Nitrogen-doped carbon coated Li3V2(PO4)3 derived from a facile in situ fabrication strategy with ultrahigh-rate stable performance for lithium-ion storage†
Cong
Wang
,
Wei
Shen
and
Haimei
Liu
*
State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: liuhm@mail.buct.edu.cn; Fax: +86 10 6442 5385; Tel: +86 10 6443 5271
First published on 23rd October 2013
Abstract
A nitrogen-doped carbon coated Li3V2(PO4)3 cathode material is prepared by using a very simple in situ approach. The Li3V2(PO4)3 particle is uniformly encapsulated by a nitrogen-doped carbon layer, where a carbon coating layer exists, nitrogen-doping also exists. N-doping on the carbon layer significantly increases the electronic conductivity of as-prepared Li3V2(PO4)3 sample, and thus, this Li3V2(PO4)3/C+N demonstrates ultrahigh rate performance and excellent cycling stability for lithium storage. When the discharging rate is increased from 0.5 C to 50 C, its capacity of 119.5 mA h g−1 decays to 110.8 mA h g−1 and an amazing capacity retention of 93% is achieved. This in situ synthetic approach of a nitrogen-doped carbon coated Li3V2(PO4)3 cathode reported herein is highly efficient and easily realized in industrial applications and moreover, is able to be extended to modify other electrode materials in which carbon-coating is indispensable, such as LiFePO4etc.
Introduction
Advanced energy storage devices and systems, such as lithium-ion batteries (LIBs), for use as the power supply in electric vehicles (EVs) or stationary energy storage for solar and wind energy as well as smart grids, are key components in the development of renewable energy, and high power and high energy density are essential to batteries for applications in the above areas.1–3 Nowadays, advanced electrode materials with high capacity and fast charge–discharge capability are critical for next generation LIBs.4–7 Recently, monoclinic lithium vanadium phosphate, Li3V2(PO4)3, has received considerable attention as LIBs cathode materials due to its advantages in terms of higher energy density, higher operating voltage, higher power density and better thermal stability as compared with LiFePO4.8–12 Moreover, among all the lithium transition metal phosphates, Li3V2(PO4)3 has the highest theoretical specific capacity of 197 mA h g−1 when on complete insert–deinsert in a potential window of 3.0–4.8 V vs. Li/Li+, even working between 3.0 and 4.3 V, a reversible capacity of 133 mA h g−1 can still be obtained.8 Therefore, Li3V2(PO4)3 is expected to be an excellent candidate cathode material, taking the place of LiFePO4, for next generation LIBs. However, Li3V2(PO4)3 has a relatively low electronic conductivity (approximately 2.4 × 10−7 S cm−1) and low Li-ion diffusion coefficient (i.e., 10−8 to 10−13 cm2 s−1),12,13 both of which significantly limit its electrochemical property and further practical applications. Therefore, numerous strategies have been attempted with the aim of modifying the electrical conductivity and diffusion coefficient of Li3V2(PO4)3, such as metal ion doping,14–17 carbon or other conductive material coating and decreasing the particle size.18–24In our previous work,25 a uniform core–shell structured Li3V2(PO4)3 coated with carbon was obtained by a momentary freeze-drying method, whose rate performance was greatly improved, and when cycled under 30 C (1 C = 133 mA g−1), a capacity of 86 mA h g−1 could be maintained. However, it is still inadequate to meet the requirements in those fields which need high discharge currents, high power densities and a long cycle life. A challenging task remains of how to further improve the rate performance and the cycling life under large charge–discharge currents. As aforementioned, carbon coating is an effective approach in ameliorating the Li3V2(PO4)3 performance.18–22 Most recently, nitrogen-doping carbon-based materials have attracted a keen interest, such as graphite, graphene and CNTs etc.,26–30 because N-doping can bring many improvements of these carbon-based materials: firstly, N-doping could modify the material structure, chemical reactivity and electronic properties;31–33 secondly, N-doping could generate numerous extrinsic defects and active sites;34 thereby, it is worth considering, if N-doping is employed in the carbon coating layer to modify the Li3V2(PO4)3, an improved electrochemical performance is expected.
In fact, using N-doped carbon coating to modify the electrode materials, such as Li4Ti5O12 and LiFePO4, has occasionally been reported to improve their electrochemical properties.35,36 Nevertheless, in these reports, the already obtained materials were just dipped into the expensive ionic liquid, after one more sintered process, a N-doped carbon layer was generated. Note that, the above method is very expensive since they used ionic liquid as the nitrogen source, in addition, the carbon-coating and nitrogen-doping might be inefficient, of low yield and incomplete, thereby, it is impossible to be employed for practical applications. In this work, a nitrogen-doped carbon coated Li3V2(PO4)3 material (referred as Li3V2(PO4)3/C+N) was prepared via a sol–gel method assisted with momentary freeze-drying, in which low-cost urea and citric acid were used as nitrogen and carbon sources, respectively. It was found that the Li3V2(PO4)3 particle is preferably encapsulated by a N-doped carbon layer, and when used as a cathode material for LIBs, this Li3V2(PO4)3/C+N material demonstrates an ultrahigh rate stable performance and excellent long cycling life under a large discharge current.
It is worthwhile noting that the present approach for preparation of Li3V2(PO4)3/C+N has several significant merits, first of all, the N-doped carbon layer on the Li3V2(PO4)3 surface is in situ fabricated, where a carbon coating layer exists, N-doping also exists; secondly, urea serves as the nitrogen source, the doping cost is obviously reduced compared with ionic liquids; thirdly, through a momentary freeze-drying process, a uniform and complete carbon coated layer is achieved; finally; the excellent structural stability of this Li3V2(PO4)3/C+N material is directly confirmed by experimental results.
Experimental
Sample preparation
Citric acid and urea in the proportions 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5, used as carbon and nitrogen sources, respectively, were initially dissolved in distilled water with continuous stirring at room temperature. Then LiOH·H2O, NH4VO3 and NH4H2PO4 in the molar ratio 3.05:2:3 were added into the above solution in sequence under constant stirring at 60 °C. The mixture was heated at 80 °C for several hours with vigorous stirring to evaporate the water until a dark blue sol was generated. The obtained sol was drop-by-drop frozen momentarily under liquid nitrogen and then the frozen-sol was adopted into the vacuum-drying process for about 24 h at about −40 °C in a freeze-drying machine. The obtained powders were ground and pre-heated at 300 °C for 4 h in the air. Finally, the pre-heated products were sintered at 850 °C for 8 h under a N2 atmosphere to yield the Li3V2(PO4)3/C+N material. In order to indicate the modification effects of the nitrogen doped carbon coating approach clearly, a controlled experiment was carried out as follows: the only carbon coated Li3V2(PO4)3 sample (referred as Li3V2(PO4)3/C), without the addition of urea, was prepared by the identical synthesis process as the above Li3V2(PO4)3/C+N material and its electrochemical performance was inspected in detail and compared with the Li3V2(PO4)3/C+N material.
:1.5, used as carbon and nitrogen sources, respectively, were initially dissolved in distilled water with continuous stirring at room temperature. Then LiOH·H2O, NH4VO3 and NH4H2PO4 in the molar ratio 3.05:2:3 were added into the above solution in sequence under constant stirring at 60 °C. The mixture was heated at 80 °C for several hours with vigorous stirring to evaporate the water until a dark blue sol was generated. The obtained sol was drop-by-drop frozen momentarily under liquid nitrogen and then the frozen-sol was adopted into the vacuum-drying process for about 24 h at about −40 °C in a freeze-drying machine. The obtained powders were ground and pre-heated at 300 °C for 4 h in the air. Finally, the pre-heated products were sintered at 850 °C for 8 h under a N2 atmosphere to yield the Li3V2(PO4)3/C+N material. In order to indicate the modification effects of the nitrogen doped carbon coating approach clearly, a controlled experiment was carried out as follows: the only carbon coated Li3V2(PO4)3 sample (referred as Li3V2(PO4)3/C), without the addition of urea, was prepared by the identical synthesis process as the above Li3V2(PO4)3/C+N material and its electrochemical performance was inspected in detail and compared with the Li3V2(PO4)3/C+N material.
Characterization
The carbon and nitrogen contents of Li3V2(PO4)3/C+N samples were measured by an organic element analyzer (Vario EL cube). The X-ray diffraction patterns of the Li3V2(PO4)3/C+N samples were determined by the D/max-Ultima III Diffractometer at 40 kV, 40 mA with Cu-Kα radiation (λ = 0.154 nm). The morphologies and particle size of Li3V2(PO4)3/C+N samples were detected by field emission scanning electron microscope (FE-SEM, Zeiss Supra 55). The HRTEM images were observed using a high resolution transmission electron microscope (HR-TEM, Hitachi H-800). The Raman spectra were achieved on a labRAM ARAMIS laser Raman spectroscopy equipped with a 514 nm Ar-ion laser. The XPS spectra were performed on a PHI Quantera SXM scanning X-ray microprobe with a 100 mm beam size, using an Al Kα (λ = 0.83 nm, hν = 1486.7 eV) X-ray source operated at 2 kV and 20 mA.Electrochemical measurement
Electrochemical performances of Li3V2(PO4)3/C+N composites were investigated using a CR2032 coin-type cell. A metallic lithium foil served as the reference and counter electrode. The work electrodes were fabricated with Li3V2(PO4)3/C+N composites, acetylene black and polyvinylidene difluoride (PVDF) at a weight ratio of 80:10:10. The slurry was pasted onto the Al foil and the electrodes were dried at 120 °C in vacuum for 12 h. The cells were assembled in an argon filled glove box. The mass loading of each electrode plate was around 2.0–2.5 mg cm−2. The mass fraction of the active material in each coin cell is 80%, so the mass loading of the active material in each coin cell is in the range of 1.6–2.0 mg cm−2. 1 M LiPF6 in ethylene carbonate (EC)–dimethyl carbonate (DMC)–ethylene methyl carbonate (EMC) with the volumetric ratio of 1:1:1 as the electrolyte, and a polypropylene micro-porous film (Cell-gard 2300) as the separator. The galvanostatic charge–discharge tests are conducted on LAND CT2001A test system (Wuhan, China). Cyclic voltammetry (CV) measurements were performed on a CHI650D (Chenhua, Shanghai) electrochemical workstation with a rate of 0.05 mV s−1. All the tests were performed in the potential ranges of 3.0–4.3 V and 3.0–4.8 V. For post-measurements of XRD and HRTEM, the Li3V2(PO4)3/C+N electrode after electrochemical cycling tests was carefully disassembled and washed with EC pure solution in an argon filled glove box, and dried at 120 °C in a vacuum oven.
Results and discussion
The above argument in the Introduction section is strongly supported by transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images of Li3V2(PO4)3/C+N. It can be clearly observed in Fig. 1a and b that a very homogeneous carbon coated layer is formed on the Li3V2(PO4)3 surface. Notably, it is not a serendipitous observation and the uniformity and integrity of this carbon layer can be further confirmed by larger-scale TEM images in Fig. S1 (ESI†). The HRTEM images and SAED patterns in Fig. 1c and d indicate the single-crystal character of the Li3V2(PO4)3 particles, and the marked lattice fringes with d-spacing of 0.247 nm, corresponding to the (−204) planes of the monoclinic Li3V2(PO4)3. It is noticeable that the thickness of the coated carbon layer is around 5–10 nm. Even more interesting is that in some selected areas (Fig. 1e and f), the carbon layer is composed of two kinds of concrete components, the inner layer closed to the Li3V2(PO4)3 has an amorphous morphology, whereas the outer layer is somewhat a graphite-like carbon since the layered structures of graphite with ∼0.34 nm can be clearly noticed.25 The XRD patterns demonstrate that as-obtained Li3V2(PO4)3/C+N is a pure phase monoclinic Li3V2(PO4)3 structure (JCPDS No: 01-072-7074) (Fig. S2a, ESI†) and average particle size is about 1–2 μm, moreover, the coated carbon layer is also noticeable in SEM images (Fig. S2b and c, ESI†).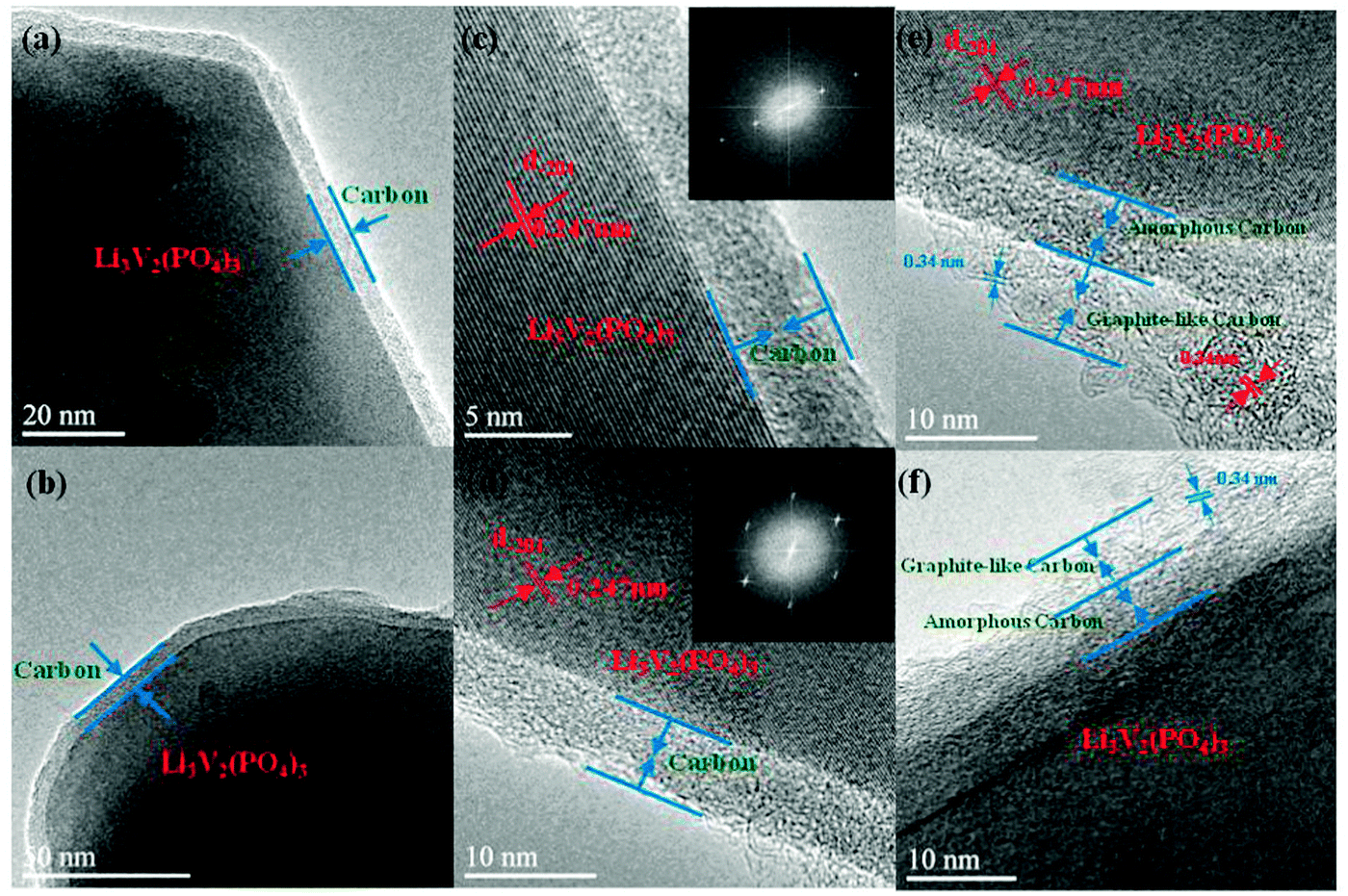 | ||
| Fig. 1 TEM and HRTEM images of Li3V2(PO4)3/C+N (inset in c and d: the Fourier transform of the corresponding SAED patterns). | ||
The successful N-doping in the carbon layer on Li3V2(PO4)3 is confirmed by Raman spectra as depicted in Fig. 2a. The intensity ratios of the D band (1350 cm−1) to the G band (1580 cm−1) for Li3V2(PO4)3/C+N which was synthesized by a freeze-drying process (referred to as LVP/C+N–F), normal Li3V2(PO4)3/C+N which was synthesized in the absence of a freeze-drying process (referred to as LVP/C+N–N) and Li3V2(PO4)3/C which was synthesized with a freeze-drying process (referred to as LVP/C–F) are 1.090, 1.075 and 1.010, respectively, implying that, whether freeze-drying is used or not, there are more defects produced by nitrogen doping in the carbon coating layer,34,37 and these defects can provide an increased number of electrochemical active sites for lithium-ion transport. As a result, the electrochemical properties of Li3V2(PO4)3/C+N will be obviously improved. Fig. 2b shows the typical high resolution XPS spectra of N1s for Li3V2(PO4)3/C+N, the peaks at binding energies of 398.9, 400.9 and 401.9 eV can be attributed to C–N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and N–O bonds,35 the existence of two different kinds of C–N bonds indicates the formation of two types of nitrogen doped carbon in Li3V2(PO4)3/C+N: pyridine-like N with a C–N bond together with a CN bond and graphite-like N with three C–N bonds.38 In addition, there is a small portion of oxidized N (N–O bond) formed. The carbon and nitrogen contents in as-obtained Li3V2(PO4)3/C+N are around 1.88 and 0.65 wt%, respectively, confirmed by organic elemental analysis.
N and N–O bonds,35 the existence of two different kinds of C–N bonds indicates the formation of two types of nitrogen doped carbon in Li3V2(PO4)3/C+N: pyridine-like N with a C–N bond together with a CN bond and graphite-like N with three C–N bonds.38 In addition, there is a small portion of oxidized N (N–O bond) formed. The carbon and nitrogen contents in as-obtained Li3V2(PO4)3/C+N are around 1.88 and 0.65 wt%, respectively, confirmed by organic elemental analysis.
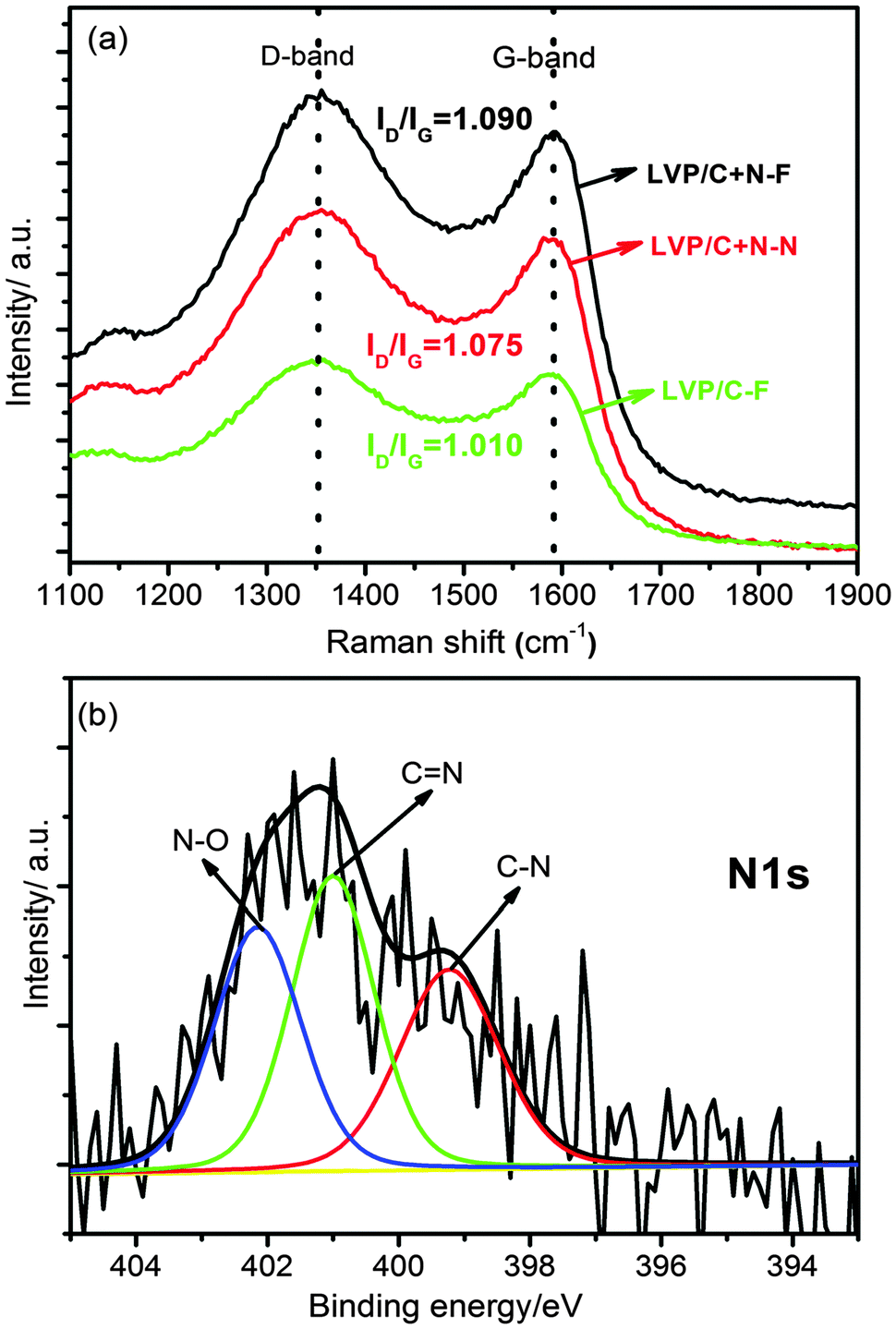 | ||
| Fig. 2 (a) Raman spectra of Li3V2(PO4)3/C+N which was synthesized with a freeze-drying process (referred as LVP/C+N–F), normal Li3V2(PO4)3/C+N (referred as LVP/C+N–N) which was synthesized in the absence of a freeze-drying process and Li3V2(PO4)3/C which was synthesized with a freeze-drying process (referred as LVP/C–F); (b) high-resolution XPS spectra of Li3V2(PO4)3/C+N which was synthesized with a freeze-drying process on N1s. | ||
The ultrahigh rate performance for lithium ion storage on present Li3V2(PO4)3/C+N was investigated by an extended rate test. Fig. 3a and b exhibit a periodic rate performance on Li3V2(PO4)3/C+N cathode material which charge–discharge at 3.0–4.3 V at various current densities. For the first two circles (Fig. 3a) that discharged from 0.5 C to 50 C (1 C = 133 mA g−1, 3.0–4.3 V), it was unexpectedly noticed that stable high discharge capacities of 119.5, 118, 115.5 and 114 mA h g−1 were achieved under low current densities of 0.5, 1, 2 and 5 C, and further demonstrates a superior rate performance with 113 mA h g−1 at 10 C, 111.9 mA h g−1 at 20 C, 111.5 mA h g−1 at both 30 and 40 C and 110.8 mA h g−1 at 50 C, namely, when the current density increased from 0.5 C to 50 C, a superior capacity retention of 93% is obtained. Compared with various Li3V2(PO4)3 samples reported so far,21,22,39–42 this Li3V2(PO4)3/C+N cathode exhibits a better capacity retention as well as higher specific capacities at high current densities from 30 C to 50 C. When continually cycled under the increased current rate until 60 C, 70 C, 80 C, 90 C and 100 C in the third cycle period (Fig. 3b), the capacities of Li3V2(PO4)3/C+N can still be retained at 106.2, 102, 99.1, 94 and 80.8 mA h g−1, respectively, clearly demonstrating its ultrahigh rate capability. Fig. 3c shows the discharge curves of the Li3V2(PO4)3/C+N at the 1st and 10th cycles under various current densities during the above rate cycling process. With the increase of the current density, an obvious potential drop is observed, owing to the electrode polarization. However, the distinct discharge plateaux and overlapped discharge curves at various current densities suggest good stability of the electrode material. Note that, in order to optimize the electrochemical properties on Li3V2(PO4)3/C+N, a smaller charge current density of 1 C was employed in the above rate performance test (Fig. 3). Nevertheless, even if the charge current densities are increased to 5 C and 10 C, the rate performance of Li3V2(PO4)3/C+N is still superior to most of the other reported Li3V2(PO4)3 (Fig. S3, ESI†).22,39–42 However, it should be pointed out that the high potential performance of the present Li3V2(PO4)3/C+N, in which all three Li+ are inserted–deinserted between the potential window of 3.0–4.8 V, is not so good as in the 3.0–4.3 V vs. Li/Li+ (Fig. S4 and S5, ESI†), indicating that further work should be focused on improving the stability of the third lithium ion.
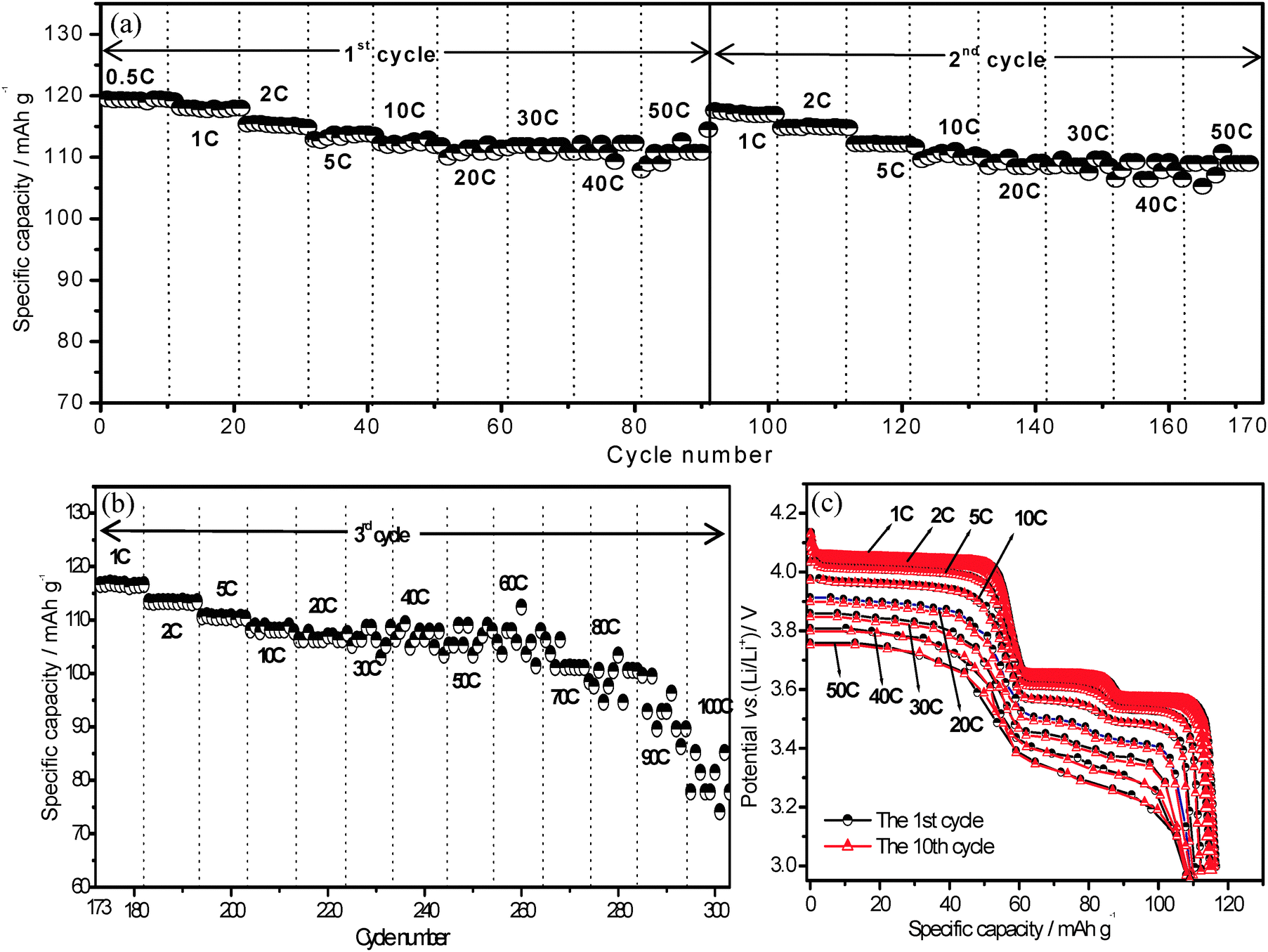 | ||
| Fig. 3 The rate performance of Li3V2(PO4)3/C+N material in the potential range of 3.0–4.3 V: (a) the 1st and 2nd cycle with the current rate from 0.5 C to 50 C, (b) continuing the 3rd cycle with the current rate from 1 C to 100 C; (c) the discharge curves of the 1st cycle and the 10th cycle for Li3V2(PO4)3/C+N at various current densities during the rate-cycling process. | ||
Another interesting feature of the present Li3V2(PO4)3/C+N is its excellent cycling performance. The cycling performances of Li3V2(PO4)3/C+N between 3.0 and 4.3 V under various current densities are shown in Fig. 4. At 1 C rate, its initial specific capacity is 119.9 mA h g−1 and still maintained at 113.1 mA h g−1 after 400 cycles, with a capacity loss of only 5.7%. When cycled at 2 C, 5 C, 10 C and 20 C, a similar result is observed (Fig. 4a). The coulombic efficiency of the Li3V2(PO4)3/C+N at 1 C and 2 C are also exhibited in Fig. 4a, both of which nearly correspond to 100%. The high efficiency further confirms the excellent electrochemical stability and reversibility of the Li3V2(PO4)3/C+N cathode materials. Even at higher current densities of 40 C, 50 C and 60 C, the discharge capacities also exhibit perfect retention from initial values of 110.8, 110.5 and 113 mA h g−1 and finally stabilized at 102.6, 100.6 and 102.6 mA h g−1 after 400 cycles (Fig. 4b), effectively demonstrating excellent cycling performance and good reversibility of the electrode material. The above conclusion is further confirmed by discharge curves at various cycles under 1 C and 20 C rates (Fig. 4c and d). As seen, whether at 1 C or at the higher 20 C, there is almost no obvious change in the curve shape until 400 cycles, suggesting the polarization on the electrode or structural aggravation is negligible during this prolonged charge–discharge process, and a similar result is also observed for 30 C (Fig. S6d, ESI†). Notably, increasing the charge current densities from 1 C to 5 C and 10 C does not bring about pronounced effects on the Li3V2(PO4)3/C+N cycling performance, in other words, even under the larger charge current densities of 5 C and 10 C, the cycling performances under discharge current densities of 40 C, 50 C and 60 C are maintained almost unchanged (Fig. S6 and S7, ESI†). In order to demonstrate the modification effects of the N-doped carbon coating approach more definitively, a comprehensive comparison of the electrochemical performances of Li3V2(PO4)3/C+N and Li3V2(PO4)3/C was conducted and displayed in Fig. S8 and S9 (ESI†). It can be clearly observed that the electrochemical properties of Li3V2(PO4)3/C+N are significantly superior to Li3V2(PO4)3/C. The ultrahigh rate performance and superior cycling stability of Li3V2(PO4)3/C+N can be attributed to several reasons, firstly, the surface N-doped carbon layer not only enhances the electronic conductivity, but also creates numerous extrinsic defects and active sites, which might aid lithium transport,34 and this has been confirmed by the results of electrochemical impedance spectroscopy (EIS) measurement (Fig. S10, ESI†) and Raman spectra; secondly, the highly ordered single-crystal feature and high-stable crystallization may facilitate lithium diffusion in the bulk phase; thirdly, the uniform and complete carbon layer on the sample surface stabilizes the interface of the electrode material; finally, the graphite-like carbon which is characteristically induced by the momentary freeze-drying process further improves the kinetic property of the sample.25
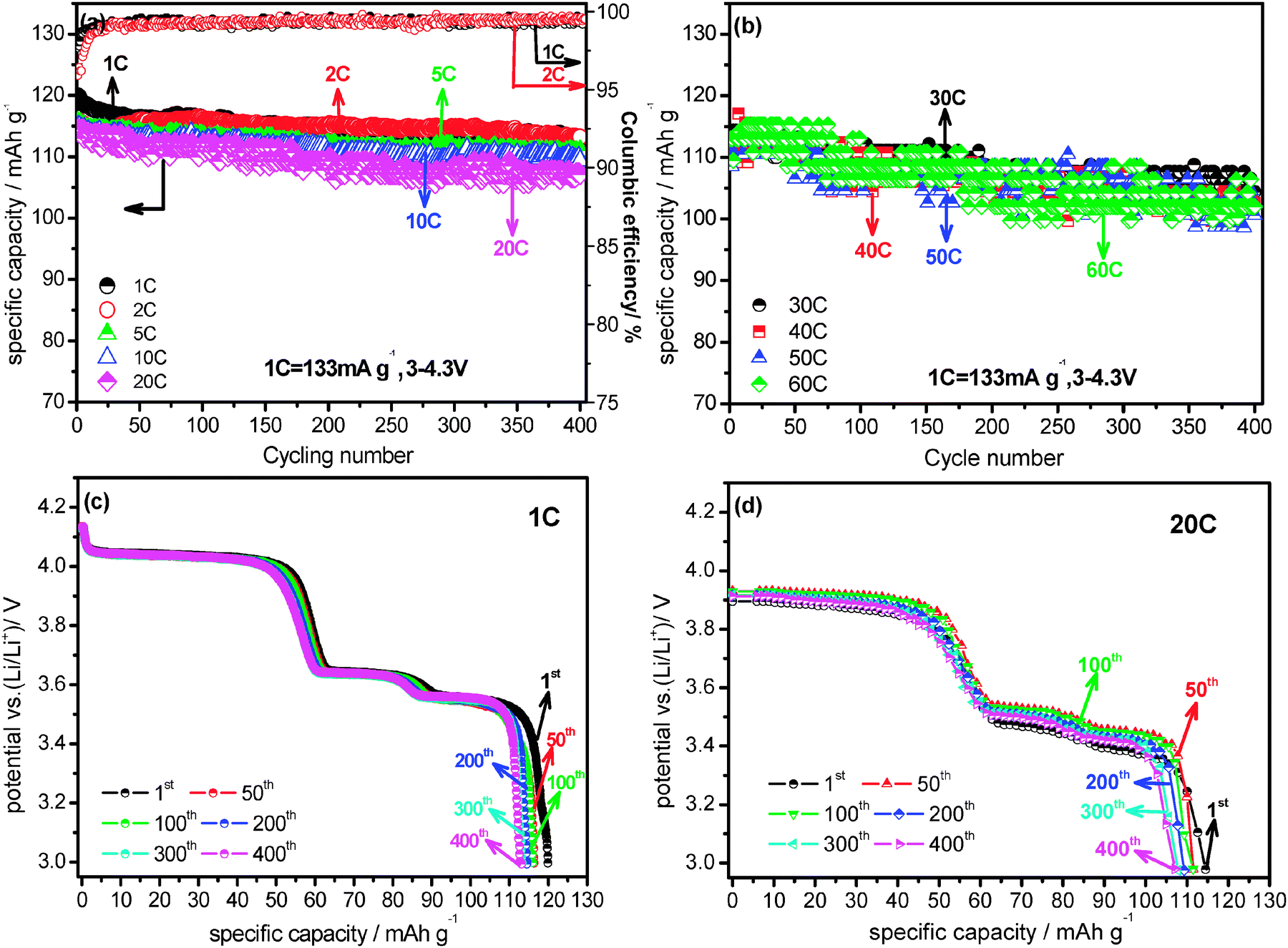 | ||
| Fig. 4 (a) Coulombic efficiency of the Li3V2(PO4)3/C+N at 1 C and 2 C; cycle performances of the Li3V2(PO4)3/C+N at various current densities: (a) 1 C to 20 C (b) 30 C to 60 C; discharge curves of the Li3V2(PO4)3/C+N at various cycles under: (c) 1 C (d) 20 C, all the tests were employed in the potential window of 3.0–4.3 V (vs. Li/Li+). | ||
The above argument about the excellent stability of this N-doped carbon coating layer and high-stable crystallization of the Li3V2(PO4)3/C+N is further and strongly supported by the XRD and TEM measurements on an electrode which had experienced a long-term cycle under extremely large current densities. Fig. 5 displays HRTEM images and XRD patterns of the Li3V2(PO4)3/C+N cathode which cycled after 500 times under 10 C, 20 C and 50 C, respectively. As shown in Fig. 5a and c, even after a long time fast Li-ion insertion–extraction process (discharged under 10 C and 50 C for 500 cycles), the carbon-coating layer can still be completely retained. Fig. 5b and d reveal clear lattice fridges of Li3V2(PO4)3/C+N with d-spacing of 0.247 nm and 0.278 nm, corresponding to (−204) and (−301) planes of monoclinic Li3V2(PO4)3, which are quite similar with the powder sample in Fig. 1c and d, and SAED patterns in the inset further confirm the single-crystal feature of the Li3V2(PO4)3/C+N cathode. Moreover, as shown in Fig. 5e, there is almost no obvious distinction among the various XRD patterns of Li3V2(PO4)3/C+N before and after the electrochemical test and all the peaks correspond to a single phase of monoclinic Li3V2(PO4)3, no peak other than Li3V2(PO4)3 is detected. Both HRTEM images and XRD patterns indicate that even after a fast charge and discharge process over a long time period, the present Li3V2(PO4)3/C+N electro-active material still maintains a highly ordered pure-phase single-crystal structure, which is most probably one of the key factors for its improved electrochemical properties.
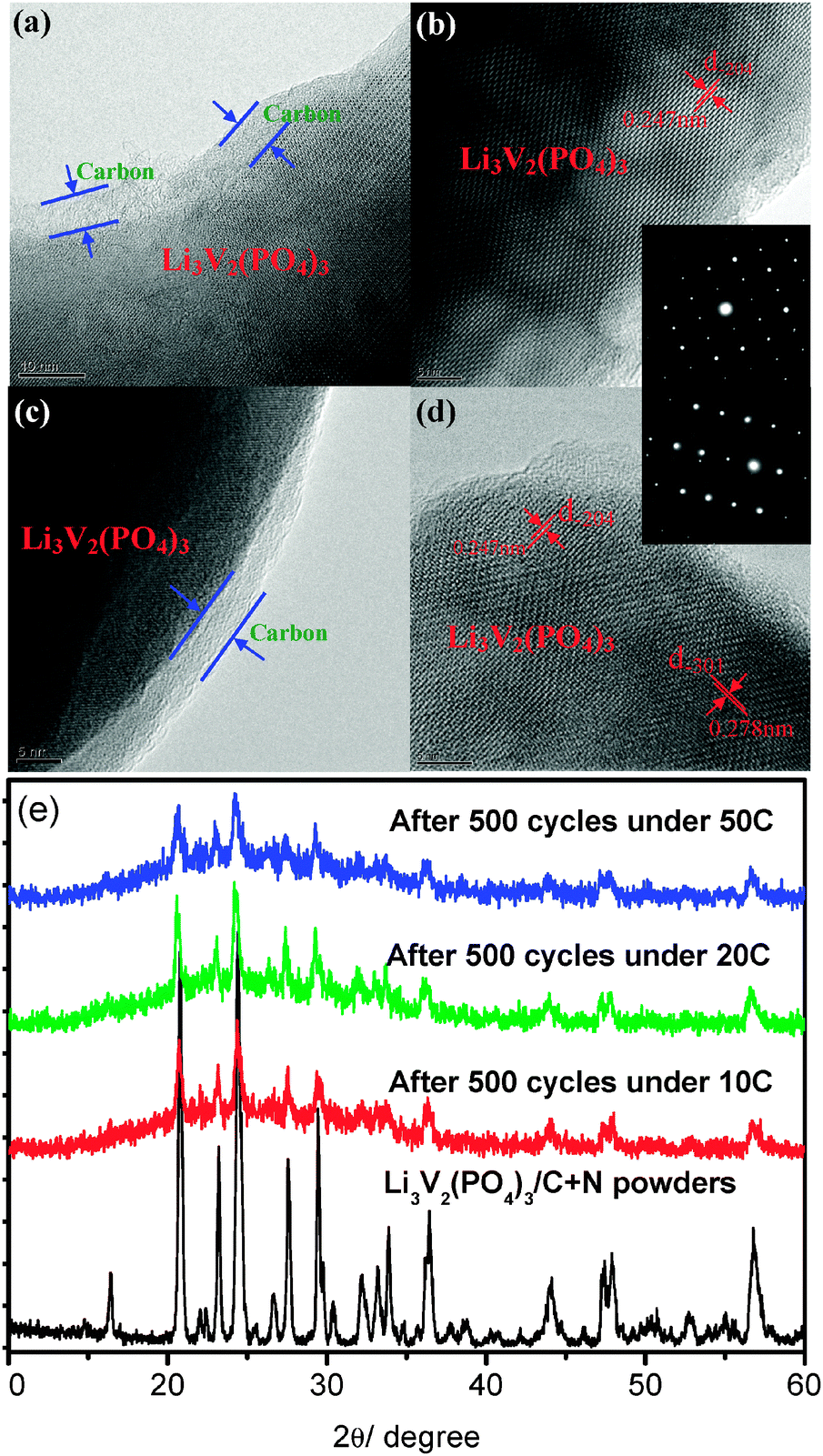 | ||
| Fig. 5 HRTEM images and XRD patterns of the Li3V2(PO4)3/C+N cathode after 500 cycles under various current densities: (a) and (b) HRTEM images after 500 cycles under 10 C; (c) and (d) HRTEM images after 500 cycles under 50 C; (e) XRD patterns. | ||
Indeed, our approach of in situ fabrication of N-doped carbon coated Li3V2(PO4)3 presented in this work is quite effective even without the assistance of the momentary freeze-drying process. For comparison, a sample of Li3V2(PO4)3/C+N was synthesized by an identical sol–gel method in the absence of a momentary freeze-drying process. The material characterization and electrochemical performance of this Li3V2(PO4)3/C+N are shown in Fig. S11 to S15 (ESI†). At the 1 C rate, its initial capacity is 115 mA h g−1 and maintains at 108.8 mA h g−1 after 400 cycles. Furthermore, even at higher current densities of 30 C, 40 C and 50 C, its discharge capacities also exhibit good retention from initial values of 102.1, 101.5 and 96 mA h g−1 and finally stabilize at 89.8, 85.6 and 81.6 mA h g−1 after 400 cycles. To the best of our knowledge, although this electrochemical performance is no better than that of the freeze-dried sample, it is still one of the best performances of a Li3V2(PO4)3 cathode material reported so far.18,19,41,42 However, as clearly observed in the HRTEM images in Fig. S15 (ESI†), compared with the freeze-dried Li3V2(PO4)3/C+N (See Fig. 1 and Fig. S1, ESI†), the carbon coated layer on the surface of the Li3V2(PO4)3/C+N sample without freeze-drying is much more incomplete and inhomogeneous. It is most likely due to the freeze drying process maintaining the microstructure of the precursor and after a sintered process, a complete and uniform N-doped carbon coated layer can be achieved. In other words, the use of freeze-drying can further enhance the modification effects of the nitrogen-doped carbon coating approach and further improve the electrochemical performance of the Li3V2(PO4)3/C+N material. Even so, this result demonstrates again that the N-doped carbon coating developed by an in situ fabrication process and derived from a urea source is quite efficient in improving the rate capability and cycling stability of Li3V2(PO4)3 cathode materials.
Conclusions
In summary, a facile in situ fabrication strategy has been developed for the preparation of N-doped carbon coated Li3V2(PO4)3 materials, of which low-cost urea and citric acid were used as nitrogen and carbon sources, respectively. The kinetic property of Li3V2(PO4)3 particles is significantly improved by this uniform and complete N-doped carbon layer and the excellent structural stability of this Li3V2(PO4)3/C+N material, therefore, this cathode material exhibits ultrahigh rate stable performance and superior cycling stability even under much large current densities. This synthetic approach reported herein is comparatively low-cost, efficient and easily realized in industrial applications and moreover, is readily extended to modify other electrode materials in which carbon-coating is indispensable, such as LiFePO4etc.Acknowledgements
This work was financially supported by the National Nature Science Foundation of China (No. 51102010, 21336003, 21371021), 111 Project (No. B07004), the Fundamental Research Funds for the Central Universities (Grant No. ZZ1232), and the Program for New Century Excellent Talents in University of China (NCET-12-0758).References
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS PubMed.
- H. M. Liu and W. S. Yang, Energy Environ. Sci., 2011, 4, 4000 CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271 CrossRef CAS PubMed.
- K. Kang, Y. S. Meng, J. Breger, C. Grey and G. Ceder, Science, 2006, 311, 977 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef CAS.
- H. Huang, S. C. Yin, T. Kerr, N. Taylor and L. F. Nazar, Adv. Mater., 2002, 14, 1525 CrossRef CAS.
- M. Y. Saidi, J. Barker, H. Huang, J. L. Swoyer and G. Adamson, J. Power Sources, 2003, 119–121, 266 CrossRef CAS.
- M. Y. Saidi, J. Barker, H. Huang, J. L. Swoyer and G. Adamson, Electrochem. Solid-State Lett., 2002, 5, A149 CrossRef CAS.
- S. C. Yin, P. S. Strobel, H. Grondey and L. F. Nazar, Chem. Mater., 2004, 16, 1456 CrossRef CAS.
- S. C. Yin, H. Grondey, P. Strobel, M. Anne and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 10402 CrossRef CAS PubMed.
- X. H. Rui, N. Ding, J. Liu, C. Li and C. H. Chen, Electrochim. Acta, 2010, 55, 2384 CrossRef CAS.
- C. Deng, S. Zhang, S. Y. Yang, Y. Gao, B. Wu, L. Ma, B. L. Fu, Q. Wu and F. L. Liu, J. Phys. Chem. C, 2011, 115, 15048 CAS.
- A. R. Cho, J. N. Son, V. Aravindan, H. Kim, K. S. Kang, W. S. Yoon, W. S. Kim and Y. S. Lee, J. Mater. Chem., 2012, 22, 6556 RSC.
- Y. G. Mateyshina and N. F. Uvarov, J. Power Sources, 2011, 196, 1494 CrossRef CAS.
- Q. Kuang, Y. M. Zhao and Z. Y. Liang, J. Power Sources, 2011, 196, 10169 CrossRef CAS.
- A. Q. Pan, J. Liu, J. G. Zhang, W. Xu, G. Z. Cao, Z. M. Nie, B. W. Arey and S. Liang, Electrochem. Commun., 2010, 12, 1674 CrossRef CAS.
- Y. Q. Qiao, J. P. Tu, X. L. Wang, D. Zhang, J. Y. Xiang, Y. J. Mai and C. D. Gu, J. Power Sources, 2011, 196, 7715 CrossRef CAS.
- C. X. Chang, J. F. Xiang, X. X. Shi, X. Y. Han, L. G. Yuan and J. T. Sun, Electrochim. Acta, 2008, 54, 623 CrossRef CAS.
- X. H. Rui, D. H. Sim, K. M. Wong, J. K. Zhu, W. L. Liu, C. Xu, H. T. Tan, N. Xiao, H. H. Hng, T. M. Lim and Q. Y. Yan, J. Power Sources, 2012, 214, 171 CrossRef CAS.
- L. J. Wang, X. X. Li, Z. Y. Tang and X. H. Zhang, Electrochem. Commun., 2012, 22, 73 CrossRef CAS.
- H. W. Liu, C. X. Cheng, X. T. Huang and J. L. Li, Electrochim. Acta, 2010, 55, 8461 CrossRef CAS.
- A. Q. Pan, D. W. Choi, J. G. Zhang, S. Q. Liang, G. Z. Cao, Z. M. Nie, B. W. Arey and J. Liu, J. Power Sources, 2011, 196, 3646 CrossRef CAS.
- C. Wang, H. M. Liu and W. S. Yang, J. Mater. Chem., 2012, 22, 5281 RSC.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321 CrossRef CAS PubMed.
- E. J. Yoo, J. J. Nakamura and H. S. Zhou, Energy Environ. Sci., 2012, 5, 6928 CAS.
- Z. Y. Lin, G. Waller, Y. Liu, M. L. Liu and C. P. Wong, Adv. Energy Mater., 2012, 2, 884 CrossRef CAS.
- P. X. Han, Y. H. Yue, L. X. Zhang, H. X. Xu, Z. H. Liu, K. J. Zhang, C. J. Zhang, S. M. Dong, W. Ma and G. L. Cui, Carbon, 2012, 50, 5498 CrossRef.
- X. F. Li, J. Liu, Y. Zhang, Y. L. Li, H. Liu, X. B. Meng, J. L. Yang, D. S. Geng, D. N. Wang, R. Y. Li and X. L. Sun, J. Power Sources, 2012, 197, 238 CrossRef CAS.
- P. A. Denis, R. Faccio and A. W. Mombru, ChemPhysChem, 2009, 10, 715 CrossRef CAS PubMed.
- B. Uchoa and A. H. C. Neto, Phys. Rev. Lett., 2007, 98, 146801 CrossRef PubMed.
- S. F. Huang, K. Terakura, T. Ozaki, T. Ikeda, M. Boero, M. Oshima, J. Ozaki and S. Miyata, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 235410 CrossRef.
- W. H. Shin, H. M. Jeong, B. G. Kim, J. K. Kang and J. W. Choi, Nano Lett., 2012, 12, 2283 CrossRef CAS PubMed.
- L. Zhao, Y. S. Hu, H. Li, Z. X. Wang and L. Q. Chen, Adv. Mater., 2011, 23, 1385 CrossRef CAS PubMed.
- S. Yoon, C. Liao, X. G. Sun, C. A. Bridges, R. R. Unocic, J. Nanda, S. Dai and M. P. Paranthaman, J. Mater. Chem., 2012, 22, 4611 RSC.
- D. Ding, X. Pan, L. Yu, Y. Cui, Y. Liang, J. Qi, W. Li, Q. Fu, X. Ma, Q. Xue, G. Sun and X. Bao, Chem. Mater., 2011, 23, 1188 CrossRef.
- H. Niwa, K. Horiba, Y. Harada, M. Oshima, T. Ikeda, K. Terakura, J. Ozaki and S. Miyata, J. Power Sources, 2009, 187, 93 CrossRef CAS.
- L. J. Wang, H. B. Liu, Z. Y. Tang, L. Ma and X. H. Zhang, J. Power Sources, 2012, 204, 197 CrossRef CAS.
- H. Wang, Y. J. Li, C. H. Huang, Y. D. Zhong and S. Q. Liu, J. Power Sources, 2012, 208, 282 CrossRef CAS.
- L. J. Wang, Z. Y. Tang, L. Ma and X. H. Zhang, Electrochem. Commun., 2011, 13, 1233 CrossRef CAS.
- L. Wang, L. C. Zhang, I. Lieberwirth, H. W. Xu and C. H. Chen, Electrochem. Commun., 2010, 12, 52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nj01021j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |