Click synthesis of symmetric bis-triazol ligands and full characterisation of their copper(II)-complexes†
Zakia
Benkhellat
a,
Mustapha
Allali
a,
Marc
Beley
a,
Emmanuel
Wenger
b,
Maxime
Bernard
c,
Nathalie
Parizel
c,
Katalin
Selmeczi
a and
Jean-Pierre
Joly
*a
aSRSMC UMR 7565 CNRS – Université de Lorraine, Faculté des Sciences, BP 70239, F-54506 Vandœuvre-lès-Nancy cedex, France. E-mail: jean-pierre.joly@sucres.uhp-nancy.fr
bCRM2, UMR 7036 CNRS – Université de Lorraine, Faculté des Sciences, BP 70239, F-54506 Vandœuvre-lès-Nancy cedex, France
cTGE Réseau National de RPE interdisciplinaire, FR-3443, Institut de Chimie, UMR 7177 CNRS – Université de Strasbourg, 4 rue Blaise Pascal, CS 90032, 67081 Strasbourg cedex, France
First published on 1st November 2013
Abstract
Eight novel ligands were prepared from a known symmetric diaza-18-crown-6 (cyclic ligand) and two commercial N,N′-dimethyl-alkyl diamines (acyclic ligands) via the Cu(I)-catalyzed Huisgen dipolar cycloaddition. All C2-symmetric isolated ligands readily formed stable crystalline 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-copper(II) complexes with cupric perchlorate. Their structural, electrochemical and physico-chemical properties were fully investigated with the help of X-ray diffraction, cyclic voltammetry, FT-IR, UV-visible, and electron paramagnetic resonance (EPR) spectroscopies. Planar – or nearly planar – arrangement of the two N3-triazole nitrogens and the two tertiary amine pivot nitrogens was found in one single four-coordinated species, in four five-coordinated species, and three six-coordinated species, with one or two solvent molecule(s), or two oxygen atoms of the crown ether, occupying the axial position(s) in the solid sate. The electron-donating or electron-withdrawing effect of the N1-substituent on the triazol was found to influence the Cu(II)/Cu(I) redox potential of all studied complexes in DMF. The EPR-spectrum of cyclic complexes in frozen DMF at 100 K exhibited two mononuclear species, one of them likely promoting the formation of dinuclear species as a minor component, whereas most acyclic complex spectra were quite similar.
:1-copper(II) complexes with cupric perchlorate. Their structural, electrochemical and physico-chemical properties were fully investigated with the help of X-ray diffraction, cyclic voltammetry, FT-IR, UV-visible, and electron paramagnetic resonance (EPR) spectroscopies. Planar – or nearly planar – arrangement of the two N3-triazole nitrogens and the two tertiary amine pivot nitrogens was found in one single four-coordinated species, in four five-coordinated species, and three six-coordinated species, with one or two solvent molecule(s), or two oxygen atoms of the crown ether, occupying the axial position(s) in the solid sate. The electron-donating or electron-withdrawing effect of the N1-substituent on the triazol was found to influence the Cu(II)/Cu(I) redox potential of all studied complexes in DMF. The EPR-spectrum of cyclic complexes in frozen DMF at 100 K exhibited two mononuclear species, one of them likely promoting the formation of dinuclear species as a minor component, whereas most acyclic complex spectra were quite similar.
Introduction
Copper is second to iron in its prevalence in redox-active metalloproteins,1 mainly as a cofactor of numerous enzymes (e.g. cytochrome-c oxidase,2 copper-thionein,3 ceruloplasmin,4 dopamine-β-mono-oxygenase,5 galactose oxidase,6 and many others). Copper is believed to play a significant role in the CNS-development of mammalians, too.7 Although this metal is of critical importance in numerous biochemical processes, free copper (i.e. uncomplexed Cu+ or Cu2+ cations) is actually cytotoxic.8,9 Therefore, versatile ligands of copper ions receive growing interest from the chemist community as models of natural enzymes10,11 (such as superoxide dismutase mimetics12), but also as new HIV-antiviral agents,13 anti-cancer agents,14,15 receptor-mediated tumour imaging agents,16,17 and catalysts of organic reactions.18 Functionalized 1,4-disubstituted 1,2,3-triazole units are versatile ligands offering several donor sites or act as bridging ligands for copper and some other rare transition metal coordination too.19–27 A large array of mono, bi, tri-, and polydentate ligands incorporating 1,4-disubstituted-1,2,3-triazoles has been synthesized since 2007.28–31 Moreover, the Cu(I)-catalyzed version of the [3+2] Huisgen32–35 azide–alkyne cycloaddition (i.e. the CuAAC ‘click reaction’)36–38 was used to synthesize ligands endowed with good complexing properties towards zinc, lead, nickel, and ammonium too.39–43 In the meantime, this powerful synthetic approach was extended to the linkage of cyclodextrines to various organic azides.44 We reported the synthesis of a new C2-symmetric macrocyclic ligand (hereafter L1, a pendant armed 18-crown-2-diaza-4-ether) endowed with good complexing properties towards copper,45 nickel and zinc cations, too.Solid-state structures revealed two different coordination modes, one symmetrical for Cu(II), quite isomorphous with the Ni(II)-analogue, versus one asymmetric for Zn(II), which were further investigated via molecular modelling. These results will be discussed elsewhere.46 During the course of this work, tetradentate ligands from a N,N′-dimethyl-1,2-diaminoethane synthesized via click chemistry were reported and their complexes with Mn2+, Ni2+, Zn2+, and Fe2+ generated. Among them, their Mn2+-complexes displayed interesting catalytic activities for the epoxidation of various terminal olefins with peracetic acid.18 We describe herein the ‘click’ syntheses of two series of new macrocyclic and acyclic ligands via the Cu(I)-catalyzed version of the Huisgen reaction between a terminal acetylene and an organic azide. The Cu(II)-complexes of these ligands were isolated (as their perchlorate salts) and characterised via different techniques: elemental analysis, FT-IR, UV-visible and EPR spectroscopies, HRMS+, high resolution X-ray diffraction (for seven over eight complexes), and cyclic voltammetry.
Results and discussion
Synthesis and characterisation of ligands
Cyclic ligands L2–4 were click-generated in good to fair yields from known N,N′-dipropargyl-diaza crown A47 and readily available aryl-azides (i.e.Scheme 1, general procedure I and ESI†).48–50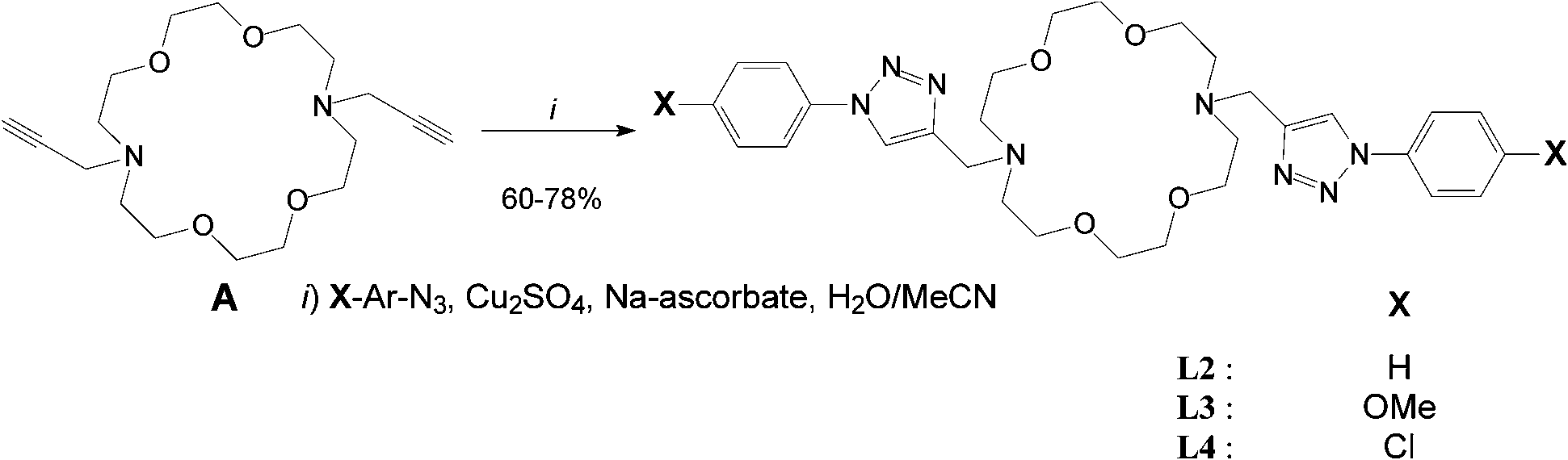 | ||
| Scheme 1 Synthesis of cyclic ligands L2–4 from diaza-crown ether A. | ||
Acyclic ligands L5–9 were isolated in fair to good yields using a slight modification of the one-pot method reported by Yan et al.51 from commercial secondary bis-N-methyl amines B and C (i.e. general procedure II and ESI†).
The tedious isolation of N,N′-dipropargyl bis-tertiary amine intermediates (after step i) did not improve overall yields (Scheme 2). Some ligands (e.g.L7 & L8) have to be chromatographed twice over silica and/or neutral alumina to ensure sufficient purity and unambiguous characterization. All ligand structures were unambiguously ascertained by 1H & 13C-NMR, FT-IR, and ES+-HRMS. 1H-NMR spectra of all ligands exhibit the expected time-averaged 2-fold symmetry and the characteristic aromatic singlet between 7.5 and 8.0 ppm for the two isochronous triazol protons at the NMR time scale (CDCl3, 298 K). Further information about the isolation and full characterisation of ligands L2–9 is provided in the ESI.†
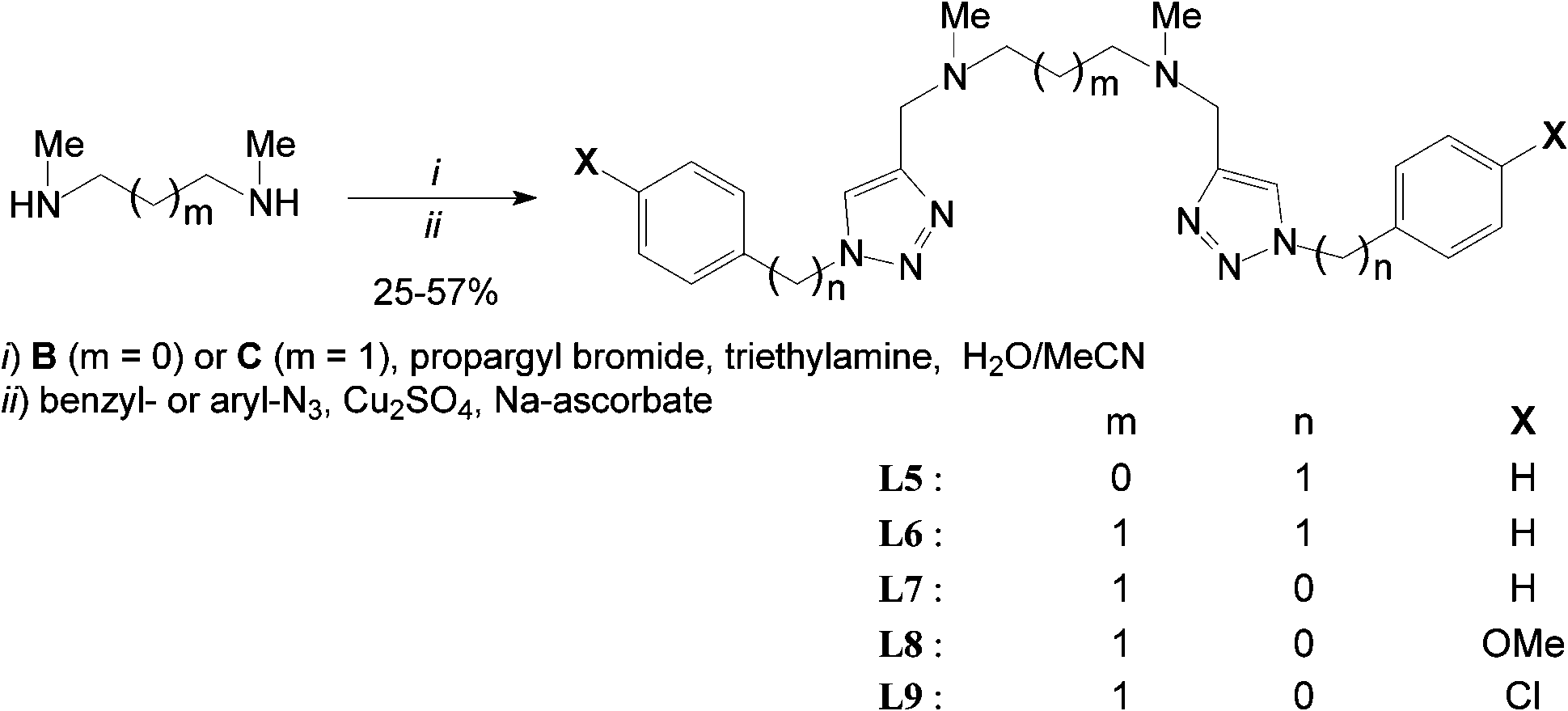 | ||
| Scheme 2 One-pot synthesis of ligands L5–9via the Cu(I)-catalyzed Huisgen reaction. | ||
Crystallographic characterization
Suitable X-ray diffraction monocrystals of 1:1 Cu(II)-complexes were isolated from ligands L2–8 and Cu(ClO4)2 hexahydrate in the presence of acetonitrile and dimethylformamide (traces) in fair to excellent yield within a week (see general procedure III and ESI†). Their crystallographic data are summarised in Tables 1 and 3.
| Complexes | [CuL1](ClO4)2 | [CuL2](ClO4)2 | [CuL3](ClO4)2 | [CuL4](ClO4)2, DMF |
|---|---|---|---|---|
| Empirical formula |
CuC32H44Cl2
N8O12 |
CuC30H40Cl2
N8O12 |
CuC32H44Cl2
N8O14 |
CuC33H45Cl4
N9O13 |
| Formula weight | 867.19 | 839.15 | 899.20 | 981.13 |
| Colour | Light blue | Blue-green | Dark green | Malachite-green |
| Cryst. size [mm] | 0.32 × 0.18 × 0.14 | 0.12 × 0.10 × 0.08 | 0.31 × 0.19 × 0.11 | 0.21 × 0.14 × 0.10 |
| Crystal system | Triclinic | Triclinic | Triclinic | Monoclinic |
| a [Å] | 9.1849(15) | 9.390(5) | 9.383(5) | 26.3848(4) |
| b [Å] | 9.2931(8) | 10.293(5) | 10.299(8) | 18.7279(3) |
| c [Å] | 11.8913(12) | 10.426(5) | 11.300(9) | 27.6457(6) |
| α [deg] | 98.876(4) | 101.979(5) | 70.19(4) | 90.00 |
| β [deg] | 108.784(5) | 108.296(5) | 66.08(3) | 143.2590(10) |
| γ [deg] | 105.480(6) | 106.798(5) | 73.30(5) | 90.00 |
| V [Å3] | 893.40(19) | 865.2(7) | 924.7(1) | 8171.8(3) |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P21/c |
| Z | 1 | 1 | 1 | 8 |
| ρ calcd [g cm−3] | 1.612 | 1.618 | 1.615 | 1.595 |
| Temp. [K] | 110(2) | 110(2) | 100(2) | 100(2) |
| R/wR | 0.0286/0.0696 | 0.0249/0.0661 | 0.0344/0.0806 | 0.0400/0.0947 |
| GOF | 1.041 | 1.047 | 1.062 | 1.025 |
| CCDC | 854682 | 853231 | 851236 | 851237 |
| Cyclic complexes | Bond distances (Å) | Bond angles (°) | ||||||
|---|---|---|---|---|---|---|---|---|
| Cu1–N1 | Cu1–N2 | Cu1–N3 | Cu1–N4 | N1–Cu1–N2 | N2–Cu1–N3 | N3–Cu1–N4 | N1–Cu1–N4 | |
| a i.e. N2–Cu–N1i bond angle, N1i and N3 being equivalent via the inversion centre cf.Fig. 1–3. b Mean values of two independent molecules. | ||||||||
| [CuL1]2+ | 1.988 | 2.114 | — | — | 80.7 | 99.3a | — | — |
| [CuL2]2+ | 2.015 | 2.096 | — | — | 81.1 | 98.9a | — | — |
| [CuL3]2+ | 2.011 | 2.107 | — | — | 81.3 | 98.7a | — | — |
| [CuL4]2+b |
1.950 | 2.178 | 1.941 | 2.166 | 80.4 | 101.3 | 82.9 | 95.2 |
The three first cyclic Cu(II)-complexes, i.e. [CuL1–3](ClO4)2, crystallised in the triclinic space group P as their perchlorate salts, with the central Cu-atom lying on the inversion centre.
However, the new [CuL2](ClO4)2 centro-symmetric complex (see Fig. 2) is quite different from [CuL1](ClO4)2,45 but very similar to [CuL3](ClO4)2 (see Fig. 3) with close bond distances and bond angles. It is noteworthy that the N1–Cu1–O1 angle was significantly smaller in [CuL1](ClO4)2 (i.e. 84.7°) than in [CuL2](ClO4)2 and [CuL3](ClO4)2 (∼87.3°).
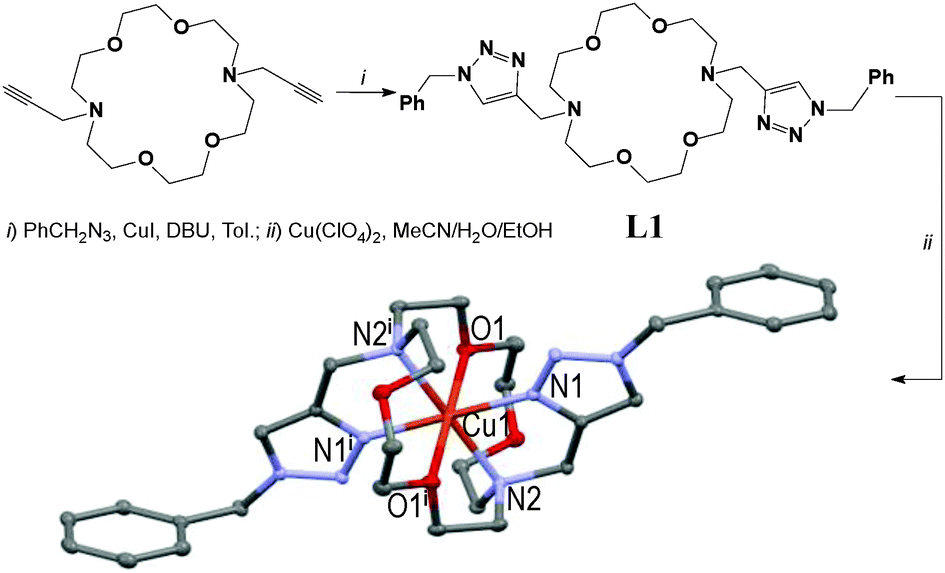 | ||
| Fig. 1 Two-step synthesis of the [CuL1](ClO4)2 centro-symmetric complex in 31% overall yield (symmetry code: (i) 1 − x, 1 − y, 1 − z).45 | ||
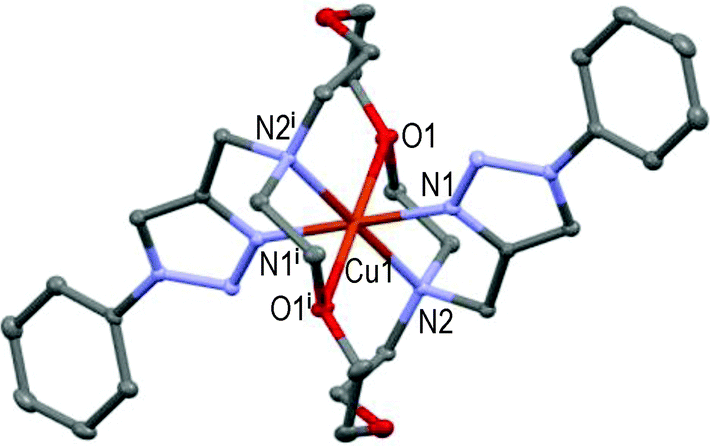 | ||
| Fig. 2 Molecular structure of [CuL2](ClO4)2 showing the hetero-atom numbering and the thermal ellipsoids at 50% probability (H-atoms and non-coordinating anions omitted for clarity; symmetry code: (i) −x, −y, −z). | ||
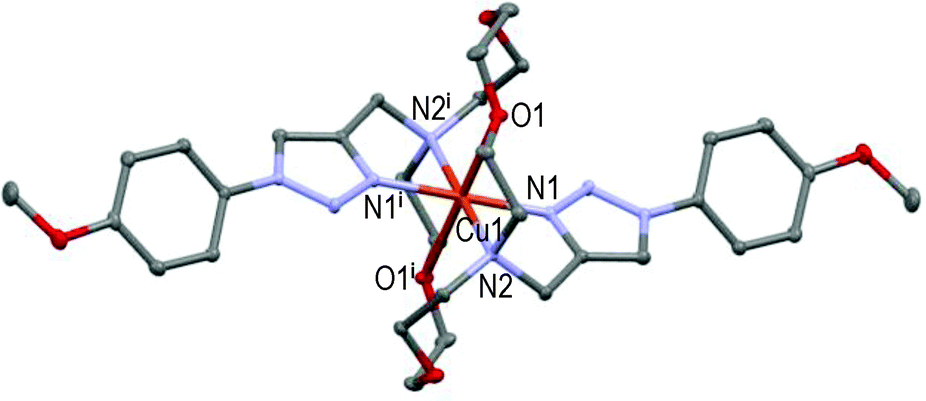 | ||
| Fig. 3 Molecular structure of [CuL3](ClO4)2 showing the hetero-atom numbering and the thermal ellipsoids at 50% probability (H-atoms and non-coordinating anions omitted for clarity; symmetry code: (i) −x, −y, 1 − z). | ||
Here again, the hexacoordinated Cu(II)-cation lies on the inversion centre in the basal plane formed by the four N-atoms and O1 occupies the axial position in the centro-symmetric complex (see Fig. 3). Strained 5-membered cycles (i.e. N1–Cu1–N2 and O1–Cu1–N2) induce bond angles diminution and a slightly distorted six-coordinated octahedral geometry. The Cu1–O1 distances fall in the range of 2.536 ± 0.011 Å in accordance with the Jahn–Teller effect.52 Replacement of the EDG-methoxy group by the EWG-chlorine atom in L4 (all else being equal) led to a completely different solid-state structure.
The tetracoordinated Cu(II) cation lies above (∼0.1 Å) the basal plane formed by N1–N2–N3–N4 in a quite distorted geometry (N1–Cu1–N2 = 80.8°, N3–Cu1–N4 = 83.1°); lone pairs of O1 and O2 oxygen atoms point towards the copper cation (i.e. red arrows: Cu1–O1 ∼2.71 Å, Cu1–O2 ∼2.79 Å), O3 and O4 atoms being too far to interfere with the metal ion (see Fig. 4).
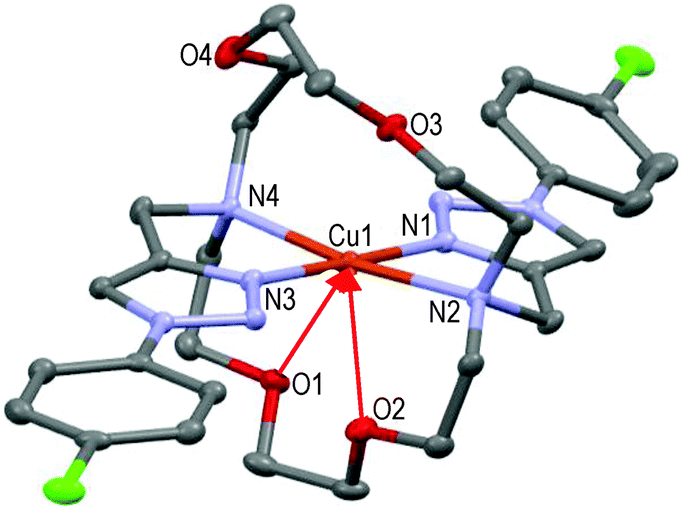 | ||
| Fig. 4 Molecular structure of one of the two independent [CuL4](ClO4)2 molecules present in the asymmetric unit showing the hetero-atom numbering and the thermal ellipsoids at 50% probability (H-atoms and non-coordinating anions omitted for clarity). | ||
| Complexes | [CuL5(MeCN)](ClO4)2 | [CuL6(ClO4)](ClO4) | [CuL7(MeCN)](ClO4)2 | [CuL8(DMF)2](ClO4)2 |
|---|---|---|---|---|
| a Flack value = 0.4745(8). | ||||
| Empirical formula |
CuC26H33Cl2
N9O8 |
CuC25H32Cl2
N8O8 |
CuC25H31Cl2
N9O8 |
CuC31H46Cl2
N10O12 |
| Formula weight | 734.06 | 707.04 | 720.03 | 885.22 |
| Colour | Deep blue | Purple | Deep purple | Blue |
| Cryst. size [mm] | 0.30 × 0.20 × 0.12 | 0.20 × 0.15 × 0.13 | 0.20 × 0.15 × 0.10 | 0.31 × 0.24 × 0.11 |
| Crystal system | Triclinic | Orthorhombic | Triclinic | Triclinic |
| a [Å] | 10.573(4) | 14.8356(4) | 13.8278(5) | 10.806(3) |
| b [Å] | 10.596(4) | 25.4907(6) | 14.1824(5) | 12.898(3) |
| c [Å] | 16.788(5) | 8.1556(2) | 16.0830(7) | 14.296(3) |
| α [deg] | 73.602(4) | 90.00 | 89.792(4) | 80.45(1) |
| β [deg] | 79.087(5) | 90.00 | 78.263(4) | 85.53(1) |
| γ [deg] | 60.693(3) | 90.00 | 87.476(2) | 73.19(1) |
| V [Å3] | 1570.4(11) | 3084.2(13) | 3085.1(2) | 1880.0(8) |
| Space group |
P |
Cmc21a |
P |
P |
| Z | 2 | 4 | 4 | 2 |
| ρ calcd [g cm−3] | 1.552 | 1.523 | 1.550 | 1.564 |
| Temp. [K] | 100(2) | 100(2) | 100(2) | 100(2) |
| R/wR | 0.0347/0.0795 | 0.0465/0.1239 | 0.0848/0.18344 | 0.0474/0.1067 |
| GOF | 1.052 | 1.192 | 1.199 | 1.063 |
| CCDC | 851241 | 851242 | 851238 | 851239 |
Selected bond distances and angles of acyclic Cu(II)-complexes from ligands L5–9 are summarised in Table 4.
| Acyclic complexes | Bond distances (Å) | Bond angles (°) | ||||||
|---|---|---|---|---|---|---|---|---|
| Cu–N1 | Cu–N2 | Cu–N3 | Cu–N4 | N1–Cu–N2 | N2–Cu–N3 | N3–Cu–N4 | N1–Cu–N4 | |
| a i.e. N2–Cu–N2iii bond angle, N2iii and N3 being equivalent by mirror symmetry cf.Fig. 7. b Mean values of two independent molecules. c See ESI for the low resolution CIF file and selected crystal data (T1). | ||||||||
| [CuL5(MeCN)]2+ | 1.984 | 2.036 | 2.036 | 1.962 | 81.5 | 86.8 | 82.4 | 103.3 |
| [CuL6(ClO4)]+ | 1.961 | 2.046 | — | — | 82.5 | 96.7a | — | — |
| [CuL7(MeCN)]2+b |
1.985 | 2.055 | 2.058 | 1.975 | 81.7 | 96.6 | 82.0 | 98.6 |
| [CuL8(DMF)2]2+ | 2.002 | 2.066 | 2.052 | 1.986 | 82.5 | 95.9 | 82.1 | 99.5 |
| [CuL9(ClO4)]+c |
1.99– | 2.07– | 2.08– | 1.97– | 82.– | 96.– | 83.– | 99.– |
Unfortunately, structure of the [CuL9(ClO4)](ClO4) complex could not be fully resolved in spite of numerous attempts under different conditions. However, its low resolution structure is given in Fig. 5 for comparison purposes in the acyclic series.
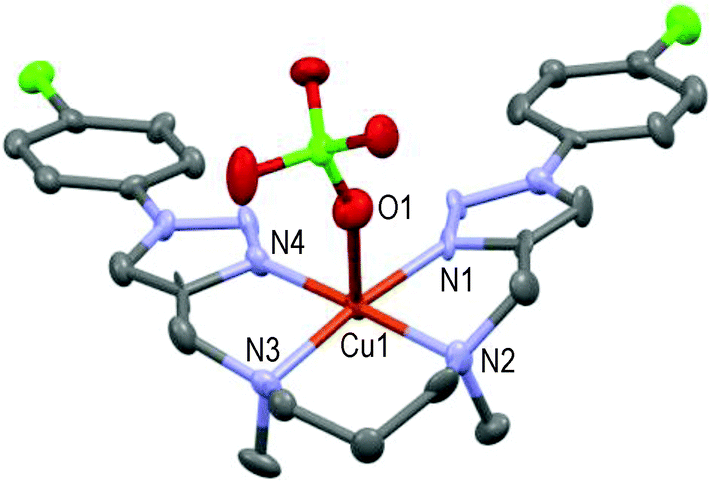 | ||
| Fig. 5 Low resolution molecular structure of [CuL9(ClO4)](ClO4) showing the hetero-atom numbering and the thermal ellipsoids at 50% probability (H-atoms and non-coordinating anions omitted for clarity). | ||
The above 1:1-complex is ‘roughly’ symmetric, with the pentacoordinated Cu(II)-ion lying approximately 0.1 Å over the basal plane formed by N1–N2–N3–N4 in a square-pyramidal geometry; one oxygen atom of the perchlorate anion occupies the axial position (Cu1–O1 ∼2.37 Å, N1–Cu1–O1 ∼94.3°) as the 5th ligand. Thanks to the 6-membered cycle (N2–Cu1–N3) the resulting structure is more relaxed than that of [CuL5(MeCN)]2+ as illustrated in Fig. 6.
 | ||
| Fig. 6 Molecular structure of [CuL5(MeCN)](ClO4)2 showing the hetero-atom numbering and the thermal ellipsoids at 50% probability (H-atoms and non-coordinating anions omitted for clarity). | ||
This mono-solvated complex is not endowed with true symmetry; N1–N2–N3–N4 forms the basal plane and N5 of one acetonitrile molecule occupies the axial position (Cu1–N5 = 2.246 Å and N1–Cu1–N5 = 97.7°) as the 5th ligand in a distorted square pyramidal geometry. Strained 5-membered cycles (i.e. N1–Cu1–N2, N2–Cu1–N3, N3–Cu1–N4) induce bond angles diminution in comparison to [CuL9(ClO4)]+ (see Fig. 5) and a significant repulsion of the pentacoordinated Cu-atom above the basal plane (∼0.34 Å). An extra carbon atom between N2 and N3 atoms in ligand L6 led to a completely different structure for the resulting Cu(II)-complex (see Fig. 7).
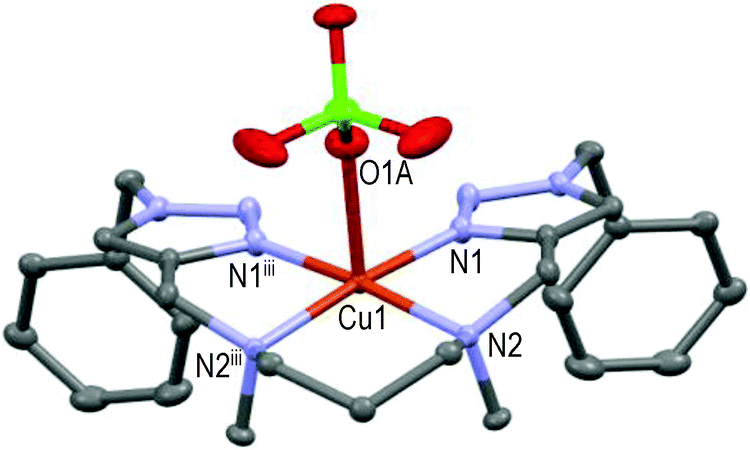 | ||
| Fig. 7 Molecular structure of [CuL6(ClO4)]ClO4 showing the hetero-atom numbering and the thermal ellipsoids at 50% probability (H-atoms and non-coordinating anions omitted for clarity; symmetry code: (iii) −x, y, z). | ||
Thanks to the 6-membered cycle (i.e. –N2–Cu1–N2iii–(CH2)3–) the mirror-symmetric [CuL6(ClO4)]+ structure is more relaxed than those of [CuL5(MeCN)]2+. The pentacoordinated Cu(II)-ion lies approximately 0.13 Å over the basal plane formed by N1–N2–N2iii–N1iii in a square-pyramidal geometry; O1A of one perchlorate anion occupies the axial position (Cu1–O1A ∼2.42 Å) as the 5th ligand. Interestingly, direct attachment of the phenyl group to the triazole moieties (all else being equal) induced a different coordination of Cu(II) by ligand L7.
The resulting 1:1-complex is here only approximately symmetric, the pentacoordinated Cu(II) cation at 0.07 Å centered over the basal plane formed by N1–N2–N3–N4 in a square-pyramidal geometry (see Fig. 8); N5 of one acetonitrile molecule occupies the axial position (N1–Cu1–N5 = 92.1°) as the 5th ligand. Lastly, the bis-solvated 1:1-complex formed from the para-methoxy-phenyl-substituted L8 ligand and Cu(II) in the presence of DMF is quite different and unique in the acyclic series (see Fig. 9).
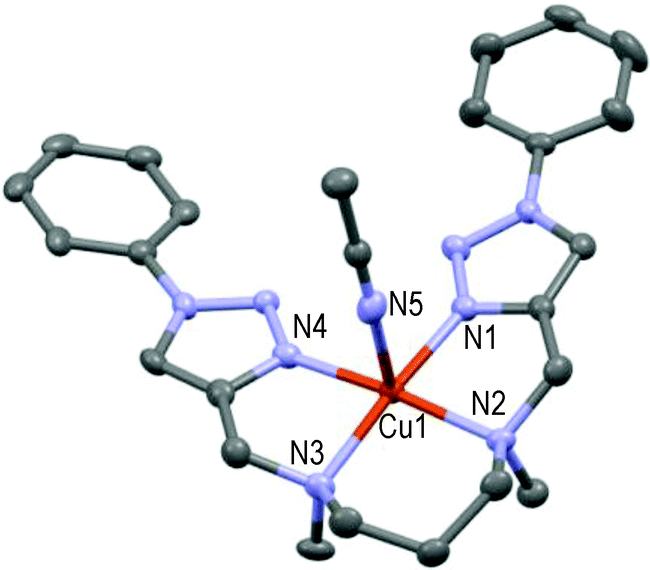 | ||
| Fig. 8 Molecular structure of one of the two independent [CuL7(MeCN)](ClO4)2 molecules present in the asymmetric unit showing the hetero-atom numbering and the thermal ellipsoids at 50% probability (H-atoms and non-coordinating anions omitted for clarity). | ||
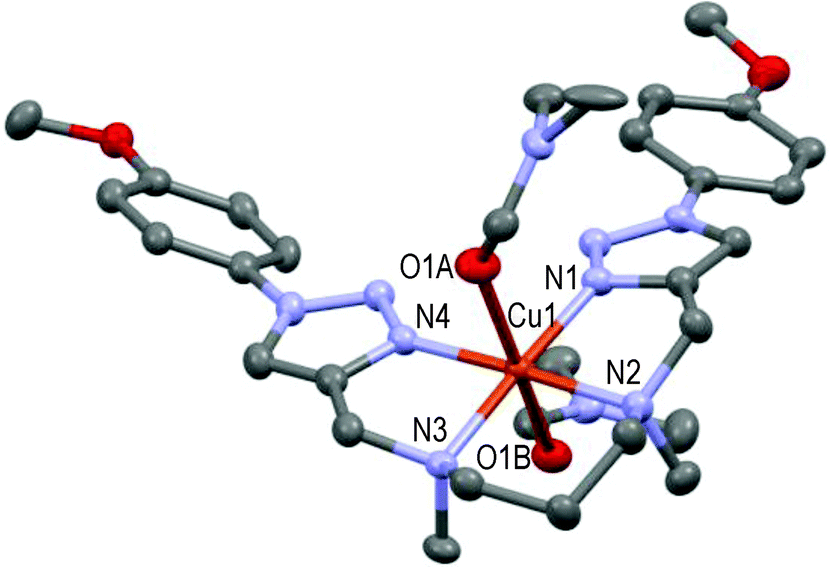 | ||
| Fig. 9 Molecular structure of [CuL8(DMF)2](ClO4)2 showing the hetero-atom numbering and the thermal ellipsoids at 50% probability (H-atoms and non-coordinating anions omitted for clarity). | ||
The hexacoordinated Cu(II) cation lies in the basal plane formed by N1–N2–N3–N4 in a distorted octahedral geometry; O1A and O1B of two DMF molecules occupy the axial positions respectively 2.367 Å and 2.548 Å apart from the copper cation, which fall in the range of reported values.53,54 This structure can be compared to that of the [CuL3](ClO4)2 complex (Fig. 3) in the cyclic series. In summary, ligands L2–9 readily form stable 1:1-Cu(II) complexes in the presence of Cu(ClO4)2 in solution. Three different geometries (i.e. tetra-, penta-, and hexa-coordination) were observed in the solid state with respect to the nature of the ligand (cyclic vs. acyclic) and the nature of the substituent on the triazoles. One or two solvent molecule(s), or one perchlorate anion, act(s) eventually as axial ligand(s) in the more open acyclic series to complete the coordination sphere.
Electrochemical studies
It is noteworthy that the formation of Cuo-species occurs at about −1.5 V as an irreversible process and that the formation of a Cu(III)-complex was not detected before the solvent oxidation which started at +1.5 V under our experimental conditions. Therefore, we studied the electrochemical behavior starting potential at +0.2 V towards negative values and kept the potential range between −0.4 and +0.8 V to avoid the irreversible formation of Cuo and the oxidation of the solvent throughout the present study. Table 5 summarizes the electrochemical results corresponding to the first cathodic peak (1st scan towards negative potential at three increasing scan rates) which was assigned to the reduction of the initial and stable geometric form, i.e. Cu(II)st to Cu(I). A reductive-controlled potential coulometry realized below the 1st cathodic peak at −0.4 V showed that the total quantity of electricity exchanged corresponds to one electron per mole of the copper complex.| Cyclic ligands | Acyclic ligands | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| L1 | L2 | L3 | L4 | L5 | L6 | L7 | L8 | L9 | |
| Epc 25 mV s−1 | −10 | +30 | +30 | +60 | −10 | −25 | −50 | −21 | +35 |
| Ipc 25 mV s−1 | −11 | −12 | −11 | −11 | −12 | −10 | −11 | −11 | −11 |
| Epc 100 mV s−1 | −20 | +10 | +20 | +45 | −30 | −45 | −65 | −50 | −10 |
| Ipc 100 mV s−1 | −22 | −24 | −22 | −22 | −24 | −20 | −21 | −22 | −22 |
| Epc 500 mV s−1 | −40 | 0 | +5 | +40 | −50 | −100 | −120 | −90 | −15 |
| Ipc 500 mV s−1 | −49 | −52 | −50 | −50 | −54 | −45 | −49 | −50 | −50 |
First cathodic peak reduction potential (i.e. Epc) analyses revealed the ligand-based electrochemical behavior of all present complexes. On the one hand, the donor or withdrawing effect of the para-substituent on the phenyl ring was found to influence more or less the reduction potential of the complexes. For instance, [CuL4]2+ and [CuL9(ClO4)]+ display a Cu(II)st → Cu(I) redox potential at more positive values than the corresponding unsubstituted complexes in cyclic and acyclic series, i.e. [CuL2]2+ and [CuL7(MeCN)]2+, respectively. Thus, the electron withdrawing effect of the chlorine substituent decreases the electronic density around the cupric ion and induces a positive shift in the reduction potential from Cu(II) to Cu(I). In the cyclic series (e.g. X = H, [CuL2]2+), the N-triazole doublets are likely more delocalized than in [CuL1]2+ (no possible π-conjugation). Accordingly, the donor effect of the triazole moiety significantly increases the electron density around the metal in [CuL1]2+ with a negative shift (ca. 35 mV) of the Epc value in comparison to [CuL2]2+. In the acyclic series, the reduction potential of [CuL7(MeCN)]2+ (m = 1, n = 0, X = H), including one acetonitrile molecule as the axial ligand, is more negative than those of [CuL8(DMF)2]2+ (m = 1, n = 0, X = OMe) and [CuL6(ClO4)]+ (m = 1, n = 1, X = H), the donor effect of the acetonitrile ligand being obviously stronger than the remote methoxy effect in [CuL8(DMF)2]2+. The mild donor effect of the methoxy group on the phenyl also induces the almost same reduction potentials (Epc ∼ 30 mV) in [CuL2]2+ (X = H) as in [CuL3]2+ (X = OMe). On the other hand, the reduction of Cu(II)st to Cu(I) is easier in [CuL5(MeCN)]2+ than in [CuL6(ClO4)]+. We attributed the higher measured Epc to the relative destabilization of Cu(II) in the more distorted geometry of [CuL5(MeCN)](ClO4)2 (m = 0) vs. [CuL6(ClO4)]ClO4 (m = 1).56 It is well-known that there are important differences in geometrical requirements for coordination of Cu(I) in comparison to Cu(II) that produce interesting dynamic processes in solution. Divalent copper preferentially forms stable hexacoordinated, or square pyramidal, complexes whereas the monovalent state induces tetrahedral coordination. The occurrence of structural rearrangements in the first coordination shell of Cu(II) to Cu(I) in the redox mechanism has been largely investigated because many processes in bio-inorganic chemistry, catalysis, and supramolecular chemistry involve electron-transfer steps in copper complexes.1 As illustrated in Table 5, the 1st reduction peak potential of each complex, i.e. Epc, depends on the scan rate since its value increases as the scan rate decreases. This behaviour suggests that the reduction of the initial and stable geometric form Cu(II)st gives intermediate metastable species, i.e. Cu(I)met, which can rearrange into a more stable form, i.e. Cu(I)st, according to a two-step EredCred reaction, that is:
| Ered step: Cu(II)st + ē ⇆ Cu(I)met |
| Cred step: Cu(I)met → Cu(I)st |
In an attempt to characterize the re-oxidation steps of Cu(I)mt and Cu(I)st, CV between −0.4 and +0.8 V was performed. The second scan was selected as a typical CV. The electrochemical behavior of [CuL4](ClO4)2 is presented below (Fig. 10) as a relevant example in the cyclic series.
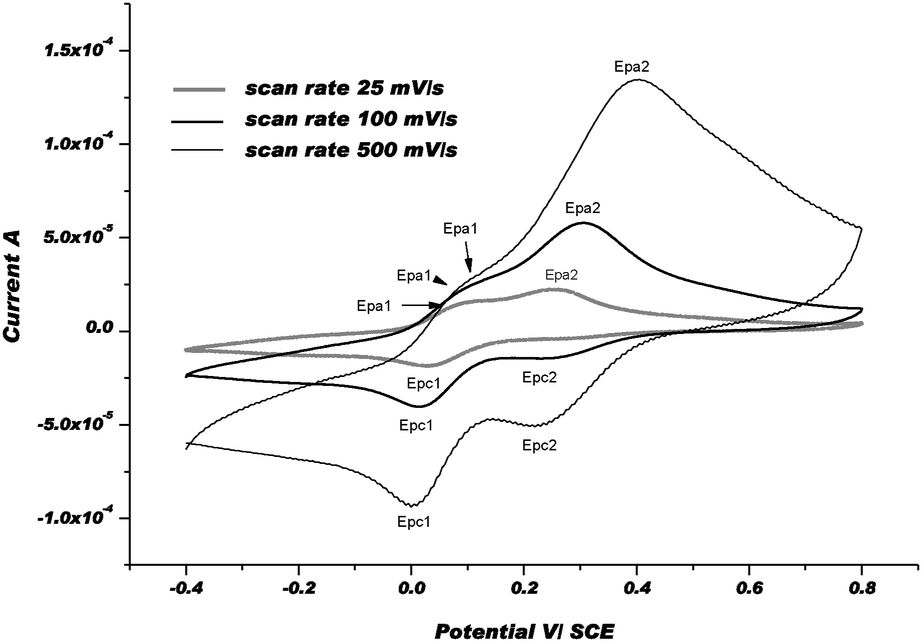 | ||
| Fig. 10 CVs of [CuL4]2+at 3 different scan rates: 25, 100, and 500 mV s−1 in Ar-purged DMF with 0.1 M Bu4NPF6 as a supporting electrolyte at rt. | ||
The positive part of CVs obviously displays two oxidation waves: a 1st wave, i.e. Epa1, which appears at about +90 mV and a second one, i.e. Epa2, at about +300 mV under our experimental conditions. After the 1st electrochemical step between +0.2 and −0.4 V (Ered), especially when the scan rate was slow, the chemical step, i.e. Cu(I)met → Cu(I)st, became more complete and the Cu(I)st/Cu(I)met ratio was much higher. For that reason, the 1st wave was assigned to the re-oxidation of Cu(I)st to Cu(II)met1 and the 2nd one to the formation of Cu(II)met2 from Cu(I)met. Thus, we postulated that the re-oxidation of Cu(I)-complexes, i.e. Cu(I)met and Cu(I)st, produces metastable Cu(II)-species via an electron transfer followed by a chemical step that is referred to as the so-called EoxCox mechanism. Indeed, after changing the scan direction at 800 mV, and since 1st reduction potential, i.e. Epc1, remains the same as in the first scan (i.e. Epc), we assumed that Cu(II)met1 and/or Cu(II)met2 relax to the initial and more stable form Cu(II)st. As shown in the CVs, the chemical rearrangement Cox is complete at 25 mV s−1. At faster scan rates, this chemical rearrangement is not complete and another reduction wave, i.e. Epc2, appears at about 0.2 V more anodic potential vs. Epc1. As described earlier, we postulate that the metastable forms of Cu(I) and Cu(II)-complexes should be coordinated by solvent molecule(s).54Fig. 11 displays the CVs obtained with [CuL8(DMF)2](ClO4)2 as a relevant complex in the acyclic series. For each scan rate, the CVs in the positive part display three re-oxidation waves. The 1st wave, i.e. Epa1 at a potential of about 110 mV vs. SCE, was attributed to the re-oxidation of Cu(I)st to Cu(II)met1. The two other waves, corresponding to Epa2 and Epa3, were ascribed to the re-oxidation of metastable Cu(I)met1 and Cu(I)met2 species formed by the reduction of Cu(II)st during the 1st scan.
 | ||
| Fig. 11 CVs of [CuL8(DMF)2]2+ at 3 different scan rates: 25, 100, and 500 mV s−1 in Ar-purged DMF with Bu4NPF6 0.1 M as a supporting electrolyte at rt. | ||
However, reversing the scan direction at 800 mV induces only one reduction peak which was clearly observed at 25 and 100 mV s−1 (i.e. Epc1). Moreover, another reduction step occurred at about +0.25 V, i.e. Epc2, at 500 mV s−1 with a very low current intensity. For each scan rate, the potential values of Epc1 were close to those summarised in Table 5. It was inferred that the chemical rearrangements after the electronic transfer may be easier and the chemical reaction constants associated with the chemical steps Cox are faster in the more flexible acyclic series than in the cyclic series. However, one should notice that these systems can be almost totally reversed at scan rates below 100 mV s−1 and 25 mV s−1 in the acyclic series and cyclic series, respectively. Comparison of these results (for the acyclic and cyclic series) with those obtained by others with analogous mononuclear Cu(II) complexes coordinated by nitrogen or sulfur atoms in the ligands show that the Cu(II)/Cu(I) couples occur within about the same potential domain, between ca. −0.4 and −1.0 V vs. SCE.54 However, chemical reorganisation steps inducing the formation of metastable intermediates, i.e. Cu(II)met and Cu(I)met, seem to be easier with diaza-crown ethers due to the weaker energy of the Cu–O bond than those of Cu–N or Cu–S bonds.55 More than two conformational metastable intermediates are generated for both oxidized and reduced species, preceding or succeeding the electron transfer steps in diaza-crown ether copper(II) complexes. Finally, the global mechanism is likely more complicated than the classical square-scheme mechanism generally proposed for electron transfer in Cu(II)/Cu(I) systems. This may be attributed mainly to the weakness of the Cu–O bond strength by comparison to the Cu–N and Cu–S bond strengths. Altogether, the present electrochemical study shows that the electron-transfer process is accompanied by bond ruptures (or formations) and changes in reversible ligand conformation particularly in the acyclic series.
EPR measurements
Acyclic complexes (i.e. from ligands L5–9) will be discussed first as they displayed much more simple EPR-spectra than cyclic ones. The X-band EPR spectra of [CuL7(MeCN)]2+ and [CuL8(DMF)2]2+ complexes recorded at 100 K in frozen DMF are presented in Fig. 12 together with simulations. | ||
| Fig. 12 EPR spectrum recorded at the X-band (10−3 M in DMF at 100 K) of [CuL7(MeCN)]2+ (left) and [CuL8(DMF)2]2+ (right); experimental (upper traces), simulation (lower traces). | ||
Fit parameters of acyclic complexes (see Table 6) are typical of a square-based coordination polyhedra56 and characteristic of a Cu(II)-ion coordinated with four N-nuclei within a planar (or near planar) configuration.57
| Complex | g Tensor | Hyperfine coupling tensor to 63Cu | |||
|---|---|---|---|---|---|
| g ⊥ | g ∥ | A(Cu)⊥ 10−4 cm−1 | A(Cu)∥ 10−4 cm−1 | g ∥/A∥ cm | |
| [CuL5(MeCN)]2+ | 2.056 | 2.289 (∼60%) | 49 | 160 | 143 |
| 2.220 (∼40%) | 163 | 136 | |||
| [CuL6(ClO4)]+ | 2.054 | 2.233 | 14 | 183 | 122 |
| [CuL7(MeCN)]2+ | 2.058 | 2.234 | 12 | 186 | 120 |
| [CuL8(DMF)2]2+ | 2.058 | 2.236 | 13 | 185 | 121 |
| [CuL9(ClO4)]+ | 2.054 | 2.237 | 9 | 192 | 116 |
Since EPR parameters may also reflect the degree of distortion of copper(II) complexes, the g∥/A∥ ratio is given as a rough estimate of coordination geometry. Values of 110–120 are reported as typical of ‘planar’ complexes while the range between 130–150 is characteristic of slight to moderate distortion and 180–250 indicates strong distortion.58 Higher frequency, e.g. the Q-band EPR spectroscopy, is well known to facilitate the interpretation of intricate spectra where several interactions are intermingled.
Although the Q-band was not required to solve the geometry of [CuL6(ClO4)]+ (i.e.Fig. 13, right), it will serve as a reference for comparison with more complicated systems. It appears that the X-band and Q-band parameters for simulations are quite consistent with the geometry of the complex in the solid state. These parameters are also very similar to those of L7 and L8 Cu(II)-complexes. The parallel components of the EPR spectra of L6, L7, L8, and L9 Cu(II)-complexes (see Table 6) confirm the rather undistorted ‘planar’ geometry in frozen solution consistent with their X-ray crystal structures. [CuL6(ClO4)]+ and [CuL7(MeCN)]2+ do not show any significant difference in their EPR parameters despite the supplementary bond between the triazole and the phenyl ring in L6 and their different coordination mode in the solid state. Yet, the Q-band EPR spectrum of [CuL5(MeCN)]2+ in frozen solution (see Fig. 13, left) requires two components for a satisfactory fit with two slightly different values of g∥. The major contribution (ca. 60%) points to an increasing axial character with an increase in g∥ and a decrease in A∥ as compared to [CuL7(MeCN)]2+, [CuL8(DMF)2]2+, and [CuL9(ClO4)]+ complexes (see S1 in ESI†). Concerning [CuL5(MeCN)]2+, the higher value of g∥/A∥ is indicative of a moderate distortion in agreement with its crystalline structure which might correspond to the more stable conformer in DMF at 100 K.59 In the cyclic series, the Q-band spectra of [CuL3]2+ and [CuL4]2+ are similar in frozen solution (see Fig. 14, left part for experimental and simulation spectra of [CuL3]2+ and ESI† S2 for [CuL4]2+). Such signals are characteristic of a mixture of binuclear species, that means species in which two close copper nuclei interact via an intermolecular way, and mononuclear copper species.60–63Fig. 14's right part illustrates the component positions of tensor g (gx, gy and gz) of binuclear species with the zero-field splitting D and the hyperfine contribution Az for two equivalent copper nuclei. The index ∥ or ⊥ is related to the mononuclear complex. Please, note that A∥ is roughly twice as high as Az in agreement with the delocalization of the electronic cloud over two copper nuclei. The value of 2D was assessed from the splitting of copper parallel lines (180 G). Within the point-dipole approximation, it corresponds to a rCu–Cu spin–spin distance of ca. 5.9 Å.
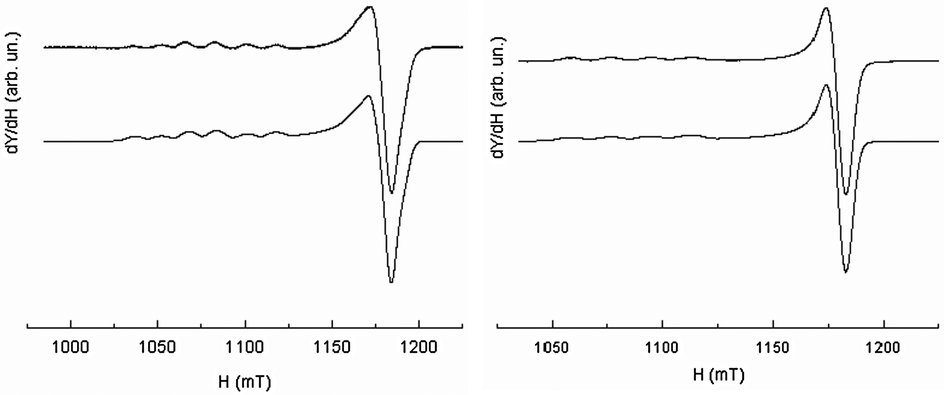 | ||
| Fig. 13 EPR spectrum recorded at the Q-band and 100 K (10−3 M in DMF at 100 K) of [CuL5(MeCN)]2+ (left) and [CuL6(ClO4)]+ (right); experimental (upper traces), simulation (lower traces). | ||
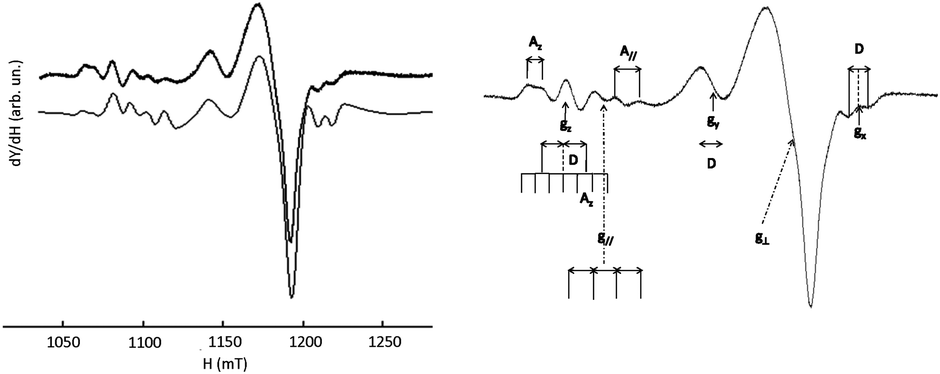 | ||
| Fig. 14 EPR spectrum of [CuL3]2+ recorded at the Q-band (10−3 M in DMF at 100 K); left: experimental (upper trace), simulation (lower trace); right: visualization of the EPR features associated with the di-nuclear complex (S = 1) labelled x, y, and z and with the mono-nuclear complex (S = 1/2) labelled ∥ and ⊥. | ||
The EPR data of mononuclear and dinuclear complexes of [CuL3]2+ in Table 7 were obtained by simulation of the entire spectrum.
| S = 1 (dinuclear species, weight: 17%) | ||||
|---|---|---|---|---|
| g z | g y | g x | A z (10−4 cm−1) | D (10−4 cm−1) |
| 2.250 | 2.115 | 2.003 | 65 | 84 |
| S = 1/2 (mononuclear complex, weight: 83%) | ||||
|---|---|---|---|---|
| g ∥ | g ⊥ | A ∥ (10−4 cm−1) | g ∥/A∥ | |
| 2.214 | 2.061 | 2.039 | 107 | 207 |
The g∥/A∥ ratio is high compared to the acyclic complexes as expected for a more distorted environment of the metal ion.59 The Q-band spectrum of [CuL2]2+ (vide infraFig. 15) results from the superimposition of two mononuclear species with very low dipolar interaction between them as revealed by a weak peak at 1163 mT (→). Such a faint contribution is not visible on the parallel part of the spectrum. Indeed, all peaks in the low field zone are equidistant.
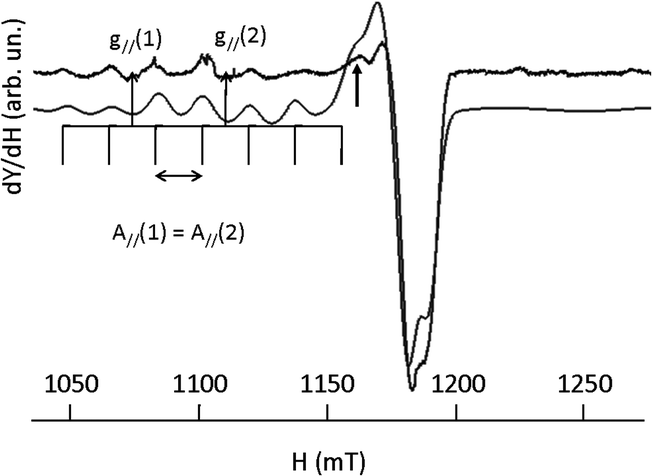 | ||
| Fig. 15 EPR spectrum of [CuL2]2+ recorded at the Q-band (10−3 M in DMF at 100 K) experimental (upper trace), simulation (lower trace). | ||
EPR data for the [CuL2]2+ complex are summarized in Table 8.
| S = 1/2 (mononuclear complex, weight: 37%) | |||
|---|---|---|---|
| g ∥ | g ⊥ | A ∥ (10−4 cm−1) | g ∥/A∥ |
| 2.258 | 2.045 | 177 | 127 |
| S = 1/2 (mononuclear complex, weight: 63%) | |||
|---|---|---|---|
| g ∥ | g ⊥ | A ∥ (10−4 cm−1) | g ∥/A∥ |
| 2.182 | 2.061 | 177 | 123 |
A tentative explanation is these solutions contain at 100 K a mixture of two more or less distorted structural species (i.e. conformers) one of them promoting the formation of dimeric species. The number and kind of bonds, and the so-called ‘crown effect’, obviously influence the flexibility and the stability of such complexes in DMF, especially at low temperatures.59,62
Conclusion
Novel ligands bearing two 1,4-substituted-triazole moieties as coordinating side-arms (i.e.L2–9) were easily prepared in one step from two commercial N,N′-dimethyl-alkyl diamines or from a known C2-symmetric N,N′-dipropargyl-diaza-18-crown-6 via the Cu(I)-catalyzed Huisgen dipolar cycloaddition. Changing the alkyl-substituent of the azide (i.e. benzyl, phenyl, p-methoxy- or p-chloro-phenyl) gave access to stable mononuclear Cu(II)-complexes with different geometries and coordinations in the solid state. Their electrochemical studies proved that the electron transfer induces intricate but reversible reorganisation of the cation coordination shell with a first cathodic peak varying within a maximum 0.16 V range. The EPR-spectrum of cyclic [CuL2]2+, [CuL3]2+ & [CuL4]2+ – and to a smaller extent that of [CuL5(MeCN)]2+ – exhibited two mononuclear species (or conformers) signals in frozen DMF at 100 K, one of them promoting the formation of dinuclear species as a minor component. Altogether these behaviours suggest that such complexes could be proposed as catalysts (e.g. for the oxidation of alcohols in carbonyl compounds or as artificial enzymes).64 Lastly, the cytotoxicity of some of these complexes towards uterine human sarcoma cells was investigated and showed promising activities.65Experimental section
Chemicals and solvents (reagent grade or better) were purchased from Sigma-Aldrich except spectrophotometric-grade DMF which was purchased from Alfa-Aesar [ref. 13808]. Anhydrous solvents were dried by usual procedures and stored over 4 Å molecular sieves under Ar before use. Chromatography was carried out using Grace silicagel (Davisil® LC60A 70–200 μm) or basic Al2O3 when stated (Sigma-Aldrich ref. 199443). 1H & 13C-NMR spectra were recorded on a Bruker AC250 spectrometer at 250 MHz for 1H and 62.9 MHz for 13C in CDCl3 if no other conditions are stated. Chemical shifts δ were measured with respect to the CH3CN signal (2.10 ppm for 1H and 1.89 ppm for 13C-NMR). CHN elemental analyses were performed using a Thermo Finnigan EA 1112 Series Flash Elemental Analyzer. ES+-HRMS spectra were obtained using a Bruker micrOTOFq mass spectrometer with capillary tensions between −1200 and −4500 V from diluted MeCN solutions. FT-IR spectra were obtained from KBr-pellets (if not other stated) on a Perkin Elmer Spectrum 1000 spectrophotometer. UV-visible spectra were recorded on a Perkin-Elmer Lambda1050 UV-Vis-NIR spectrophotometer using a 1 cm-optical-pathlength cell at T = 298 K. X-ray structures were determined using a Bruker-Nonius Kappa APEXII or a Agilent SuperNova diffractometer equipped with a low temperature device using liquid N2 to perform XRD intensity measurements near 100 K. X-ray source was a sealed tube giving Mo Kα radiation (λ = 0.71073 Å). Diffraction pattern frames were processed with Apex or CrysAlis software. The structures were solved by direct methods using the SIR software package66a and refined by full matrix least-squares on F squared using SHELXL software.66b In both crystal structure models, non-hydrogen atoms were located in the difference Fourier syntheses and were refined with anisotropic thermal parameters. Observed hydrogen atoms were placed at calculated positions using a riding model. Cyclic voltammetry (CV) was performed using a Radiometer PST006 potentiostat using a conventional three-electrode cell at rt. The KCl calomel electrode (SCE) was separated from the test compartment using a bridge tube. The test solution was dimethylformamide containing 10−1 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte. The working electrode was a 10 mm Pt wire and the counter-electrode a 1 cm2 vitreous carbon disc. 5.0 × 10−4 M fresh solution of each studied compound was used and purged for 5 min with Ar before each measurement. All potentials were quoted versus SCE. Under these conditions the redox potential of the couple Fc+/Fc was found to be 0.47 V. EPR spectra were recorded using a continuous-wave EMXplus spectrometer (Bruker Biospin GmbH, Germany) equipped with a high sensitivity resonator (4119HS-W1, Bruker). The spectrometer was tuned such that settings (modulation coils, incident microwave power) were not distorting the EPR signal (X-band Larmor frequency ∼9.3 GHz and Q-band Larmor frequency ∼34 GHz). Measurements were carried out in frozen 10−3 M DMF (ACS reagent ≥ 99.8% GC) solutions of copper(II) complexes held at 100 K with liquid nitrogen. Simulations were generated with the EasySpin free software.67General procedure I
Synthesis of ligands L2–4. To a stirred suspension of N,N′-bis-propargyl-diaza-18-6 crown ether A49 (500 mg, 1.47 mmol) in a mixture of water–acetonitrile (1:1, 25 mL) at rt were added the aryl-azide (2.1 eq.), Na-ascorbate (117 mg, 0.4 eq.), and CuSO4·5H2O (74 mg, 0.2 eq.) under Ar. The resulting brownish solution was stirred overnight at rt under Ar. The reaction mixture was filtered, extracted with CH2Cl2 (4 × 90 mL), and the organic phases were combined, washed with 5% aq. NH4OH (2 × 20 mL), brine (20 mL), water (3 × 20 mL), dried over MgSO4, and finally evaporated under reduced pressure. The crude material was purified by liquid chromatography on silica (and/or alumina) with mixtures of CH2Cl2–MeOH as eluents.
General procedure II
Synthesis of ligands L5–9. A solution of symmetric sec. N,N′-dimethylamine (B or C, 5.0 mmol), triethylamine (1.5 mL, 2.15 eq.), and propargyl bromide (80 wt% in toluene, 1.2 mL, 2.15 eq.) in a mixture of acetonitrile–water (1:1, 90 mL) was stirred for 90 min at rt under Ar. The organic azide (2.15 eq.), Na-ascorbate (0.4 g, 0.4 eq.), and CuSO4·5H2O (0.25 g, 0.2 eq.) were then added and the resulting brownish solution was stirred overnight at rt under Ar. The reaction mixture was filtered on a sintered glass, extracted with CH2Cl2 (4 × 90 mL), and the organic phases were combined, washed with 5% aq. NH4OH (2 × 20 mL), brine (20 mL), water (3 × 20 mL), dried over MgSO4, and finally evaporated under reduced pressure. The crude resulting gum was purified by liquid chromatography (LC) on silica or alumina with mixtures of CH2Cl2–MeOH as eluents.
General procedure III
Synthesis of Cu-complexes. Warning: perchlorates are potentially explosive compounds. While we have experienced no problems during our syntheses, extreme care must be taken when working with perchlorate complexes or salts and only small quantities should be handled! Cu(ClO4)2·6H2O (1.2 eq.) was dissolved in a solution of the ligand (50.0 mg) in a mixture of acetonitrile–water–EtOH (3 mL, 1:1:1) at rt. The resulting suspension was stirred for 2 h at 70 °C, allowed to cool to rt, filtered through a small cotton pad, concentrated to half, diluted with two drops of DMF (ca. 30 mg), and finally stored for several days in the dark at 4 °C. Crystallographic-suitable monocrystals were isolated by filtration on a sintered glass, rinsed with dist. H2O, CH2Cl2, and finally dried at rt in the dark.
Acknowledgements
M. A. gratefully acknowledges financial support from the French National Research Agency via a post-doctoral grant (ANR-09-BLAN-0180-01). The authors warmly thank the ‘Institut Jean Barriol’ for X-ray diffraction measurement facilities and Dr Slimane Dahaoui (radio-crystallographic measurements and structure determination), Mr François Dupire for measuring HRMS, Mr Stéphane Parant for recording UV-Vis and Mrs Sandrine Adach for performing elemental analysis.Notes and references
- D. B. Rorabacher, Chem. Rev., 2004, 104, 651–697 CrossRef CAS PubMed.
- G. C. M. Steffens, R. Biewald and G. Buse, Eur. J. Biochem., 1987, 164, 295–300 CrossRef CAS.
- C. F. Wright, D. H. Hamer and K. McKenney, J. Biol. Chem., 1988, 263, 1570–1574 CAS.
- B. S. Magdoff-Fairchild, F. M. Lovell and B. W. Low, J. Biol. Chem., 1969, 244, 3497–3499 CAS.
- S. S. Hasnain, G. P. Diakun, P. F. Knowles, N. Binsted, C. D. Garner and N. J. Blackburn, Biochem. J., 1984, 221, 545–548 CAS.
- F. Thomas, Eur. J. Inorg. Chem., 2007, 2379–2404 CrossRef CAS.
- V. Desai and S. G. Kaler, Am. J. Clin. Nutr., 2008, 88, 855S–858S CAS.
- W. Kaim and J. Rall, Angew. Chem., Int. Ed. Engl., 1996, 35, 43–60 CrossRef CAS.
- J. Nelson, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2010, 106, 235–254 RSC.
- C. J. Fritchie, J. Biol. Chem., 1973, 248, 7516–7521 CAS.
- I. A. Koval, K. Selmeczi, C. Belle, C. Philouze, E. Saint-Aman, I. Gautier-Luneau, A. M. Schuitema, M. van Vliet, P. Gamez, O. Roubeau, M. Lüken, B. Krebs, M. Lutz, A. L. Spek, J.-L. Pierre and J. Reedijk, Chem.–Eur. J., 2006, 12, 6138–6150 CrossRef CAS PubMed.
- A. L. Abuhijleh and J. Khalaf, Eur. J. Med. Chem., 2010, 45, 3811–3817 CrossRef CAS PubMed.
- S. García-Gallego, M. J. Serramía, E. Arnaiz, L. Díaz, M. A. Muñoz-Fernández, P. Gómez-Sal, M. F. Ottaviani, R. Gómez and F. J. de la Mata, Eur. J. Inorg. Chem., 2011, 1657–1665 CrossRef.
- B. S. Creaven, B. Duff, D. A. Egan, K. Kavanagh, G. Rosair, T. Thangella, R. Venkat and M. Walsh, Inorg. Chim. Acta, 2010, 363, 4048–4058 CrossRef CAS PubMed.
- E. N. da Silva Jr, M. A. B. F. de Moura, A. V. Pinto, M. C. F. R. Pinto, M. C. B. V. de Souza, A. J. Araújo, C. Pessoa, L. V. Costa-Lotufo, R. C. Montenegro, M. O. de Moraes, V. F. Ferreir and M. O. F. Goulart, J. Braz. Chem. Soc., 2009, 20, 635–643 CrossRef PubMed.
- O. Jacobson, I. D. Weiss, L. Szajek, J. M. Farber and D. O. Kiesewetter, Bioorg. Med. Chem., 2009, 17, 1486–1493 CrossRef CAS PubMed.
- M. Shokeen and T. J. Wadas, Med. Chem., 2011, 7, 413–429 CrossRef CAS.
- E. Hao, Z. Wang, L. Jiao and S. Wang, Dalton Trans., 2010, 39, 2660–2666 RSC.
- J. Xiang, Y.-G. Yin and P. Mei, Inorg. Chem. Commun., 2007, 10, 1168–1171 CrossRef CAS PubMed.
- P. S. Donnelly, S. D. Zanatta, S. C. Zammit, J. M. White and S. J. Williams, Chem. Commun., 2008, 2459–2461 RSC.
- T. L. Mindt, C. Schweinsberg, L. Brans, A. Hagenbach, U. Abram, D. Tourwé, E. Garcia-Garayoa and R. Schibli, ChemMedChem, 2009, 4, 529–539 CrossRef CAS PubMed.
- G. F. Manbeck, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2011, 50, 3431–3441 CrossRef CAS PubMed.
- C. B. Anderson, B. Christopher, A. B. S. Elliott, J. E. M. Lewis, C. J. McAdam, K. C. Gordon and J. D. Crowley, Dalton Trans., 2012, 41, 14625–14632 RSC.
- W. Yang and Y. Zhong, Chin. J. Chem., 2013, 31, 329–338 CrossRef CAS.
- C. B. Anderson, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Organometallics, 2013, 32, 788–797 CrossRef CAS.
- D. Wang, D. Denux, J. Ruiz and D. Astruc, Adv. Synth. Catal., 2013, 355, 129–142 CrossRef CAS.
- J. M. Fernandez-Hernandez, J. I. Beltran, V. Lemaur, M.-D. Galvez-Lopez, C.-H. Chien, F. Polo, E. Orselli, R. Froehlich, J. Cornil and L. De Cola, Inorg. Chem., 2013, 52, 1812–1824 CrossRef CAS PubMed ; and literature cited therein.
- B. M. J. M. Suijkerbuijk, B. N. H. Aerts, H. P. Dijkstra, M. Lutz, A. L. Spek, G. van Koten and R. J. M. Klein Gebbink, Dalton Trans., 2007, 1273–1276 RSC.
- J. D. Crowley and D. A. McMorran, Top. Heterocycl. Chem., 2012, 28, 31–84 CAS.
- P.-Y. Jin, P. Jin, Y.-A. Ruan, Y. Ju and Y.-F. Zhao, Synlett, 2007, 3003–3006 CAS.
- G. E. Kostatis, P. Xydias, E. Nordlander and J. C. Plakatouras, Inorg. Chim. Acta, 2012, 383, 327–331 CrossRef PubMed.
- R. Huisgen, G. Szeimies and L. Mœbius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS.
- J. Bastide, J. Hamelin, F. Texier and Y. Vo Quang, Bull. Soc. Chim. Fr., 1973, 7–8, 555–2579 Search PubMed.
- R. Huisgen, Pure Appl. Chem., 1989, 61, 613–628 CrossRef CAS.
- Q. Wang, S. Chittaboina and H. N. Barnhill, Lett. Org. Chem., 2005, 2, 293–301 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., 2001, 113, 2056–2075 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., 2002, 114, 2708–2711 CrossRef.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675–696 RSC.
- E. Tamanini, A. Katewa, L. M. Sedger, M. H. Todd and M. Watkinson, Inorg. Chem., 2009, 48, 319–324 CrossRef CAS PubMed.
- E. Tamanini, K. Flavin, M. Motevalli, S. Piperno, L. A. Gheber, M. H. Todd and M. Watkinson, Inorg. Chem., 2010, 49, 3789–3800 CrossRef CAS PubMed.
- L.-D. Li, C. S. B. Gomes, P. T. Gomes and M. T. Duarte, Dalton Trans., 2011, 40, 3365–3380 RSC.
- A. Späth, E.-M. Rummel and B. König, Molbank, 2010, M663 CrossRef PubMed.
- Z. Jia, R. K. Singh and D. Wang, Biomedical Applications of “Click”-Modified Cyclodextrinsin Click Chemistry in Glycoscience: New Developments and Strategies, ed. Z. J. Witczak and R. Bielski, John Wiley & Sons, Inc., 2013, ch. 11 Search PubMed.
- J.-P. Joly, M. Beley, K. Selmeczi and E. Wenger, Inorg. Chem. Commun., 2009, 12, 382–384 CrossRef CAS PubMed.
- Submitted for publication.
- V. J. Gatto, K. A. Arnold, A. M. Viscariello, S. R. Miller, C. R. Morgan and G. W. Gokel, J. Org. Chem., 1986, 51, 5373–5384 CrossRef CAS.
- F. Moulin, Helv. Chim. Acta, 1952, 35, 167–180 CrossRef CAS.
- I. Wilkening, G. del Signore and C. P. R. Hackenberger, Chem. Commun., 2010, 47, 349–351 RSC.
- A. Zarei, A. R. Hajipour, L. Khazdooz and H. Aghaei, Tetrahedron Lett., 2009, 50, 4443–4445 CrossRef CAS PubMed.
- Z.-Y. Yan, Y.-B. Zhao, M.-J. Fan, W.-M. Liu and Y.-M. Liang, Tetrahedron, 2005, 61, 9331–9337 CrossRef CAS PubMed.
- M. V. Veidis, G. H. Schreiber, T. E. Gough and G. J. Palenik, J. Am. Chem. Soc., 1969, 91, 1859–1860 CrossRef CAS.
- F.-Y. Dong, Y.-T. Li, Z.-Y. Wu, J.-M. Dou and D.-Q. Wang, J. Chem. Crystallogr., 2007, 37, 445–449 CrossRef CAS PubMed.
- A. Cheansirisomboon, S. Youngme, C. Pakawatchai and N. Chaichit, Mendeleev Commun., 2010, 20, 109–110 CrossRef CAS PubMed.
- N. M. Villeneuve, R. R. Schroeder, L. A. Ochrymowycz and D. B. Rorabacher, Inorg. Chem., 1997, 36, 4475–4483 CrossRef CAS PubMed.
- E. V. Rybak-Akimova, A. Y. Nazarenko, L. Chen, P. W. Krieger, A. M. Herrera, V. V. Tarasov and P. D. Robinson, Inorg. Chim. Acta, 2001, 324, 1–15 CrossRef CAS.
- E. Tamari, S. E. J. Rigby, M. Motevalli, M. H. Todd and M. Watkinson, Chem.–Eur. J., 2009, 15, 3720–3728 CrossRef PubMed.
- A. W. Addison, Spectroscopic and redox trends from model copper complexes, in Copper Coordination Chemistry: Biochemical and Inorganic Perspectives, ed. K. D. Karlin and J. Zubieta, Adenine Press, New York, 1983 Search PubMed.
- V. C. da Silveira, G. F. Caramori, M. P. Abbott, M. B. Gonçalves, H. M. Petrilli and A. M. da Costa Ferreira, J. Inorg. Biochem., 2009, 103, 1331–1341 CrossRef PubMed.
- S. S. Eaton, G. R. Eaton and C. K. Chang, J. Am. Chem. Soc., 1985, 107, 3177–3184 CrossRef CAS.
- L. J. Singh, N. S. Devi, S. P. Devi, W. B. Devi, R. K. Hemakumar Singh, B. Rajeswari and R. M. Kadam, Inorg. Chem. Commun, 2010, 13, 365–368 CrossRef CAS PubMed.
- D. da G. J. Batista, P. B. da Silva, L. Stivanin, D. R. Lachter, R. S. Silva, J. Felcman, S. R. W. Louro, L. R. Teixeira, M. de Nazaré and C. Soeiro, Polyhedron, 2011, 30, 1718–1725 CrossRef CAS PubMed.
- J. A. Weil and J. R. Bolton, in Electron Paramagnetic Resonance, Elementary Theory and Practical Applications, ed. J. A. Weil and J. R. Bolton, John Wiley & Sons, Inc., 1993, ch. 6.3 Search PubMed.
- F. Rosati and G. Roelfes, ChemCatChem, 2010, 2, 916–927 CrossRef CAS.
- Q. A. De Paula, J.-P. Joly, K. Selmeczi, P. Colepicolo, G. Indig, A. M. Da Costa Ferreira, Eurobic 11 – Granada (Spain) Sept. 12–16, 2012.
- (a) A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and G. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS; (b) G. M. Sheldrick, SHELXS97 and SHELXL97, University of Göttingen, Germany, 1997 Search PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and full characterisation of L2–9 ligands including ES-HRMS+ spectra of all reported complexes; EPR spectra (Q-band at 100 K) of [CuL9(ClO4)]ClO4 (S1) and [CuL4](ClO4)2 (S2) complexes in PDF format; low resolution crystallographic data & structure refinement parameters of the [CuL9(ClO4)](ClO4) complex and T1 in CIF format. CCDC 853231, 851236–851242, 854682. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj00570d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |