocenylphenol and the alkyne (1) were not active against the HeLa cell line, having high IC50 values (IC50 = 22.91 μM and 33.11 μM, respectively). In the case of benzyl triazole 3a, an IC50 value of 41.71 μM was observed against the HeLa cell lines. However, in the case of the ferrocene–carbohydrate triazoles, it was observed that, with the exception of the compounds derived from ribose and glucose sugars (3f and 3h, respectively), the triazoles derived from xylose (3b–e) and galactose (3g) did not show any cytotoxicity (IC50 > 100 μM) towards the tested cell lines. Although 4-ferrocenylphenol and the alkyne (1) exhibited cytotoxicity towards the HeLa cell lines, all of the ferrocene–carbohydrate triazoles were found to be non-toxic towards this cell line. The triazoles derived from ribose and glucose, showed lower cytotoxicity against the HeLa cells (IC50 values of 61.79 and 36.71 μM, respectively) compared to 4-ferrocenyl phenol and the alkyne (1).
The ribose triazole (3f) exhibited moderate cytotoxicity against the hormone dependent breast cancer cell line MCF-7 (IC50 = 9.0 μM), but exhibited low toxicity against the hormone independent breast cancer cell line MDA-MB-231 (IC50 = 41.18 μM). The glucose triazole (3g) exhibited similar cytotoxicity against the MCF-7 and MDA-MB-231 breast cancer lines (IC50 = 19.91 μM and 19.07 μM, respectively).
In order to determine the role of the linker, the IC50 values of ferrocene–carbohydrate conjugates 3d, 3f and 3g were compared with the IC50 values of their previously reported amide triazole analogs38 (Table 4). Triazoles 3f and 3g demonstrated cytotoxicity towards the HeLa cell line, whereas their amide triazole analogs were found to be non-toxic towards this cell line. In a similar manner, the glucose triazole 3g, exhibited significant toxicity on MDA-MB-231 and MCF-7 breast cancer cells. On the other hand, in the case of the breast cancer cell lines, poor cytotoxicity was observed for the 3-O-benzyl xylofuranose and ribofuranoside triazoles (3d and 3f respectively), compared to their amide-triazole analogues.
| Carbohydrate scaffold | Linker | IC50 (μM) | ||
|---|---|---|---|---|
| HeLa | MDA-MB-231 | MCF-7 | ||
| a Ref. 38. | ||||
| 3-O-Benzyl xylofuranose | Amide-triazolea | >100 | 2.82 | 11.31 |
| Phenoxymethyl triazole (3d) | >100 | >100 | >100 | |
| O-Methyl ribofuranoside | Amide-triazolea | >100 | 7.31 | 3.18 |
| Phenoxymethyl triazole (3f) | 61.79 | 41.18 | 9.02 | |
| Per-O-acetyl glucopyranoside | Amide-triazolea | >100 | >100 | >100 |
| Phenoxymethyl triazole (3g) | 36.71 | 19.07 | 19.91 | |
The biological studies carried out on all of the (4-ferrocenylphenoxy)methyl triazole conjugates indicated that their hydrophobic nature and the lack of any component capable of forming hydrogen bonds adjacent to the ferrocene core were detrimental for their cytotoxic activity. Unlike amides (found in amino acids and peptides), the aryl groups significantly hamper the biological properties of the conjugates. The cytotoxicity data obtained from this study indicate that the linker and the carbohydrate scaffold play specific roles in the exhibition of cytotoxicity in the ferrocene–carbohydrate conjugates. Further work in this area is being conducted in order to investigate the effect of aromatic heterocyclic units on the biological activity of ferrocene–carbohydrate conjugates.
Conclusions
Ferrocene–carbohydrate triazoles containing a hydrophobic phenoxymethyl linker were synthesized. The crystal structure of the ferrocenyl alkyne was solved using a single crystal X-ray diffraction study. The compounds demonstrated characteristic transitions in their UV-Visible spectra. The compounds exhibited variable electrochemical behaviour in aprotic and protic solvents like dichloromethane, DMSO and DMSO–buffer solution. In dichloromethane solution, a quasi-reversible oxidation with high peak separation potentials was observed, whereas, in coordinating solvents such as DMSO and DMSO–buffer solutions, a reversible oxidation process was observed. Furthermore, the compounds had different diffusion coefficient values in the DMSO and DMSO–buffer solutions (pH = 7.4). The higher diffusion coefficients of these phenoxymethyl triazole linked ferrocene–carbohydrate conjugates under physiological conditions demonstrate no appreciable solvent induced stabilization. The variable diffusion coefficients of the compounds in the buffer solution indicated that the aryl moiety could impart its hydrophobic nature onto the compounds. The poor cytotoxicity of the compounds may be attributed to this hydrophobic nature. It was observed that only two compounds exhibited cytotoxicity with an IC50 value > 15 μM. In conclusion, this study demonstrates the effect of replacing a hydrogen bonding group adjacent to the ferrocene unit with a hydrophobic aryl moiety on the electrochemical and biological behaviour of ferrocene–carbohydrate conjugates. In order to investigate the importance of hydrogen bonding sources in ferrocene–carbohydrate conjugates, further design of such conjugates are under progress.Acknowledgements
This work is financially supported by the in-house project MLP-0008 of CSIR-IICT. S. B. D. and P. S. are grateful to the Council of Scientific and Industrial Research (CSIR), New Delhi for the award of research fellowships.Notes and references
- A. Nguyen, A. Vessiéres, E. A. Hillard, S. Top, P. Pigeon and G. Jaouen, Chimia, 2007, 61, 716 CrossRef CAS.
- P. Messina, E. Labbé, O. Buriez, E. A. Hillard, A. Vessiéres, D. Hamels, S. Top, G. Jaouen, Y. M. Frapart, D. Mansuy and C. Amatore, Chem.–Eur. J., 2012, 18, 6581 CrossRef CAS PubMed and the references cited there in.
- D. Dive and C. Biot, ChemMedChem, 2008, 3, 383 CrossRef CAS PubMed.
- M. Navarro, W. Castro and C. Biot, Organometallics, 2012, 31, 5715 CrossRef CAS.
- P. F. Salas, C. Herrmann and C. Orvig, Chem. Rev., 2013, 113, 3450 CrossRef CAS PubMed.
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 12, 1 Search PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3 CrossRef CAS PubMed.
- C. Ornelas, New J. Chem., 2011, 35, 1973 RSC.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931 CrossRef CAS PubMed.
- S. Martić, M. Labib, P. O. Shipman and H.-B. Kraatz, Dalton Trans., 2011, 40, 7264 RSC.
- D. Osella, M. Ferrali, P. Zanello, F. Laschi, M. Fontani, C. Nervi and G. Cavigiolio, Inorg. Chim. Acta, 2000, 306, 42 CrossRef CAS.
- W. E. Geiger, Organometallics, 2007, 26, 5738 CrossRef CAS.
- A. Monney and M. Albrecht, Coord. Chem. Rev., 2013, 257, 2420 CrossRef CAS PubMed.
- S. Behesthi, A. Lataoifeh and H.-B. Kraatz, J. Organomet. Chem., 2011, 696, 1117 CrossRef PubMed.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278 CrossRef CAS PubMed.
- R. Chelmowski, S. D. Köster, A. Kerstan, A. Prekelt, C. Grunwald, T. Winkler, N. Metzler-Nolte, A. Terfort and C. Wöll, J. Am. Chem. Soc., 2008, 130, 14952 CrossRef CAS PubMed.
- G. Gasser, N. Hüsken, S. D. Köster and N. Metzler-Nolte, Chem. Commun., 2008, 3675 RSC.
- N. Hüsken, G. Gasser, S. D. Köster and N. Metzler-Nolte, Bioconjugate Chem., 2009, 20, 1578 CrossRef PubMed.
- Á. Mooney, A. J. Corry, C. Ní Ruairc, T. Mahgoub, D. O'Sullivan, N. O'Donovan, J. Crown, S. Varughese, S. M. Draper, D. K. Rai and P. T. M. Kenny, Dalton Trans., 2010, 39, 8228 RSC.
- Á. Mooney, A. J. Corry, D. O'Sullivan, D. K. Rai and P. T. M. Kenny, J. Organomet. Chem., 2009, 694, 886 CrossRef PubMed.
- A. G. Harry, J. Murphy, W. E. Butler, R. Tiedt, Á. Mooney, J. C. Manton, M. T. Pryce, N. O'Donovan, N. Walsh, J. Crown, D. K. Rai and P. T. M. Kenny, J. Organomet. Chem., 2013, 734, 86 CrossRef CAS PubMed.
- J. M. Casas-Solvas, A. Vargas-Berenguel, L. F. Capitan-Vallvey and F. Santoyo-Gonzalez, Org. Lett., 2004, 6, 3687 CrossRef CAS PubMed.
- J. M. Casas-Solvas, E. Ortiz-Salmerón, J. J. Giménez-Martínez, L. García-Fuentes, L. F. Capitán-Vallvey, F. Santoyo-González and A. Vargas-Berenguel, Chem.–Eur. J, 2009, 15, 710 CrossRef CAS PubMed.
- C. Förster, M. Kovaĉević, L. Bariŝić, V. Rapić and K. Heinze, Organometallics, 2012, 31, 3683 CrossRef.
- T. Itoh, S. Shirakami, N. Ishida, Y. Yamashita, T. Yoshida, H.-S. Kim and Y. Wataya, Bioorg. Med. Chem. Lett., 2000, 10, 1657 CrossRef CAS.
- C. G. Hartinger, A. A. Nazarov, V. B. Arion, G. Giester, M. Jakupec, M. Galanski and B. K. Keppler, New J. Chem., 2002, 26, 671 RSC.
- V. Zsoldos-Máldy, A. Csámpai, R. Szabá, E. Mészéros-Alapi, J. Pásztor, F. Hudecz and P. Sohár, ChemMedChem, 2006, 1, 1119 CrossRef PubMed.
- A. Hottin, F. Dubar, A. Steenackers, P. Delannoy, C. Biot and J.-B. Behr, Org. Biomol. Chem., 2012, 10, 5592 CAS.
- A. Hottin, D. W. Wright, A. Steenackers, P. Delannoy, F. Dubar, C. Biot, G. J. Davies and J.-B. Behr, Chem.–Eur. J., 2013, 19, 9526 CrossRef CAS PubMed.
- C. L. Ferreira, C. B. Ewart, C. A. Barta, S. Little, V. Yardley, C. Martins, E. Polishchuk, P. J. Smith, J. R. Moss, M. Merkel, M. J. Adam and C. Orvig, Inorg. Chem., 2006, 45, 8414 CrossRef CAS PubMed.
- C. Herrmann, P. F. Salas, B. O. Patrick, C. de Kock, P. J. Smith, M. J. Adam and C. Orvig, Dalton Trans., 2012, 41, 6431 RSC.
- C. Herrmann, P. F. Salas, J. F. Cawthray, C. de Kock, B. O. Patrick, P. J. Smith, M. J. Adam and C. Orvig, Organometallics, 2012, 31, 5736 CrossRef CAS.
- C. Herrmann, P. F. Salas, B. O. Patrick, C. de Kock, P. J. Smith, M. J. Adam and C. Orvig, Organometallics, 2012, 31, 5748 CrossRef CAS.
- R. Trivedi, S. B. Deepthi, L. Giribabu, B. Sridhar, P. Sujitha, C. G. Kumar and K. V. S. Ramakrishna, Eur. J. Inorg. Chem., 2012, 2267 CrossRef CAS.
- R. Trivedi, S. B. Deepthi, L. Giribabu, B. Sridhar, P. Sujitha, C. G. Kumar and K. V. S. Ramakrishna, Appl. Organomet. Chem., 2012, 26, 369 CrossRef CAS.
- S. B. Deepthi, R. Trivedi, L. Giribabu, P. Sujitha and C. G. Kumar, Dalton Trans., 2013, 42, 1180 RSC.
- V. Weinmayr, J. Am. Chem. Soc., 1955, 77, 3009 CrossRef CAS.
- Bruker 2001, SAINT (Version 6.28a) & SMART (Version 5.625), Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- D. J. Roberts, D. Nolan, G. M. ÓMáille, G. W. Watson, A. Singh, I. Ledoux-Rak and S. M. Draper, Dalton Trans., 2012, 41, 8850 RSC.
- Y. L. K. Tan, P. Pigeon, E. A. Hillard, S. Top, M. Plamont, A. Vessiéres, M. J. McGlinchey, H. Müller-Bunz and G. Jaouen, Dalton Trans., 2009, 10871 RSC.
- W.-Y. Wong, K.-Y. Ho, S.-L. Ho and Z. Lin, J. Organomet. Chem., 2003, 683, 341 CrossRef CAS.
- W.-Y. Wong, G.-L. Lu, L. Liu, J.-X. Shi and Z. Lin, Eur. J. Inorg. Chem., 2004, 2066 CrossRef CAS.
- E. Hillard, A. Vessiéres, L. Thouin, G. Jaouen and C. Amatore, Angew. Chem., Int. Ed., 2006, 45, 285 CrossRef CAS PubMed.
- N. Chavain, H. Vezin, D. Dive, N. Touati, J.-F. Paul, E. Buisine and C. Biot, Mol. Pharmacol., 2008, 5, 710 CrossRef CAS PubMed.
- M. Chahma, J. S. Lee and H.-B. Kraatz, J. Organomet. Chem., 2002, 648, 81 CrossRef CAS.
- J. L. Kerr, J. S. Landells, D. S. Larsen, B. H. Robinson and J. Simpson, J. Chem. Soc., Dalton Trans., 2000, 1411 RSC.
- T. Morikita and T. Yamamoto, J. Organomet. Chem., 2001, 637, 809 CrossRef.
- M. Navarra, M. Celano, J. Maiuolo, S. Schenone, M. Botta, A. Angelucci, P. Bramanti and D. Russo, BMC Cancer, 2010, 10, 602 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, UV-Visible and CV spectra of the compounds. CCDC 925958 contains supplementary crystallographic data for the structure. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj01022h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |