Ratiometric fluorescent Zn2+ and In3+ receptors of fused pyrazine with an aminopropanol chain in acetonitrile†
Katarzyna
Ostrowska
*a,
Alicja
Kaźmierska
a,
Maria
Rąpała-Kozik
b and
Justyna
Kalinowska-Tłuścik‡
a
aFaculty of Chemistry, Jagiellonian University, R. Ingardena 3, PL-30060 Kraków, Poland. E-mail: ostrowsk@chemia.uj.edu.pl; Fax: +48-12-63-40-515
bFaculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Gronostajowa 7, PL-30387 Kraków, Poland
First published on 11th October 2013
Abstract
A series of new potential intramolecular charge transfer ICT fluorescent receptors of Zn2+, Cd2+, Hg2+, Ga3+, In3+, and Tl3+ ions based on N-aryl or N-alkyl variously fused pyridopyrrolopyrazine or pyrrolo[2,3-b]quinoxaline with an integrated donor group, such as 3-aminopropanol, were synthesized and verified. Among [N, N, O] tridentate donors 4a, 6a–d, 10a, 12e, and 14e, only the integrated fluorophore-receptors with the nitrogen atom at the N-5 position of heterocyclic systems, pyrido[2,3-b]pyrrolo[2,3-e]pyrazine 6a, selectively respond to zinc and indium with significant fluorescent enhancement and different mechanisms of binding. The first example of Z to E interconversion of enaminone forms of a ligand responsible for Zn2+ complex fluorescence enhancement was documented.
Introduction
One of the most challenging fields in organic and supramolecular chemistry is development of the fluorescent molecular sensors, especially for heavy and transition metal ions.1–11 Current fluorescent molecular sensors for metal ions usually contain a fluorophore and a receptor, which can form an integrated or a spaced fluorophore–receptor system. Receptors include N-, O-, or S-donor groups, which are responsible for selective metal ion binding. The fluorophores with conjugated π-electron systems change their photophysical properties upon complexation, signalling binding with metal ions. The emitted signals are detected as the enhancement of fluorescent emission with a red or blue shift of the emitted band or the quenching of the fluorescence. Azaheteroarenes, containing one or more heterocyclic nitrogen atoms, such as pyridine,12,13 quinoline,14–20 quinoxaline,21,22 acridine,23–27 indole,28–30 and carbazole31,32 are usually used as fluorogenic sensor components. These heterocyclic systems with separated or integrated receptor units containing amino, imino, amido, hydroxy, alkoxy, carbonyl, or carboxyl groups and a metal ion form five- or six-membered chelate rings with binding mode 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1, or even 3:1.33 The connection between the ionophore and the fluorophore is essential for signalling and recognition function. The cation-induced photophysical changes are mainly based on photoinduced electrons, protons or charge transfer and excimer formation. The general classification of fluorescent sensors is connected with the origin of photoinduced changes such as photoinduced electron transfer cation sensors (PET) or photoinduced charge transfer cation sensors (ICT). Usually the cation sensors (ICT) contain conjugated electron-donating and electron-withdrawing groups forming an intramolecular push–pull electronic system. During the formation of the complex the metal cation interacts with the donor or acceptor moiety, which affects the efficiency of intramolecular charge transfer upon excitation by light.
:1, 2:1, or even 3:1.33 The connection between the ionophore and the fluorophore is essential for signalling and recognition function. The cation-induced photophysical changes are mainly based on photoinduced electrons, protons or charge transfer and excimer formation. The general classification of fluorescent sensors is connected with the origin of photoinduced changes such as photoinduced electron transfer cation sensors (PET) or photoinduced charge transfer cation sensors (ICT). Usually the cation sensors (ICT) contain conjugated electron-donating and electron-withdrawing groups forming an intramolecular push–pull electronic system. During the formation of the complex the metal cation interacts with the donor or acceptor moiety, which affects the efficiency of intramolecular charge transfer upon excitation by light.
8-arylsulphonamide (Zinquin)13 or 8-carboxamide derivatives of quinoline (AQZ)15 are the most widely used ICT fluorescent sensors for zinc cations in biological samples (Fig. 1). Sensors contain electron-acceptor amido groups and electron-donors with the N-1 quinoline nitrogen atom. When the metal ion is bound to an electron-withdrawing amido group, the complexation is associated with the deprotonation of the amine leading to an enhanced ICT process from the donor to the acceptor upon excitation by light. The replacement of hydrogen by the zinc ion and the formation of a five-membered chelate ring broaden the conjugation of the π-system resulting in Stokes red-shift of the absorption spectra. In addition to this shift a 300-fold increase in their fluorescence intensity upon chelation of zinc is observed.34
 | ||
| Fig. 1 General structures of the studied chemosensors. | ||
We previously designed ICT integrated fluorophore–receptor sensors 1a–4a for zinc ions, with (E/Z)-3-[(alkylamino)phenyl-methylidene]-pyrrolo[2,3-b]quinoxaline as the fluorogenic component and amino, hydroxyl, aminoalkyl or hydroxyalkyl groups as additional receptors (Fig. 1).35 Among these [N, N, N] or [N, N, O] tridentate ligands 1a–4a, compound 3a showed the ratiometric turn-on fluorescence response toward zinc cations within the working zinc concentration range of Kd = 3.77 ± 0.51 μM and a binding stoichiometry of 1:1. This push–pull (E/Z)-enamine undergoes deprotonation while binding zinc cations, yielding a red-shifted excitation wavelength with induced charge transfer signal transduction (ICT).35
The electronegativity of the oxygen atom in the carbonyl group of imide causes the delocalization of N-1 or enamine nitrogen's lone pairs into the π system, leading to formation of zwitterions A or B, respectively (Scheme 1). Form B is stabilised by an internal hydrogen bond. The positive charge on iminium ions in form A may be modulated by electron-donating or electron-withdrawing character of these groups attached to N-1. The electron-donating group could decrease and stabilize positive charge on iminium ions, or the electron-withdrawing group could increase the positive charge and enhance the dipole moment of 4a. Deprotonation and coordination of the zinc atom, more electropositive than hydrogen, to the nitrogen of the conjugated enaminone moiety and the N-4 nitrogen donor of quinoxaline changes the distribution of electronic charge in the push–pull system. Formation of the six-membered chelate ring extends π-conjugation resulting in a cation-induced red shift of the absorption spectrum and fluorescence emission upon excitation by light.
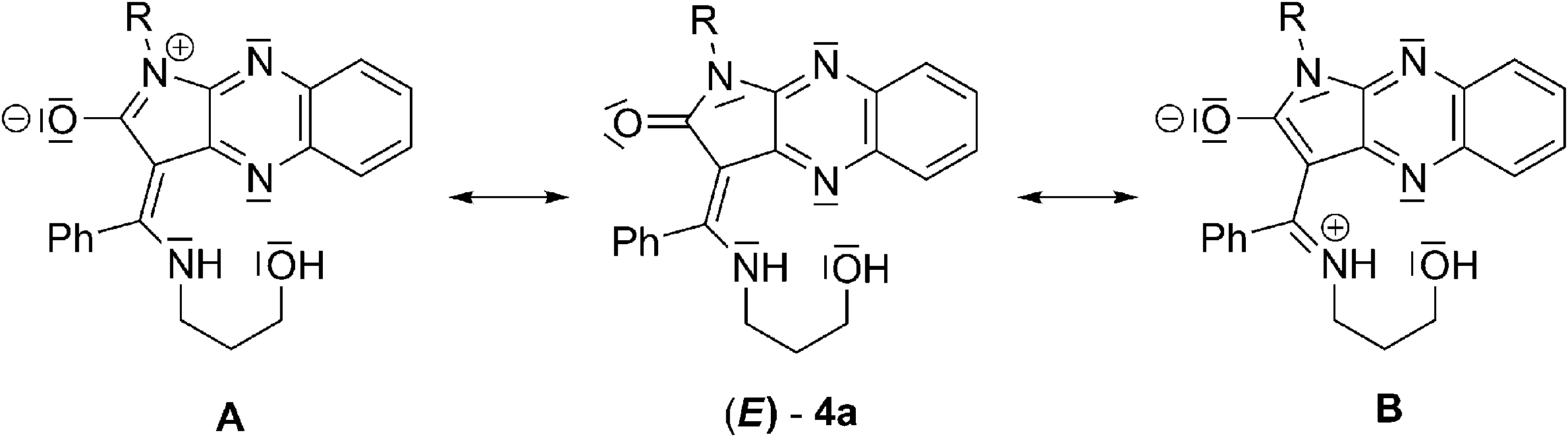 | ||
| Scheme 1 Zwitterionic resonance structures of receptor 4a. | ||
In this paper, we report the syntheses and photophysical properties of various new fluorophores: aryl or alkylphenyl substituted at the N-1 position of pyridopyrrolopyrazine (6),36 (10), (12),37 and pyrrolo[2,3-b]quinoxaline-7-carboxylic acid (14),37 integrated with 3-aminopropan-1-ol upon complexation with biologically12–20,25,26,28,31,38–40 and environmentally20,24,27,32,38,39 important metal ions, such as Zn2+, Cd2+, Hg2+, Ga3+, In3+, and Tl3+.
Results and discussion
Ligands 6, 10, 12, and 14 were efficiently synthesised according to our previously published procedure, using ketones 5, 9a, 11f, 11e and salt of the latter or a regioisomeric mixture of keto acids 13 with 3-aminoalkohol as reagents (Scheme 2).35 Regioisomer 9a was obtained using a multistep method exploiting the exo-enol tautomerism of 7a in CH2Cl2 to furnish 3-amino-N-(2-amino-3-pyridyl)-pyrrolidinedione 8a, which was then cyclized in EtOH. Aminoalcohol was used in excess, acting as both reagent and solvent and reacting regioselectively at the benzoyl carbon atom. The reaction was carried out at room temperature for 5 days while the product precipitated. To shorten the reaction time for 6a, we modified this procedure by adding propanol to the reaction mixture and heating the starting materials at 50 °C for 3 h. Compounds 6c and 14f crystallise with one molecule of aminopropanol. | ||
| Scheme 2 N-Alkyl enamination of 3-benzoylpyridopyrrolopyrazine 5a–d, 9a, 11e,f and pyrrolo[2,3-b]quinoxaline-7-carboxylic acid derivatives 13e,f. | ||
According to our previous results, we obtained mixtures of the E/Z diastereoisomers of 6a–6d, 10a, 12e, 12f and 14e, 14f, with E-isomers predominating in solution (Table 1). The presence of a nitrogen atom at N-5 for 6 effectively shifts the E/Z equilibrium in CDCl3 to E-form, plausibly due to formation of an additional intramolecular hydrogen bond between the N-5 nitrogen atom and the hydroxyl group in solution. In the case of the mixture of regioisomeric 6- and 7-carboxylic acids of 3-benzoylpyrrolo[2,3-b]quinoxaline 13, only the regioisomeric 7-substituted products 14e and 14f were isolated.
| Yield [%] | E/Z ratio CDCl3 | E/Z ratio DMSO-d6 | E/Z ratio CD3CN | |
|---|---|---|---|---|
| a Compound is barely soluble in CDCl3. | ||||
| 6a Ar = Ph | 98 | 10:1 |
2.7:1 |
2.6:1 |
| 6b Ar = C6H4(4-Cl) | 98 | 8.7:1 |
3.2:1 |
|
| 6c Ar = C6H4(4-Me) | 82 | 8.7:1 |
3:1 |
|
| 6d Ar = C6H4(4-OEt) | 51 | 7.1:1 |
2.9:1 |
|
| 10a Ar = Ph, R = (CH2)3OH | 75 | 2:1 |
1.9:1 |
|
| 12e Ar = Bn | 35 | 2:1 |
2.1:1 |
|
| 12f Ar = CH2CH2Ph | 21 | 2.6:1 |
2.2:1 |
|
| 14e Ar = Bn | 42 | 2.2:1 |
||
| 14f Ar = CH2CH2Ph | 31 | 2.8:1 |
||
X-ray structure analysis41–48 of (E)-6a and (E)-12e showed the stabilisation of the configuration in the solid. For both structures, an intramolecular hydrogen bond N30–H30⋯N4 is observed H30⋯N4 = 2.16(3) Å, N30⋯N4 = 2.837(3) Å and N30–H30⋯N7 = 137(3)° for 6a (Fig. 2) and H30⋯N4 = 2.162(17) Å, N30⋯N4 = 2.894(1) Å and N30–H30⋯N7 = 138.0(14)° for 12e (Fig. 3).
 | ||
| Fig. 2 Crystal structure of 6a as a monohydrate, showing the atom labelling scheme. Displacement ellipsoids of non-hydrogen atoms are drawn at the 30% probability level. H atoms are presented as small spheres with an arbitrary radius. Dashed lines represent a hydrogen bond system observed within the asymmetric unit. | ||
 | ||
| Fig. 3 Crystal structure of 12e, specifically, the more abundant conformation with an occupancy of 88.4%, showing the atom labelling scheme. Displacement ellipsoids of non-hydrogen atoms are drawn at the 30% probability level. H atoms are presented as small spheres of an arbitrary radius. The dashed line represents an intramolecular hydrogen bond. | ||
The absorption and emission data for the studied compounds are listed in Table 2. The quantum yields (Φn) for compounds 5a,d, 6a,d and 9a, 10a, 12e,f, and 14e,f were determined in chloroform and acetonitrile using 1 mM stock solution prepared in DMSO. The lifetimes (τn) for all of the analysed compounds were obtained in chloroform. The solubility of the compounds was the basis of the solvent selection. In principle, no significant differences were observed in the absorption and emission spectra in the two solvents for compounds 6a, 9a, 10a, 12e, and 14e. The UV/Vis spectra of all enamines 6, 10a, 12e,f, and 14e,f exhibited two maxima (376–388 and 393–402 nm) with equal molar absorption coefficients.
| λ abs [nm] | ε [103 M−1 cm−1] | λ em [nm] | Φ n , | τ n , [s] | |
|---|---|---|---|---|---|
| a n.f.: non-detectable fluorescent emission. b The fluorescence quantum yields (Φn) were estimated from the corrected fluorescence spectra using 9,10-diphenylanthracene in cyclohexane (Φs = 0.90) as the standard. c Average of three measurements. d Measurements in CHCl3. e Measurements in CH3CN. | |||||
| 5a | 396d | 24.16 ± 1.71 | 456 | 0.023 ± 0.0006 | 5.38 × 10−10 ± 1.01 × 10−11 |
| 5b | 400d | 26.84 ± 1.54 | 456 | 0.007 ± 0.0010 | 5.08 × 10−10 ± 7.58 × 10−12 |
| 5c | 396d | 29.62 ± 2.31 | n.f. | — | n.f. |
| 5d | 396d | 30.18 ± 2.53 | n.f. | — | n.f. |
| 6a | 388, 408d | 28.25 ± 2.31 | 444 | 0.0077 ± 0.0005 | 5.36 × 10−10 ± 7.92 × 10−11 |
| 6b | 388, 408d | 22.69 ± 2.88 | 444 | 0.0083 ± 0.0004 | 2.17 × 10−10 ± 1.01 × 10−11 |
| 6c | 388, 408d | 14.35 ± 2.69 | 444 | 0.0073 ± 0.0005 | 2.57 × 10−10 ± 4.05 × 10−12 |
| 6d | 388, 408d | 27.87 ± 1.77 | 444 | 0.0027 ± 0.0008 | 2.21 × 10−10 ± 7.60 × 10−12 |
| 9a | 396e | 27.61 ± 1.72 | 457 | 0.0560 ± 0.0008 | 8.34 × 10−10 ± 1.24 × 10−11 |
| 10a | 382, 398e | 31.17 ± 1.99 | 440 | 0.0021 ± 0.0005 | 3.27 × 10−10 ± 5.75 × 10−11 |
| 12e | 376, 393e | 30.80 ± 5.07 | 438 | 0.0018 ± 0.0006 | 1.03 × 10−10 ± 2.51 × 10−12 |
| 12f | 376, 393e | 28.54 ± 3.30 | 438 | 0.0015 ± 0.0004 | 1.01 × 10−10 ± 2.89 × 10−11 |
| 14e | 376, 392e | 28.25 ± 1.38 | 438 | 0.0010 ± 0.0005 | 3.92 × 10−10 ± 7.09 × 10−12 |
| 14f | 386, 402e | 31.40 ± 1.83 | 440 | 0.0021 ± 0.0004 | 1.74 × 10−10 ± 9.12 × 10−11 |
All enamines exhibit weak fluorescence properties regardless of the various azaheteroarene fluorophores. To verify the sensing abilities of integrated fluorophore–receptor sensors for divalent and trivalent Zn2+, Cd2+, Hg2+, Ga3+, In3+, Tl3+ ions, we performed a detailed spectrophotometric study. The most effective turn-on fluorescence response among N-aryl or N-alkyl variously fused pyridopyrrolopyrazine or pyrrolo[2,3-b]quinoxaline for Zn2+ and In3+ is exhibited by 6a (Fig. 4a and b). Trivalent indium and divalent zinc ions have comparable Pauling's ionic radius and electronegativity. Additionally these ions have confined d10 electron shells. Thus, the trivalent indium cation should form a more stable complex with 6a in comparison with divalent zinc ions.49 Different emission profiles for 6a-Zn2+ and 6a-In3+ suggest, however, different mechanisms of complexation responsible for fluorescence enhancement.
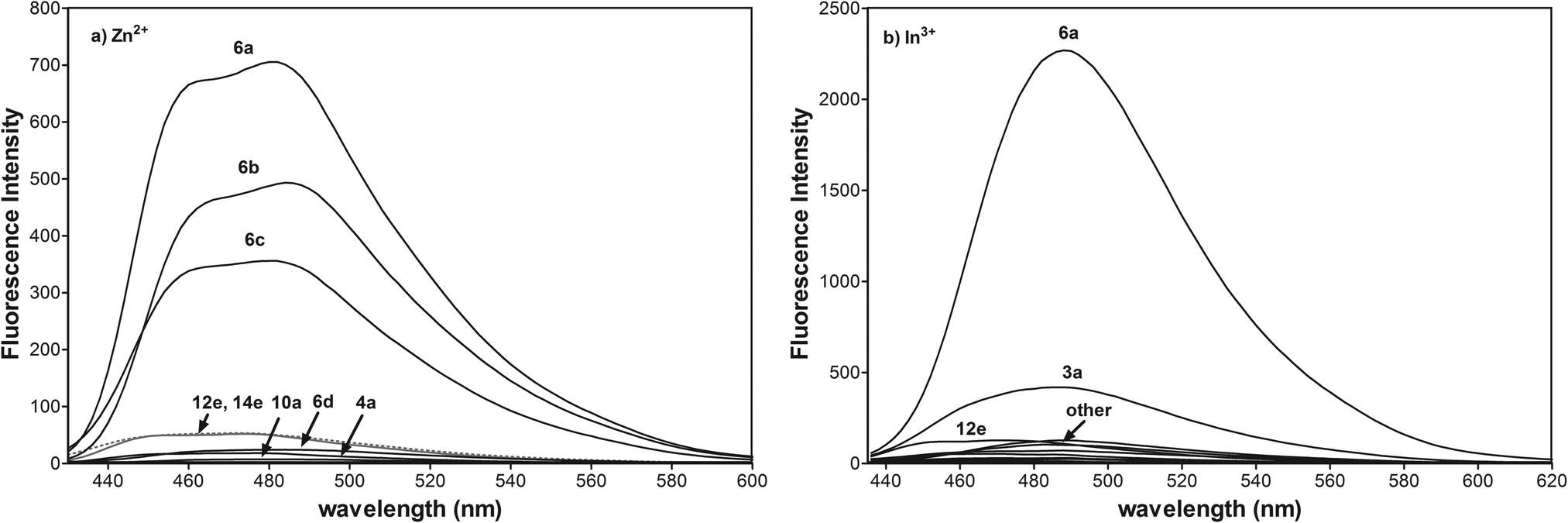 | ||
| Fig. 4 (a) Fluorescence spectra of 4a, 6a–d, 10a, 12e, and 14e (10 μM) in CH3CN with 1 equiv. of Zn2+ (λex = 415 nm, voltage 400 V), (b) fluorescence spectra of 3a, 6a, 10a, 12e, and 14e (10 μM) in CH3CN with 1 equiv. of In3+ (λex = 425 nm, voltage 550 V). | ||
We detected very low fluorescence response of all azaheteroarenes to Cd2+, and Hg2+ ions (ESI†) and lack of any changes for Ga3+, Tl1+, and Tl3+ ions.
The [N, N, O] tridentate ligands 6a, 6b are fluorescent for zinc ions, most likely due to the electronic effect of the most electron-withdrawing phenyl and 4-chlorophenyl groups at the N-1 position and the additional integrated nitrogen donor at the N-5 position of the fused heterocyclic system. The presence of the less electron-withdrawing methyl and ethoxyl group at the 4-substituted phenyl substituent for 6c and 6d amplifies the electron-donating character of the amide nitrogen atom. That reduces the dipole moment of the fluorophore and degree of charge transfer upon excitation of light which is in accordance with the literature.50
In contrast to the photooptical properties of reported N-methylnaphthalimide51,52 or N-methylindole53 derivatives, the electron-donor group at the N-1 position, such as benzyl for 12e and 14e reduces the degree of charge transfer responsible for fluorescence enhancement. The possible explanation for that difference in photooptical properties of fused N-alkyl imides may be connected not only with the electron-donating character of alkyl substituents but also with the restriction of π-conjugation. It seems probable that extended π-conjugation of the pyridopyrrolopyrazine with the aryl group at the N-1 position is essential for switching upon binding the zinc ion. However, confirmation of this photooptical phenomenon requires additional study. Other pyridopyrrolopyrazine derivatives 10a and pyrrolo[2,3-b]quinoxaline 4a as [N, N, O] tridentate ligands do not exhibit a turn-on fluorescence response. The quite similar behaviour of these tridentate ligands is not surprising because the receptor component, 3-aminopropanol and lack of additional donor nitrogen at N-5, are the same.
To study the influence of anions on the fluorescent spectra of 6a-Zn, we performed experiments between 6a (10 μM, Fig. 5) and different salts such as ZnSO4, ZnCl2, Zn(NO3)2, Zn(OTf)2, Zn(OAc)2, and Zn(acac)2 (10 μM). The experiment showed that the fluorescent enhancement of 6a-Zn increases in accordance with the basicity of anions. When anions are bound to metals in complexes their basicity may modulate the fluorescence intensity via changing the electron density on the donor group of the fluorophore. Furthermore, acetate and acetylacetonate anions may also interfere with the fluorescent probe, despite the fact that 6a-Zn(OAc)2 and 6a-Zn(acac)2 have similar fluorescent profiles to 6a-ZnCl2.
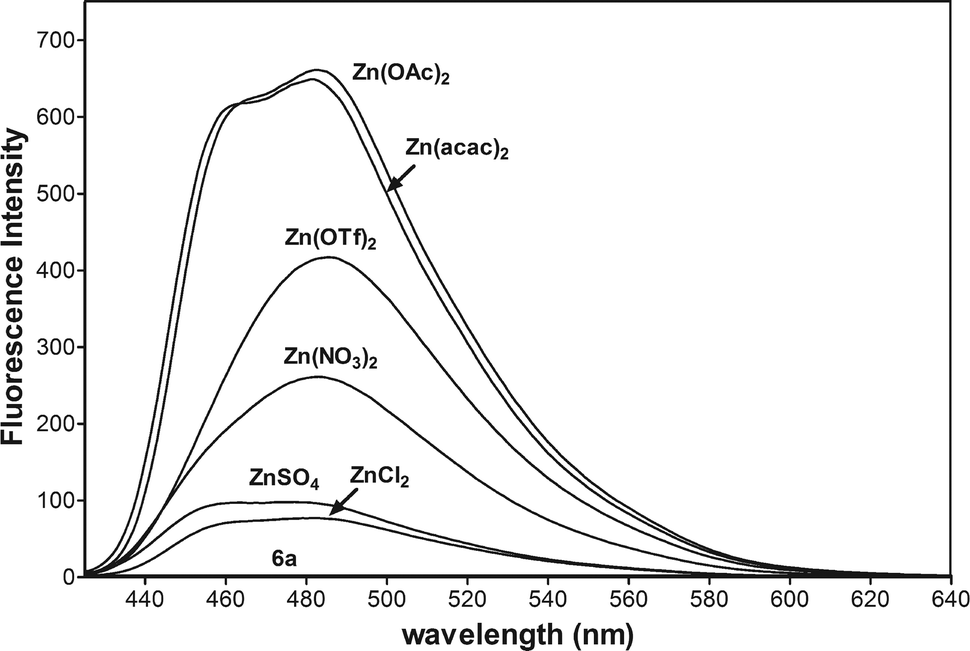 | ||
| Fig. 5 Fluorescence spectra of 6a (10 μM) in CH3CN with 1 equiv. of Zn(acac)2, Zn(OAC)2, Zn(OTf)2, Zn(NO3)2, ZnSO4, and ZnCl2 (λex = 415 nm, voltage 400 V). | ||
The basicity of anions should also affect the equilibrium of complex formation. The stronger bases such as acetate and acetylacetonate anions may more easily deprotonate the ligand 6a than other weaker bases under study, especially the chloride anion, so the stronger bases should shift the equilibrium towards the formation of products. The fluorescence titration data confirmed that emissions for 6a-Zn at λex = 415 nm are saturated with 1.2 equiv. of Zn(acac)2, 1.6 equiv. of Zn(OAc)2, and 2.0 equiv. of ZnCl2, respectively (Fig. 6a, ESI†).
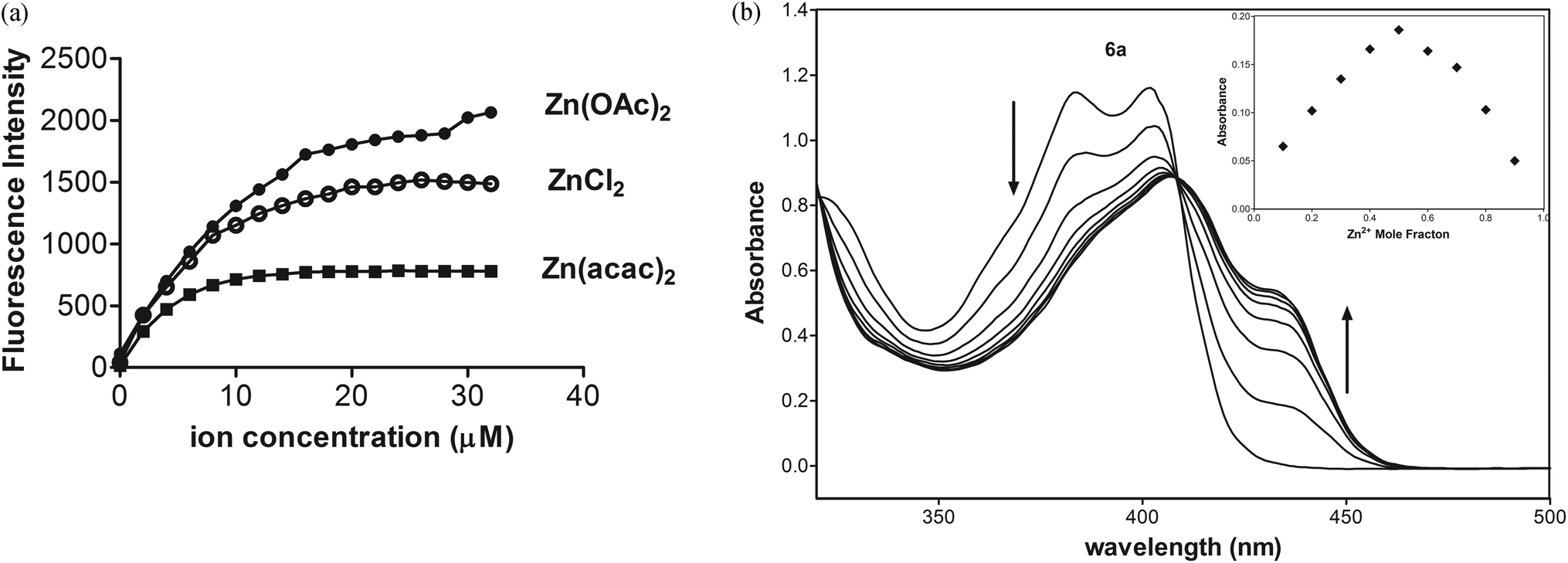 | ||
| Fig. 6 (a) Increase in fluorescence intensity of 6a (10 μM) in CH3CN as the function of Zn2+ concentration for Zn(acac)2 at λex/λem = 415/482 nm (400 V), Zn(OAc)2 at λex/λem = 415/482 nm (450 V), and for ZnCl2λex/λem = 415/464 nm (550 V). (b) Absorption titration of 6a (50 μM) in CH3CN with 0.2 equiv. of Zn(OAc)2. Inset: Job's plot for the binding of 6a with Zn(OAc)2 at λ = 435 nm in CH3CN. | ||
As shown in Fig. 6b, UV-Vis spectra of 6a exhibited two maximal absorptions at 385 and 403 nm. Upon addition of Zn(OAc)2, the absorbance at these peaks decreased, whereas the new two absorption peaks appeared at 407 and 435 nm with an isosbestic point at 409 nm. The photometric titration for 6a-Zn is saturated with 0.6 equiv. of Zn2+.
To determine the binding stoichiometry of 6a with Zn(acac)2, the absorption of 6a at 435 nm was plotted as a function of the molar fraction of 6a under a constant total concentration of 6a and Zn2+. The resulting Job plot is shown in Fig. 6b. The maximum absorbance was reached when the molar fraction was 0.5, which indicates a 1:1 ratio for 6a-Zn.
To evaluate the E/Z equilibrium during the formation of 6a-Zn and confirm its binding mode, a detailed 1H NMR titration was performed (Scheme 3, Fig. 7, ESI†). We used zinc acetylacetonate as a counter salt in 1H NMR titration because the characteristic chemical shifts of the CH proton for the enol tautomer of acetylacetonate makes it possible to track the progress of complex formation.
 | ||
| Scheme 3 Proposed binding modes of 6a with Zn(acac)2. | ||
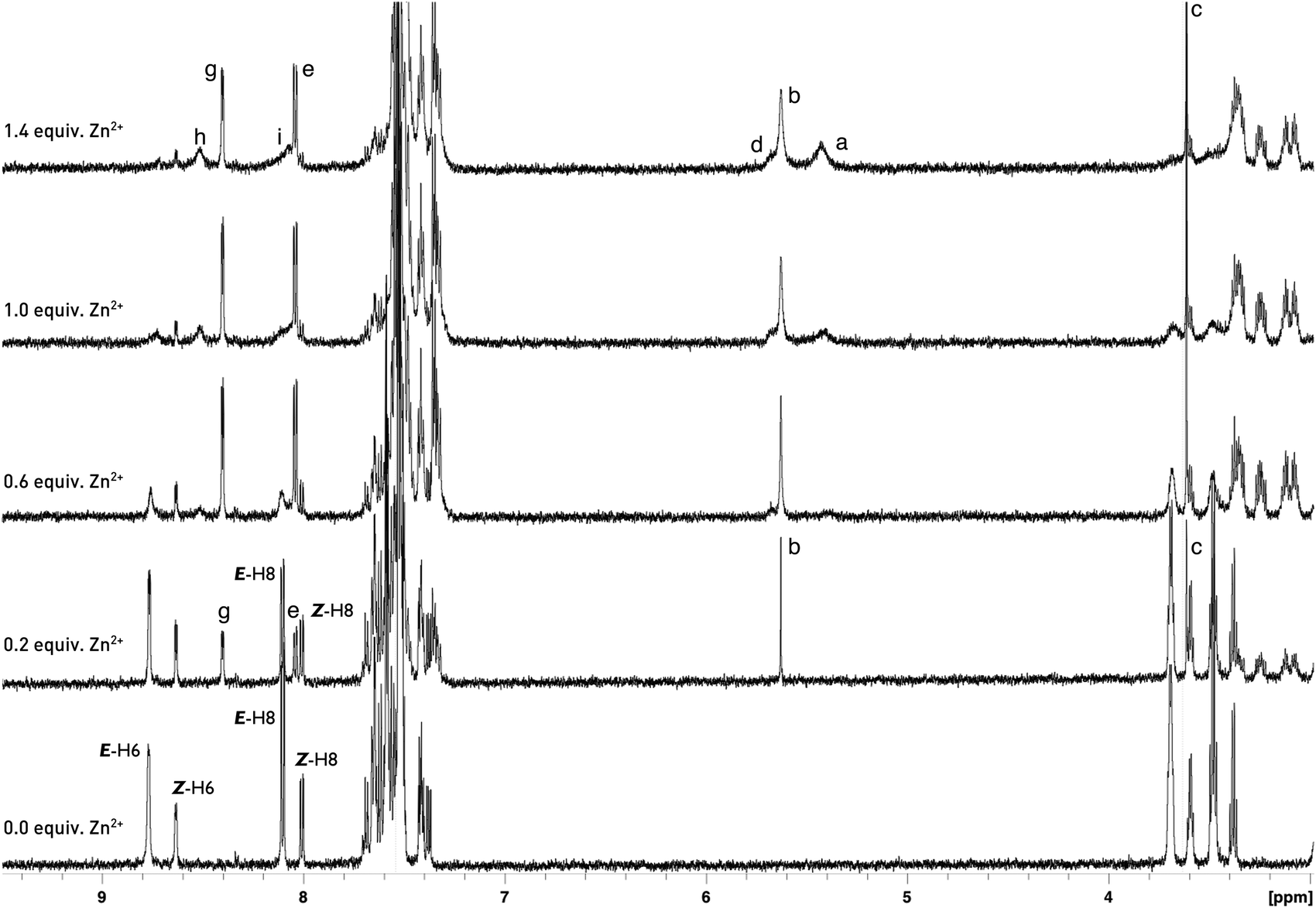 | ||
| Fig. 7 Partial 1H NMR spectra (600 MHz) of 6a (5 mM) in the presence of different concentrations of Zn(acac)2 in CD3CN. (Please see the ESI† for partial 1H NMR spectra of 6a in the region 1–4 ppm.) | ||
Upon gradual addition of 0–0.6 equiv. of Zn(acac)2 to a CD3CN solution of (E/Z)-6a, a new set of signals were observed with the simultaneous reduction of the NH signal, indicating the formation of a complex. The protons (E/Z)-H-6 of the free ligand are upfield shifted from δE-H-6 = 8.78 ppm and δZ-H-6 = 8.62 ppm to δ = 8.40 ppm. Simultaneously, the protons (E)-H-8 and (Z)-H-8 are upfield and downfield shifted from δE-H-8 = 8.11 ppm and δZ-H-8 = 8.01 ppm to δ = 8.03 ppm, respectively (Fig. 7). These signals were attributed to the H-6 and H-8 of the complex. The CH and CH2 protons of two tautomeric forms of acetylacetone also appeared at chemical shifts of δ = 5.63 and δ = 3.61 ppm, respectively. The presence of only one signal in the alkene region of the chemical shifts proved the formation of an L2Zn complex with a 2:1 binding mode. Further addition of zinc ions from 0.6 to 1.4 equiv. revealed the reorganisation of the complex to 6a-Zn-acac with a 1:1 binding mode of the ligand and zinc. This reorganisation was indicated by the appearance of the new set of broadened signals. In the diagnostic alkene region two additional signals appeared at δ = 5.67 and 5.45 ppm with the simultaneous increase of population of CH2 for the keto form of acetylacetone relative to the populations of H-6 in the L2Zn complex at 8.40 ppm and 6a-Zn-acac at 8.49 ppm. The protons at 5.67 ppm and 5.45 ppm were attributed to the CH fragment of acetylacetone chelating the zinc in 6a-Zn-acac and CH for zinc(II) acetylacetonate, respectively. The 1H NMR study shows that the Z diastereoisomer of 6a during the addition of zinc ion isomerises into its E-form, which coordinates the metal cation simultaneously with deprotonation. The upfield shifted signals for (E/Z)-H-6 reveal that the nitrogen atom at the N-5 position in pyrido[2,3-b]pyrrolo[2,3-e]pyrazine derivatives 6a does not directly coordinate the zinc cation during binding but probably facilitates the deprotonation and formation of complex (E)-6-Zn-acac.
Reorganisation of the complex from L2Zn to L-Zn-acac is not observed in fluorometric and photometric titrations. However, the 1H NMR experiment indicated that two equilibria were estabilished during the formation of the complex with binding mode 1:1.
Quite different equilibria are revealed by 1H NMR titration for E/Z-6a with InCl3 (ref. 54 and 55) (Scheme 4, Fig. 8). After addition of 0.4 equiv. of indium ions probably two separate complexes with E- and Z-diastereisomers of 6a, respectively, are formed. Interconversion of Z-6a to E-form and deprotonation during binding are not observed. Metal ions are consumed immediately and two sets of new signals appear indicating formation of the L3M system. One set shows broadened signals for diagnostic protons H-6, H-7 and H-8, because of long range spin coupling of these protons with the 115In isotope. These signals are shifted to 9.92, 8.55 and 8.43 ppm. Plausibly the indium ion binds to the N-5 nitrogen donor of the fluorophore. That coordination is possible for the Z-diastereoisomer of 6a because the N-5 nitrogen atom is easily accessible. The Z-NH proton is upfield shifted and overlaps with the E-NH-form. The second set shows the three-spin system ABM with three different coupling constants JH-6,H-7 = 4.9 Hz, JH-7,H-8 = 8.1 Hz, and JH-6,H-8 = 1.4 Hz. Protons H-6, H-7, and H-8 are upfield shifted to 8.95, 8.76, and 7.98 ppm, respectively. After addition of 0.6 equiv. of salt the new equilibria start to establish with two broadened set of signals in the ratio 2:1 indicating reorganisation and formation of the two new complexes with two N–H signals. Probably, two indium ions prefer binding to E/Z-6a at N-5 and N-9 nitrogen donors and form E-6a-In2 and Z-6a-In2. All attempts to isolate these complexes failed.
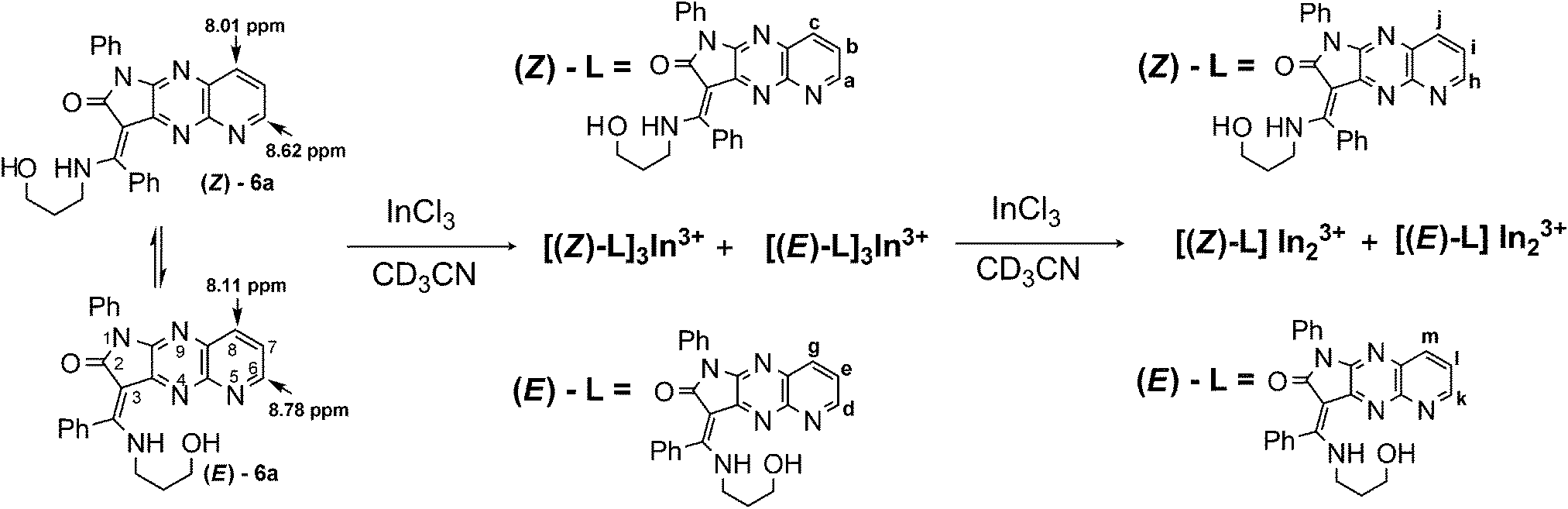 | ||
| Scheme 4 Proposed binding modes of 6a with InCl3. | ||
 | ||
| Fig. 8 Partial 1H NMR spectra (600 MHz) of 6a (5 mM) in the presence of different concentrations of InCl3 in CD3CN. | ||
As shown in Fig. 9a, upon addition of InCl3 to 6a, the absorbance decreased and the new broaden absorption peak appeared at 427 nm with an isosbestic point at 408 nm. The spectral change for 6a-In terminates by the addition of 2 equiv. of In3+. The fluorescence titration data show that emissions for 6a-In at λex = 425 nm appear after addition of 0.6 equiv. of metal ions and they are saturated with 3 equiv. of InCl3 (Fig. 9b). This indicates that 6a coordinates first with In3+, producing non-emissive complexes. Further addition of these ions leads to the formation of emissive complexes.
 | ||
| Fig. 9 (a) Absorption titration of 6a (50 μM) in CH3CN with 0.2 equiv. of InCl3. (b) Fluorescence titration (λex = 425 nm) of 6a (10 μM) in CH3CN with InCl3. The concentration of In3+ was increased from 0 to 36 μM. Inset: increase in fluorescence intensity at λem = 488 nm as a function of In3+ concentration. | ||
The metal ions except gallium and thallium affect the absorption spectral profile of 6a indicating the coordination of the ligand with Zn2+, Cd2+, Hg2+, and In3+ ions (Fig. 10a).
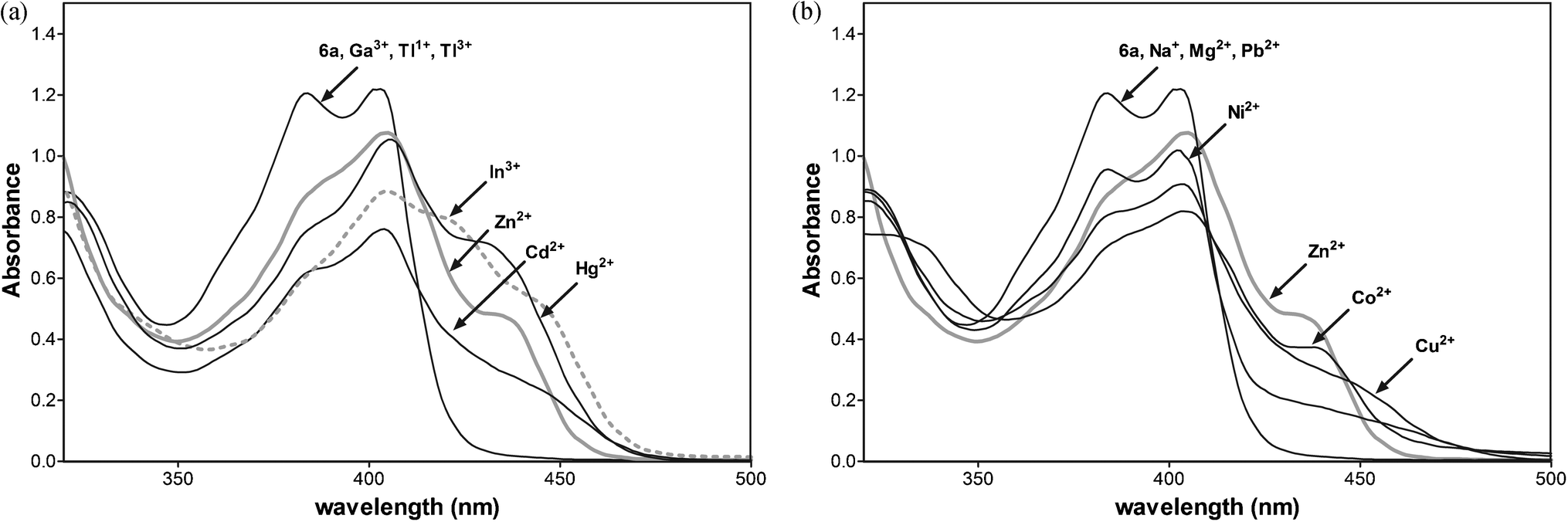 | ||
| Fig. 10 (a) Absorption spectra of 6a (50 μM) in CH3CN with 1 equiv. of Zn2+, Cd2+, Hg2+, Ga3+, In3+, Tl3+ and Tl1+. (b) Absorption spectra of 6a (50 μM) in CH3CN with 1 equiv. of Zn2+, Na+, Mg2+, Pb2+, Ni2+, Co2+ and Cu2+. | ||
The selectivity of 6a towards Zn2+ and other metal ions was examined, and the results are shown in Fig. 11. An extremely low fluorescence emission intensity relative to that for Zn2+ was observed upon the addition of Cd2+, Co2+, Hg2+, Mg2+, Na+, Ni2+, and Pb2+ metal cations.
 | ||
| Fig. 11 (a) Fluorescence spectra of 6a (10 μM) in CH3CN with 1 equiv. of Cd2+, Co2+, Hg2+, Mg2+, Na+, Ni2+, Pb2+, Zn2+, and 3 equiv. of In3+ (λex = 415 nm). (b) Metal ion selectivity profiles of 6a (10 μM) in CH3CN in the presence of various metal ions (10 μM) at λex/λem = 415/480 nm. | ||
To study the influence of other metal ions on the binding of Zn2+ to 6a, we performed competitive experiments between Zn2+ (10 μM) and Cd2+, Cu2+, Co2+, Hg2+, Mg2+, Na+, Ni2+ Pb2+, and In3+ (10 μM, Fig. 11a and b). The addition of Hg2+, Na+, and Pb2+ have a negligible effect on Zn2+ sensing. The fluorescence of 6a with Zn2+ is slightly quenched by Co2+, Cd2+, and Hg2+ ions. The great impact on the fluorescence emission of 6a with Zn2+ has the presence of one equiv. of indium ions, quenching the emission. Three equiv. of In3+ enhance the fluorescence emission with the changes in the profile similar to 6a-In indicating the displacement of zinc by the indium metal ion.
Additionally, the absorption data detected for 6a and the metal ions (Fig. 10b) show that the addition of Na+, Mg2+, and Pb2+ does not affect the absorption spectral profile of 6a. Thus, these metal cations most likely do not bind to the receptor and do not compete in complexation with 6a. The most significant changes, indicating chelation, are observed upon the addition of Zn2+, Cd2+, Co2+, Cu2+, Hg2+and Ni2+ metal ions. The addition of 1 equiv. of Cd2+, Co2+, or Hg2+ into the complex 6a with Zn2+ slightly reduces the emission peak at 480 nm, indicating that the displacement of Zn2+ from the complex by Cd2+, Co2+, or Hg2+ ions is unfavourable. The competition experiment shows excellent selectivity for Zn2+ over all metal ions under study except In3+.
The fluorescence properties of the new complexes are solvent-dependent, and the new complexes are non-fluorescent in the presence of water and in DMSO. To understand this phenomenon, we measured the 1H NMR spectra of 6a with 0.5, 1.0, 1.5, and 2.0 equiv. of Zn(acac)2 in DMSO-d6. All spectra, even that for the highest zinc cation concentration, show four sets of signals: E-, Z-diastereoisomers of 6a with E/Z-NH signals and 2:1 and 1:1 complexes of 6a-Zn. This polar aprotic solvent enhances solvation by hindering the deprotonation of the enamino group and thus the further formation of the complex (ESI†).
Conclusions
We have designed and verified the sensing abilities of integrated azaheteroarene fluorophores, such as N-aryl or N-alkyl pyrido[2,3-b]pyrrolo[2,3-e]pyrazine (6), pyrido[2,3-b]pyrrolo[3,2-e]pyrazine (10), pyrido-[3,4-b]pyrrolo[3,2-e]pyrazine (12), and pyrrolo[2,3-b]quinoxaline-7-carboxylic acid (14), integrated with 3-aminopropan-1-ol upon complexation with Zn2+, Cd2+, Hg2+, Ga3+, In3+, and Tl3+ metal ions. We have shown that the [N, N, O] tridentate ligands 6a, 6b, and 6c with the nitrogen atom at N-5 and N-aryl groups selectively respond to zinc and 6a to indium ions in acetonitrile. The benzyl and (4-ethoxy)phenyl groups at the N-1 position weaken the acceptor character of the amide carbonyl group in fluoroionophore systems and quench ICT fluorescence enhancement for 6d, 12e, and 14e. An additional nitrogen donor at N-7 or N-8 changes the electron density in ICT azaheteroarene indicators and that should also influence the dipole moment of fluorophores. Pyrido[2,3-b]pyrrolo[3,2-e]pyrazine derivative 10a with the nitrogen atom at N-8 does not exhibit fluorescence emission with Zn2+, Cd2+, Hg2+, Ga3+, In3+, and Tl3+ metal ions. 1H NMR titration showed two different mechanisms of binding Zn2+ and In3+ to 6a. The Z-diastereoisomer of 6a during the addition of the zinc ion isomerises to its E-form, which coordinates metal cations simultaneously with deprotonation in acetonitrile. Indium ions form two separate complexes with (E)-6a and (Z)-6a without deprotonation. Although ligands under study do not operate in aqueous media, further functionalisation of the receptor unit will allow us to obtain sensors with desirable physicochemical properties which will be reported in due course.Experimental
Materials and instrumentation
All of the solvents for the UV and fluorescence spectra were obtained from Sigma-Aldrich or Merck and were of spectroscopic purity. The solution metal ions were prepared from ZnCl2, ZnSO4, Zn(NO3)2, Zn(OTf)2, Zn(acac)2, Zn(OAc)2, Ga(acac)3, InCl3, TlNO3, Tl(OAc)3, Cu(acac)2, Ni(acac)2, Co(acac)2, Cd(acac)2, Hg(OAc)2, NaOAc, Pb(OAc)2, Mg(OAc)2. Melting points were determined on a Boetius PHMK 05 melting point apparatus. IR spectra were measured on a Thermo Scientific Nicolet IR200 FT-IR. The 1H NMR and 13C NMR spectra were recorded using Bruker Avance III 600 and II 300 spectrometers at 300 K. The chemical shifts (δ) are reported in parts per million (ppm) on a δ scale downfield from TMS. The 1H NMR spectra were referenced internally to the residual proton resonance in CDCl3 (δ 7.26 ppm), DMSO-d6 (δ 2.49 ppm), and CD3CN (δ 1.96 ppm). The 13C NMR spectra were referenced to CDCl3 (δ 77.0 ppm), or, DMSO-d6 (δ 39.7 ppm). The coupling constants (J) are reported in Hertz (Hz). Mass spectra were recorded on a Finnigan Mat 95 (EI, 70 eV) and ESI spectrometers. Microanalyses were performed with a Vario Micro Tube CHNS; the results agreed with the calculated values.For the determination of ε and quantum yields indicated in Table 2, the analysed compounds were diluted in selected solvents to prepare 20 μM stock solutions: chloroform for 5 and 6 and acetonitrile for 9a, 10a, 12e, 12f, 14e, and 14f using a 1 mM stock solution prepared in DMSO. The fluorescence lifetimes were determined using a 20 μM stock solution in chloroform. The absorption spectra of compounds 5, 6, 9a, 10a, 12e, 12f, 14e, and 14f (Table 2) were recorded on a Microplate Reader Infinite M200 (Tekan) spectrophotometer in 1 cm cells at 25 °C.
The fluorescence measurements for all analysed compounds were done using a Hitachi F-4500 spectrofluorometer. All spectra were recorded at 25 °C with an excitation slit width of 5 nm, an emission slit of 10 nm and a PMT voltage of 700 V.
The fluorescence quantum yields of the analysed compounds were determined using 9,10-diphenylanthracene (Φs = 0.90 in cyclohexane) as a standard. The fluorescence lifetimes were determined using a time-correlated single-photon counting system based on the Horiba Jobin Yvon IBH lifetime spectrofluorometer system components, which consisted of a picosecond single-photon-detection TBX-04 module integrated with a fast photomultiplier, high-voltage power supply, GHz preamplifier and picosecond-timing discriminator. A picosecond pulsed laser diode (NanoLED-11, Horiba Jobin, Yvon) was used as a light source. The specimens were excited at 372 nm with a 1 MHz repetition rate. The fluorescence decays were observed through a cut-off filter of 475 nm (Andover Corporation Optical Filter). To avoid pulse pile-up, the power of the diode was adjusted to an appropriately low level using a neutral gradient filter. The excitation pulse diode laser profile, required for the deconvolution analysis, was measured using a diluted glycogen suspension without a filter. For the data acquisition and decay analysis, the Jobin Yvon IBH data station and DAS6 software were used. All measurements were performed at 25 °C, and each recorded lifetime was averaged from five independent decay measurements.
The Job plots for 6a with Zn(acac)2 and absorption spectra of 6a (50 μM) in CH3CN with respective 1 equiv. of metal cations were recorded on a Hitachi U-3900H spectrophotometer. The fluorescence measurements (Fig. 4, 5, 6a, 9b, 11a and b) were also recorded using a F-7000 FL spectrophotometer (25 °C, excitation and emission slit width of 5 nm, 400 V and 550 of PMT voltage). The fluorescence spectra were obtained using a 1 mM stock solution prepared in DMSO for compounds 4a, 10a, 12e, and 14e.
Substrates 5a–d, 7a, 11e,f, and 13e,f were synthesised according to previously reported procedures.35,36
X-ray structure analysis of 6a and 12e
Crystals of 6a were obtained from acetonitrile solution by slow evaporation of the solvent under ambient conditions. The crystal suitable for X-ray diffraction experiment was a yellow plate of dimensions: 0.3 × 0.1 × 0.03 mm. X-ray diffraction data were collected at 130 K using a Nonius Brucker KappaCCD diffractometer with MoKα radiation (λ = 0.71073 Å), with the software Collect.41 Data were processed using HKL SCALEPACK42 and HKL DENZO.42 The phase problem was solved by direct methods using SHELXS-9743 program. The model parameters were refined by full-matrix least-squares on F2 using SHELXL-97.43 All non-hydrogen atoms were refined anisotropically. H atoms attached to N and O atoms were located on the difference Fourier map. These in the hydroxyl group and the water molecule were refined using the DFIX restraint with a target bond length of 0.82 Å, within an assumed estimated standard deviation of 0.02 Å. For the water molecule the DANG restraint was applied with an H1w⋯H2w distance of 1.35 Å, within an assumed estimated standard deviation of 0.04 Å. Positions of hydrogen atoms attached to C atoms were calculated with C–H = 0.95 Å for aromatic, C–H = 0.99 Å for methylene and were refined using the riding model with the isotropic displacement parameter Uiso = 1.2 Ueq of the parent atom. Crystal data: moiety formula C25H21N5O2 H2O, Mr = 441.48, monoclinic, space group P21/c, a = 11.2202(2) Å, b = 6.1966(4) Å, c = 31.8009(10) Å, β = 109.409(2)°, V = 2085.48(15) Å3, Z = 4, Dc = 1.406 g cm−3, μ(MoKα) = 0.10 mm−1, F(000) = 928; 14239 collected reflections, 4696 unique (Rint = 0.057), 3177 observed (I > 2σ(I)). For 313 refined parameters, the final R1 = 0.062 for reflections with F2 > 2σ(F2), wR2 = 0.186 for all unique reflections, S = 1.04.
Crystals of 12e were obtained from DMSO-d6 solution by slow evaporation of the solvent under ambient conditions. The crystal suitable for X-ray diffraction experiment was a yellow prism of dimensions: 0.45 × 0.3 × 0.3 mm. X-ray diffraction data were collected at 110 K using a SuperNova diffractometer with MoKα radiation (λ = 0.71073 Å). Data were processed using CRYSALISPro.44 The phase problem was solved using direct methods with SHELXS-9743 program. The model parameters were refined by full-matrix least-squares on F2 using SHELXL-97.43 All non-hydrogen atoms were refined anisotropically. H atoms attached to N and O atoms were located on the difference Fourier map and refined independently. Positions of hydrogen atoms attached to C atoms were calculated with C–H = 0.95 Å for aromatic, C–H = 0.99 Å for methylene and were refined using the riding model with the isotropic displacement parameter Uiso = 1.2 Ueq of the parent atom. Crystal data: moiety formula C26H23N5O2, Mr = 437.49, monoclinic, space group P21/c, a = 9.1766(1) Å, b = 11.1148(1) Å, c = 21.7846(2) Å, β = 106.341(1)°, V = 2132.19(4) Å3, Z = 4, Dc = 1.363 g cm−3, μ(MoKα) = 0.09 mm−1, F(000) = 920; 32725 collected reflections, 5395 unique (Rint = 0.032), 4494 observed (I > 2σ(I)). For 323 refined parameters, the final R1 = 0.039 for reflections with F2 > 2σ(F2), wR2 = 0.103 for all unique reflections, S = 1.03.
WinGX 45 software was used to prepare materials for publication. Figures showing asymmetric units of 6a and 12e were made using ORTEP-3 for Windows.46 For calculation of the weighted least-squares planes through selected atoms program PARST47,48 was used. Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 890197 and 890198 for structure of 12e and 6a, respectively.
General procedure: synthesis of enaminone 6a–d, 10a, 12e, 12f, 14e, and 14f
A benzoyl-azaheteroarene derivative 5, 9, 11, or 13 (0.55 mmol) was added to the amine (2 mL). The mixture was left for 5 days, and then diluted with cold H2O (100 mL). The precipitated solid was collected by filtration, washed with H2O, and crystallised from acetonitrile.:1 (CDCl3), E/Z = 2.7:1 (DMSO-d6), E/Z = 2.6:1 (CD3CN). IR (ATR): 3335, 3207, 3070, 2927, 2875, 1704, 1596 cm−1. 1H NMR (300.13 MHz, DMSO-d6): δ = 1.69 (quint, 2H, J = 6.0 Hz, Z-CH2), 1.76 (quint, 2H, J = 6.0 Hz, E-CH2), 3.31 (s, H2O) 3.41 (m, 6H, E/Z-CH2NH, Z-CH2OH), 3.54 (q, 2H, J = 6.0 Hz, E-CH2OH), 4.62 (t, 2H, J = 6.0. Hz, E/Z-OH), 7.37–7.62 (m, 22H, E/Z-H-Ar), 8.04 (d, J = 8.2 Hz, Z-H-8), 8.14 (dd, 1H, J = 1.8 Hz, J = 8.2 Hz, E-H-8), 8.58 (d, 1H, J = 4.3 Hz, Z-H-6), 8.74 (dd, 1H, J = 1.8 Hz, J = 4.3 Hz, E-H-6), 10.69 (br. s, 1H, Z-NH), 10.89 (br. s, 1H, E-NH). 13C NMR (75.47 MHz, DMSO-d6): δ = 32.91, 42.05, 57.85, 89.49, 122.29, 127.39, 127.49, 128.01, 128.54, 128.80, 130.15, 130.52, 133.53, 136.03, 148.29, 165.17, 166.48, 173.50. MS-ESI: m/z 424 (M+ + 1). Anal. calcd for C25H21N5O2 + H2O: C, 68.01; H, 5.25; N, 15.86%. Found: C, 67.99; H, 4.95; N, 15.71%.
:1 (CDCl3), E/Z = 3.2:1 (DMSO-d6). IR (ATR): 3331, 3207, 3062, 2957, 2923, 1702, 1593 cm−1; 1H NMR (300.13 MHz, DMSO-d6); δ = 1.69 (quint, 2H, Z-CH2), 1.75 (quint, 2H, J = 6.3 Hz, E-CH2), 3.43 (m, 4H, E/Z-CH2NH2), 3.56 (m, 4H, E/Z-CH2OH), 4.35 (t, 1H, E-OH), 4.62 (t, 1H, Z-OH), 7.37–7.70 (m, 20H, E/Z-H-Ar), 8.03 (1H, d, J = 8.1 Hz, Z-H-8), 8.05 (1H, dd, J = 8.0 Hz, J = 1.7 Hz, E-H-8), 8.58 (1H, dd, J = 4.5 Hz, J = 1.7 Hz, Z-H-6), 8.75 (1H, dd, J = 4.5 Hz, J = 1.7 Hz, E-H-6), 10.69 (br. s, 1H, Z-NH), 10.89 (br. s, 1H, E-NH). 13C NMR (75.47 MHz, DMSO-d6) δ = 32.88, 42.10, 57.85, 89.41, 122.34, 128.01, 128.56, 128.79, 128.90, 130.20, 130.45, 131.70, 132.42, 133.30, 136.04, 146.45, 148.19, 148.44, 164.93, 166.54. MS-ESI: m/z 458 (M+ + 1). Anal. calcd for C25H20ClN5O2 + H2O: C, 63.09; H, 4.66; N, 14.71%. Found: C, 63.17; H, 4.46; N, 14.85%.
:1 (CDCl3), E/Z = 3:1 (DMSO-d6). IR (ATR): 3435, 3282, 3040, 2925, 2890, 1709, 1607 cm−1. 1H NMR (300.13 MHz, CDCl3) for crude product 6c + NH2(CH2)3OH, H2O: δ = 1.62 (m, 2H, J = 5.8 Hz, CH2), 1.90 (m, 4H, J = 5.8 Hz, E/Z-CH2), 2.36 (s, 3H, E-CH3), 2.44 (s, 3H, Z-CH3), 2.63 (br. s, 4H, E/Z-OH, H2O) 2.97 (t, 2H, J = 5.8 Hz, CH2NH2), 3.44 (br. m, 2H, Z-CH2NH), 3.56 (t, 2H, J = 5.8 Hz, E-CH2NH), 3.78–3.88 (m, 6H, J = 5.8 Hz, E/Z-CH2-OH; CH2OH), 7.23–7.66 (m, 20H, E/Z-H-Ar), 8.09 (dd, 1H, J = 1.8 Hz, J = 8.1 Hz, Z-H-5), 8.19 (dd, 1H, J = 1.8 Hz, J = 8.1 Hz, E-H-7), 8.69 (dd, 2H, J = 4.4 Hz, J = 1.8 Hz, E/Z-H-6), 10.77 (br. s, 1H, Z-NH), 11.09 (br. s, 1H, E-NH). 13C NMR (75.47 MHz, CDCl3) for crude product 6c + NH2(CH2)3OH, H2O: δ = 21.17, 32.53, 33.73, 41.43, 42.29, 59.12, 63.41, 90.27, 121.78, 126.59, 127.56, 128.74, 129.43, 130.25, 130.44, 134.15, 136.91, 137.34, 147.28, 147.68, 148.54, 149.25, 166.14, 166.78. 1H NMR (300.13 MHz, DMSO-d6) 6c: δ = 1.69 (quint, 2H, J = 6.0 Hz, Z-CH2), 1.76 (quint, 2H, J = 6.0 Hz, E-CH2), 2.36 (s, 3H, E-CH3), 2.44 (s, 3H, Z-CH3), 2.63 (br. s, 4H, E/Z-OH, H2O) 2.97 (t, 2H, J = 5.8 Hz, CH2NH2), 3.44 (br. m, 2H, Z-CH2NH), 3.56 (t, 2H, J = 5.8 Hz, E-CH2NH), 3.78–3.89 (m, 6H, J = 5.8 Hz, E/Z-CH2-OH; CH2OH), 7.23–7.66 (m, 20H, E/Z-H-Ar), 8.09 (dd, 1H, J = 1.8 Hz, J = 8.1 Hz, Z-H-5), 8.19 (dd, 1H, J = 1.8 Hz, J = 8.1 Hz, E-H-7), 8.69 (dd, 2H, J = 4.4 Hz, J = 1.8 Hz, E/Z-H-6), 10.58 (br. s, 1H, Z-NH), 11.09 (br. s, 1H, E-NH). MS-ESI: m/z 438 (M+ + 1). Anal. calcd for C26H23N5O2 + H2O: C, 68.56; H, 5.53; N, 15.37%. Found: C, 68.93; H, 5.28; N, 14.90%.
:1 (CDCl3), E/Z = 2.9:1 (DMSO-d6). IR (ATR): 3445, 3275, 3076, 2978, 2937, 1714, 1642 cm−1. 1H NMR (300.13 MHz, CDCl3) for crude product 6d: δ = 1.37–1.48 (m, 6H, J = 6.0 Hz, E/Z-CH3), 1.85–1.97 (m, 4H, J = 6.0 Hz, E/Z-CH2), 3.46 (q, 2H, J = 6.0 Hz, Z-CH2NH), 3.56 (q, 2H, J = 6.0 Hz, E-CH2NH), 3.80 (t, 2H, J = 5.8 Hz, Z-CH2OH) 3.87 (t, 2H, J = 5.8 Hz, E-CH2OH), 4.01–4.14 (m, 4H, J = 6.0 Hz, E/Z-CH2O), 6.95 (d, 2H, J = 8.7 Hz, E-H-3′/5′), 7.07 (d, 2H, J = 8.7 Hz, Z-H-3′/5′) 7.38–7.65 (m, 20H, E/Z-H-Ar), 8.09 (dd, 1H, J = 1.9 Hz, J = 8.1 Hz, Z-H-5), 8.18 (dd, 1H, J = 1.8 Hz, J = 8.1 Hz, E-H-7), 8.70 (dd, 1H, J = 4.5 Hz, J = 1.8 Hz, E-H-6), 8.75 (dd, 1H, J = 4.4 Hz, J = 1.9 Hz, Z-H-6), 10.77 (br. s, 1H, Z-NH), 11.06 (br. s, 1H, E-NH). 13C NMR (75.47 MHz, CDCl3) for crude product 6d: δ = 14.77, 32.61, 42.33, 59.25, 59.50, 63.65, 90.26, 114.75, 115.03, 121.79, 125.57, 127.57, 128.01, 128.73, 130.25, 130.41, 134.16, 136.81, 147.36, 147.77, 148.49, 149.27, 158.11, 166.28, 166.72. MS-ESI: m/z 468 (M+ + 1). Anal. calcd for C27H25N5O3: C, 69.36; H, 5.39; N, 14.98%. Found: C, 69.13; H, 5.28; N, 14.90%.
:1 (CDCl3), E/Z = 1.9:1 (DMSO-d6). IR (ATR): 3235, 3059, 2862, 1711, 1609, 1587, 1561 cm−1. 1H NMR (300.13 MHz, CDCl3): δ = 1.83 (m, 2H, Z-CH2), 1.90 (m, 2H, E-CH2), 2.05 (br. s, 2H, E/Z-OH), 3.43–3.54 (m, 4H, E/Z-CH2NH), 3.77 (t, 2H, J = 5.8 Hz, Z-CH2OH), 3.83 (t, 2H, J = 5.8 Hz, E-CH2OH), 7.28–7.74 (m, 22H, E/Z-H-Ar), 8.16 (dd, 1H, J = 1.7 Hz, J = 8.1 Hz, E-H-5), 8.66 (dd, 1H, J = 1.7 Hz, J = 4.4 Hz, 1H, Z-H-7), 8.74 (dd, 1H, J = 1.7 Hz, J = 4.4 Hz, 1H, E-H-7), 10.59 (br. s, 1H, Z-NH), 10.74 (br. s, 1H, E-NH). 13C NMR (75.47 MHz, CDCl3): δ = 32.32, 32.62, 42.01, 59.77, 90.16, 121.58, 126.72, 127.19, 127.63, 128.02, 128.68, 128.76, 128.99, 130.30, 132.74, 133.90, 135.20, 137.02, 148.86, 161.07, 166.08. MS-ESI: m/z 423 (M+ + 1). 424; anal. calcd for C25H21N5O2 + H2O: C, 70.91; H, 5.0; N, 16.54%. Found: C, 70.92; H, 5.03; N, 16.29%.
:1 (CDCl3), E/Z = 2.1:1 (DMSO-d6). IR (ATR): 3411, 3241, 3060, 2937, 2870, 1703, 1612 cm−1. 1H NMR (300.13 MHz, CDCl3): δ = 1.87 (m, 2H, J = 6.0 Hz, Z-CH2), 1.92 (m, 2H, J = 6.0 Hz, E-CH2; 2H, E/Z-OH), 3.46 (q, 2H, J = 6.0 Hz, Z-CH2-NH), 3.51 (q, 2H, J = 6.0 Hz, E-CH2-NH), 3.79 (t, 2H, J = 6.0 Hz, Z-CH2-OH), 3.85 (t, 2H, J = 6.0 Hz, E-CH2-OH), 5.05 (s, 2H, E-CH2Bn), 5.23 (s, 2H, Z-CH2Bn), 7.20–7.63 (m, 22H, E/Z-H-Ar), 8.36 (d, 1H, J = 5.6 Hz, Z-H-6), 8.52 (d, 1H, J = 5.6 Hz, E-H-6), 9.13 (s, 1H, Z-H-8), 9.20 (s, 1H, E-H-8), 10.61 (t, 1H, Z-NH), 10.78 (t, 1H, E-NH). 13C NMR (75.47 MHz, CDCl3): δ = 32.56, 41.94, 42.08, 59.75, 104.89, 119.99, 127.43, 127.59, 127.87, 128.41, 128.60, 128.69, 128.81, 130.25, 130.50, 136.98, 151.19, 166.33, 166.65. MS-ESI: m/z 438 (M+ + 1). Anal. calcd for C27H25N5O2 + H2O: C, 69.07; H, 5.80; N, 14.92%. Found: C, 68.77; H, 5.61; N, 14.73%.
:1 (CDCl3), E/Z = 2.2:1 (DMSO-d6). IR (ATR): 3414, 3056, 2944, 2826, 2748, 1697, 1609 cm−1. 1H NMR (300.13 MHz, CDCl3): δ = 1.87 (quint, 2H, J = 6.0 Hz, Z-CH2), 1.93 (quint, 2H, J = 6.0 Hz, E-CH2; 2H, E/Z-OH; H2O), 3.00–3.05 (m, 2H, E-CH2Ph), 3.12–3.18 (m, 2H, Z-CH2Ph), 3.47 (q, 2H, J = 6.0 Hz, Z-CH2NH), 3.51 (q, 2H, J = 6.0 Hz, E-CH2NH), 3.80 (t, 2H, J = 6.0 Hz, Z-CH2-OH), 3.87 (t, 2H, J = 6.0 Hz, E-CH2-OH), 4.07–4.12 (m, 2H, E-CH2N), 4.25–4.31 (m, 2H, Z-CH2N), 7.25–7.63 (m, 22 H, E/Z-H-Ar), 8.36 (d, 1H, J = 5.6 Hz, Z-H-6), 8.52 (d, 1H, J = 5.6 Hz, E-H-6), 9.13 (s, 1H, Z-H-8), 9.19 (s, 1H, E-H-8), 10.59 (t, 1H, Z-NH), 10.75 (t, 1H, E-NH). 13C NMR (75.47 MHz, CDCl3): δ = 32.31, 32.55, 34.25, 39.81, 42.10, 59.76, 90.94, 120.00, 126.35, 127.53, 127.92, 128.39, 128.48, 128.82, 128.94, 130.46, 138.56, 144.21, 149.14, 151.16, 166.49. MS-ESI: m/z 452 (M+ + 1). Anal. calcd for C27H25N5O2 + H2O: C, 69.07; H, 5.80; N, 14.92%. Found: C, 68.77; H, 5.61; N, 14.73%.
:1 (DMSO-d6). IR (ATR): 3211, 3058, 3034, 2927, 2878, 1690, 1614, 1591, 1573 cm−1. 1H NMR (300.13 MHz, DMSO-d6): δ = 1.71 (quint, 2H, J = 6.3 Hz, Z-CH2), 1.77 (quint, 2H, J = 6.3 Hz, E-CH2), 3.35–3.39 (m, 4H, Z/E-CH2NH), 3.47 (t, 2H, Z-CH2OH), 3.58 (t, 2H, E-CH2OH), 4.65 (s, 1H, Z-OH), 4.76 (s, 1H, E-OH), 4.93 (s, 2H, E-CH2Bn), 5.15 (s, 2H, Z-CH2Bn), 7.18–7.65 (m, 21H, E/Z-H-Ar), 7.84 (dd, 1H, J = 8.5 Hz, J = 1.9 Hz, Z-H-7), 7.95 (d, 1H, J = 8.5 Hz, Z-H-8), 8.02 (dd, 1H, J = 8.5 Hz, J = 1.7 Hz, E-H-7), 8.25 (d, 1H, J = 1.9 Hz, Z-H-5), 8.31 (d, 1H, J = 1.7 Hz, E-H-5), 10.50 (t, 1H, J = 5.6 Hz, Z-NH), 10.75 (t, 1H, J = 5.6 Hz, E-NH), 13.00 (s, 2H, COOH). 13C NMR (75.47 MHz, DMSO-d6): δ = 32.31, 32.53, 41.28, 41.43, 42.25, 42.42, 58.27, 58.34, 89.44, 89.72, 125.60, 125.87, 127.30, 127.67, 127.85, 128.06, 128.21, 128.35, 128.53, 128.60, 129.13, 130.09, 130.21, 130.62, 130.94, 137.10, 137.33, 137.49, 137.69, 141.39, 142.14, 146.93, 147.00, 165.56, 165.61, 165.80, 167.23, 169.44. MS-ESI: m/z 481 (M+). Anal. calcd for C28H25N4O4: C, 69.84; H, 5.23; N, 11.64%. Found: C, 69.85; H, 4.95; N, 11.62%.
:1 (DMSO-d6). IR (ATR): 3350, 3210, 3031, 2928, 2870, 1672, 1585, 1557 cm−1. 1H NMR (300.13 MHz, DMSO-d6) for 14f + NH2(CH3)2OH: δ = 1.73 (m, 6H, J = 6.85 Hz, CH2, E/Z-CH2), 2.87 (t, 4H, J = 7.04 Hz, E/Z-CH2Ph), 2.96 (t, 2H, J = 7.7 Hz, CH2NH2), 3.10 (t, 1H, J = 7.5 Hz, OH), 3.33 (m, 4H, E/Z-CH2NH), 3.44–3.59 (m, 8H, E/Z-CH2OH, E/Z-CH2N), 3.39 (t, 2H, CH2OH), 4.19 (t, 1H, E/Z-OH), 7.09–7.64 (m, 21H, E/Z-H-Ar), 7.82 (d, 1H, J = 8.5 Hz, E-H-8), 7.85 (dd, 1H, J = 1.9 Hz, Z-H-7), 8.02 (dd, 1H, J = 8.5 Hz, J = 1.7 Hz, E-H-7), 8.23 (d, 1H, J = 1.9 Hz, Z-H-5), 8.31 (d, 1H, J = 1.7 Hz, E-H-5), 10.36 (s, 1H, Z-NH), 10.58 (s, 1H, E-NH). 13C NMR (75.47 MHz, DMSO-d6): δ = 31.36, 32.44, 33.67, 36.79, 42.02, 58.24, 89.73, 125.97, 126.39, 126.76, 128.14, 128.52, 128.76, 129.92, 130.83, 136.95, 137.79, 138.80, 139.36, 145.71, 146.52, 164.59, 165.65, 169.32. MS-ESI: m/z 493 (M+ − 1). Anal. calcd for C32H37N5O6 (14f + NH2(CH3)2OH + H2O): C, 65.40; H, 6.35; N, 11.92%. Found: C, 65.46; H, 6.30; N, 11.89%.
General procedure: synthesis of 2H-pyrido[2,3-b]pyrrolo[3,2-e]pyrazin-2-one 9
Triethylamine (172 mg, 0.23 mL, 2 mmol) was added to a solution of 7a (498 mg, 1.7 mmol) in EtOH at rt. The mixture was stirred for 2 h and then EtOH was evaporated. The precipitate was dissolved in CH2Cl2 (30 mL). SOCl2 (212 mg, 0.13 mL, 1.78 mmol) was added to an ice-cooled stirred CH2Cl2 solution at 0 °C. After 1 h, the mixture was warmed to rt, and stirred for 24 h; then H2O (100 mL) was added to the stirred mixture. The CH2Cl2 phase was separated and dried (MgSO4). Aromatic 2,3-diaminopyridine (1.7 mmol) was added to the CH2Cl2 phase. The mixture was stirred for 5 days at rt. The precipitate 8a was collected and dissolved in EtOH (50 mL) and then refluxed for 3 hours. The precipitate 9a was collected and crystallised from EtOH.Acknowledgements
We thank Grzegorz Trębacz for the quantum yield measurements and Anna Ostrowska for preparing Fig. 4–11. The research was carried out using the equipment purchased thanks to the financial support from the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00-12-023/08).References
- P. de Silva, H. Q. N. Gunaratne, Th. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef PubMed.
- J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551–8588 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443–3480 CrossRef CAS PubMed.
- E. L. Que, D. W. Domaille and Ch. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed.
- C. Lodeiro, J. L. Capelo, J. C. Mejuto, E. Oliveira, H. M. Santos, B. Pedras and C. Nuñez, Chem. Soc. Rev., 2010, 39, 2948–2976 RSC.
- G. Aragay, J. Pons and A. Merkoçi, Chem. Rev., 2011, 111, 3433–3458 CrossRef CAS PubMed.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- Y. Jeong and J. Yoon, Inorg. Chim. Acta, 2012, 381, 2–14 CrossRef CAS PubMed.
- M. Dutta and D. Das, TrAC, Trends Anal. Chem., 2012, 32, 113–132 CrossRef CAS PubMed.
- M. Formica, V. Fusi, L. Giorgi and M. Micheloni, Coord. Chem. Rev., 2012, 256, 170–192 CrossRef CAS PubMed.
- S. C. L. Pinheiro, I. M. Raimundo Jr, M. C. Moreno-Bondi and G. Orellana, Anal. Bioanal. Chem., 2010, 398, 3127–3138 CrossRef PubMed.
- M. C. Kimber, I. B. Mahadevan, S. F. Lincoln, A. D. Ward and E. R. T. Tiekink, J. Org. Chem., 2000, 65, 8204–8209 CrossRef CAS PubMed.
- V. V. S. Mummidivarapu, K. Tabbasum, J. P. Chinta and Ch. P. Rao, Dalton Trans., 2012, 41, 1671–1674 RSC.
- Y. Zhang, X. Guo, W. Si, L. Jia and X. Qian, Org. Lett., 2008, 10, 473–476 CrossRef CAS PubMed.
- X. Zhou, B. Yu, Y. Guo, X. Tang, H. Zhag and W. Liu, Inorg. Chem., 2010, 49, 4002–4007 CrossRef CAS PubMed.
- H. Zhang, Q.-L. Wang and Y.-B. Jiang, Tetrahedron Lett., 2007, 48, 3959–3962 CrossRef CAS PubMed.
- J. Jiang, H. Jiang, X. Tang, L. Yang, W. Dou, W. Liu, R. Fang and W. Liu, Dalton Trans., 2011, 40, 6367–6370 RSC.
- X. Zhou, Y. Lu, J.-F. Zhu, W.-H. Chan, A. W. M. Lee, P.-Sh. Chan, R. N. S. Wong and N. K. Mak, Tetrahedron, 2011, 67, 3412–3419 CrossRef CAS PubMed.
- L. Xue, Ch. Liu and H. Jiang, Org. Lett., 2009, 11, 1655–1658 CrossRef CAS PubMed.
- T. Ghosh, B. G. Maiya and A. Samanta, Dalton Trans., 2006, 795–801 RSC.
- E. Korin, B. Cohen, Ch.-Ch. Zeng, Y.-Sh. Xu and J. Y. Becker, Tetrahedron, 2011, 67, 6252–6258 CrossRef CAS PubMed.
- B. Wilson, L. Gude, M.-J. Fernández, A. Lorente and K. B. Grant, Inorg. Chem., 2005, 44, 6159–6173 CrossRef CAS PubMed.
- Y. Wang, X.-Y. Hu, L. Wang, Zh.-B. Shang, J.-B. Chao and W.-J. Jin, Sens. Actuators, B, 2011, 156, 126–131 CrossRef CAS PubMed.
- M. S. Park, K. M. K. Swamy, Y. J. Lee, H. N. Lee, Y. J. Jang, Y. H. Moon and J. Yoon, Tetrahedron Lett., 2006, 47, 8129–8132 CrossRef CAS PubMed.
- C. Bazzicalupi, A. Bencini, I. Matera, S. Puccioni and B. Valtancoli, Inorg. Chim. Acta, 2012, 381, 162–169 CrossRef CAS PubMed.
- H. N. Lee, H. N. Kim, K. M. K. Swamy, M. S. Park, J. Kim, H. Lee, K.-H. Lee, S. Park and J. Yoon, Tetrahedron Lett., 2008, 49, 1261–1265 CrossRef CAS PubMed.
- M. Taki, Y. Watanabe and Y. Yamamoto, Tetrahedron Lett., 2009, 50, 1345–1347 CrossRef CAS PubMed.
- A. Rocha, M. M. B. Marques and C. Lodeiro, Tetrahedron Lett., 2009, 50, 4930–4933 CrossRef CAS PubMed.
- H.-H. Wu, Y.-L. Sun, Ch.-F. Wan, Sh.-T. Yang, Sh.-J. Chen, Ch.-H. Hu and A.-T. Wu, Tetrahedron Lett., 2012, 53, 1169–1172 CrossRef CAS PubMed.
- J. Zhang, H. Cui, M. Hojo, Sh. Shuang and Ch. Dong, Bioorg. Med. Chem. Lett., 2012, 22, 343–346 CrossRef CAS PubMed.
- P.-E. Danjou, J. Lyskawa, F. Delattre, M. Becuwe, P. Woisel, S. Ruellan, S. Fourmentin and F. Cazier-Dennin, Sens. Actuators, B, 2012, 171–172, 1022–1028 CrossRef CAS PubMed.
- B. Wang and E. V. Anslyn, in Chemosensors, ed. D. Lee, A John Wiley & sons. Inc. Publication, Singapore, 2011, pp. 41–64 Search PubMed.
- M. S. Nasir, Ch. J. Fahrni, D. A. Suhy, K. J. Kolodsick, Ch. P. Singer and T. V. O'Halloran, J. Biol. Inorg. Chem., 1999, 775–783 CrossRef CAS.
- K. Ostrowska, E. Piegza, M. Rąpała-Kozik and K. Stadnicka, Eur. J. Org. Chem., 2012, 3636–3646 CrossRef CAS.
- K. Jamroży, K. Szymoniak and K. Ostrowska, Heterocycles, 2008, 75, 2275–2282 CrossRef.
- K. Ostrowska, K. Szymoniak, M. Szczurek, K. Jamroży and M. Rąpała-Kozik, Tetrahedron, 2011, 67, 5219–5227 CrossRef CAS PubMed.
- R. Satapathy, Y.-H. Wu and H.-Ch. Lin, Org. Lett., 2012, 14, 2564–2567 CrossRef CAS PubMed.
- Q. Zhao, R.-F. Li, Sh.-K. Xing, X.-M. Liu, T.-L. Hu and X.-H. Bu, Inorg. Chem., 2011, 50, 10041–10046 CrossRef CAS PubMed.
- J. Du, J. Fan, X. Peng, H. Li and Sh. Sun, Sens. Actuators, B, 2010, 144, 337–341 CrossRef CAS PubMed.
- Nonius, COLLECT, Nonius BV, Delft, The Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, Macromolecular Crystallography, Part A, C. W. Carter, Jr and R. M. Sweet, ed., Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- Agilent, CrysAlis PRO, Agilent Technologies UK Ltd, Yarnton, England, 2011 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- L. J. Farrugia, ORTEP-3 for Windows - a version of ORTEP-III with a Graphical User Interface (GUI), J. Appl. Crystallogr., 1997, 30, 565–568 CrossRef CAS.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
- M. Nardelli, A. Musatti, P. Domiano and G. Andreetti, Ric. Sci., 1965, 15(II-A), 807 Search PubMed.
- G. A. Lawrance, Introduction to Coordination Chemistry, ed. A. John, Wiley & sons. Inc., Publication, 2010, pp. 127–131 Search PubMed.
- Ch.-Sh. Choi, K.-S. Jeon, W.-Ch. Jeong and K.-H. Lee, Bull. Korean Chem. Soc., 2010, 31, 1375–1376 CrossRef CAS.
- A. Demeter, T. Bérces, L. Biczók, V. Wintgens, P. Valat and J. Kossanyi, J. Phys. Chem., 1996, 100, 2001–2011 CrossRef CAS.
- P. Valat, V. Wintgens, J. Kossanyi, L. Biczok, A. Demeter and T. Berces, J. Am. Chem. Soc., 1992, 114, 946–953 CrossRef CAS.
- S. Periyaraja, A. B. Mandal and P. Shanmugam, Org. Lett., 2011, 13, 4980–4983 CrossRef CAS PubMed.
- Q.-Y. Cao, J. F. Zhang, W. X. Ren, K. Choi and J. S. Kim, Tetrahedron Lett., 2011, 52, 4464–4467 CrossRef CAS PubMed.
- D. Y. Han, J. M. Kim, J. Kim, H. S. Jung, Y. H. Lee, J. F. Zhang and J. S. Kim, Tetrahedron Lett., 2010, 51, 1947–1951 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 2D fluorescence spectra of 4a, 4a-Hg, 6a, 6a-Zn, 6a-In, 6a-Hg, 12e, and 14e. Fluorescence spectra of 4a, 6, 10a, 12e, and 14e (10 μM) in CH3CN with 1 equiv. of Cd2+. Fluorescence spectra of 4a, 6, 10a, 12e, and 14e (10 μM) in CH3CN with 1 equiv. of Hg2+. Fluorescence titration of 6a with Zn(acac)2, Zn(OAc)2, and ZnCl2. 1H and 13C NMR spectra of compounds 6a–d, 8a, 9a, 10a, 12e,f, and 14e,f. Partial 1H NMR titration spectra of 6a with Zn(acac)2 in CD3CN and DMSO-d6. Conformational and intermolecular interaction analysis of crystal structures of 6a and 12e. CCDC 890198 and 890197. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj00750b |
| ‡ For crystal structure information, e-mail: kalinows@chemia.uj.edu.pl. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |