DOI:
10.1039/C3NJ00913K
(Paper)
New J. Chem., 2014,
38, 146-154
Uniform Lu2O3 hollow microspheres: template-directed synthesis and bright white up-conversion luminescence properties
Received
(in Victoria, Australia)
9th August 2013
, Accepted 3rd October 2013
First published on 4th October 2013
Abstract
Well-dispersed, uniform Lu2O3 hollow microspheres have been successfully fabricated via a urea-based precipitation method in the presence of colloidal PS microspheres as templates, followed by subsequent heat treatment. The structure, morphology, formation process, and fluorescent properties are well investigated using various techniques. The results indicate that the hollow microspheres can be well indexed to the cubic Lu2O3 phase. The hollow Lu2O3 microspheres with a uniform diameter of about 2.2 μm maintain the spherical morphology and good dispersion of the PS spheres template. The shell of the hollow microspheres consists of numerous nanocrystals with the thickness of approximately 20 nm. Moreover, the possible formation mechanism of evolution from the PS spheres to the amorphous precursor and to the final hollow Lu2O3 microspheres has also been proposed. Under 980 nm laser diode excitation, Lu2O3:Yb3+/Tm3+, Lu2O3:Er3+ and Lu2O3:Yb3+/Er3+ products are mainly dominated by blue, green and red light emissions, respectively. The ratio of the intensity of green luminescence to that of red luminescence decreases with an increase of the concentration of Yb3+ in Lu2O3:Er3+ samples. Furthermore, the UC white light was successfully obtained in the Lu2O3:Yb3+/Er3+/Tm3+ system by adjusting the relative doping concentration of Yb3+, Er3+ and Tm3+. The obtained UC white light has CIE-x = 0.3478 and CIE-y = 0.3143, which are very close to the standard equal energy white light illuminate (x = 0.33, y = 0.33). Because of abundant luminescent colors from RGB to white in Lu2O3:Yb3+/Er3+/Tm3+ samples under 980 nm laser diode (LD) excitation, they can potentially be used as fluorophores in the fields of color displays, backlights, UC lasers, photonics, and biomedicine.
1. Introduction
The development of nano/micromaterials with controllable size and morphology has been extensively pursued, not only because of the fundamental scientific interest, but also because of their technological applications.1,2 Among the various nano/micromaterials, hollow nano/microspheres currently represent one of the fastest growing areas in comparison with other structural features.3,4 Owing to their distinct low effective density, high specific surface area, and encapsulation ability, the hollow spheres are considered to be promising materials in many fields such as catalysis,5 photonic devices,6 electrochemical cells,7 and biotechnology.8 Recently, many efforts have been made in the development of different methods for the design and preparation of nano- or microsized hollow spheres. Template-directed synthesis, with hard templates (including silica, carbon spheres, and metal colloids)9–13 and soft templates (consisting of vesicles, emulsion droplets, micelles, and gas bubbles),14–17 has been demonstrated to be an effective approach for preparing inorganic hollow spheres. Among various hard templates, polymer latex particles, especially polystyrene (PS) spheres, have been reported to be effective templates for the preparation of hollow spherical inorganic materials such as TiO2,18,19 SiO2,20 Ta2O5,21 and ZnS.22 However, to the best of our knowledge, no report has been devoted to the preparation of the hollow lutetium oxide spheres by using PS colloidal spheres as the template.
Much research attention has been paid to the synthesis of lanthanide compounds, because they can be used as high performance phosphors, catalysts, and other functional materials due to their novel electronic, optical, and chemical properties.23–26 Because of the interesting physical properties of the lutetium oxide (Lu2O3), such as high melting point, phase stability, and low thermal expansion,27–29 it always serves as an excellent candidate for lanthanide ion substitution. Also, lutetium (Lu) may be a more favorable cation than yttrium (Y) for trivalent lanthanide dopant emission due to the intensity-borrowing mechanism mixing the 4f and 5d orbitals of the Ln3+ ions via the lattice valence band levels.30 Ln3+-doped Lu2O3 (Ln3+ = Eu3+, Tb3+, Er3+, Ho3+, Sm3+) materials have been proven to be important phosphors as reported in the previous literature.31,32 Up to now, different morphologies of Lu2O3 have been prepared by various synthesis methods. For example, one-dimensional Lu2O3 phosphors and three-dimensional flower-like Lu2O3 have been synthesized through the hydrothermal methods.33 Two-dimensional Lu2O3 epitaxial films have also been fabricated via pulsed laser deposition. The single-crystalline and monodisperse cubic Lu2O3 nanocrystals were synthesised via a nonhydrolytic approach.34 However, to the best of our knowledge, there have been few reports on the synthesis of uniform and well-dispersed Ln3+-doped Lu2O3 hollow spheres and their corresponding white up-conversion luminescence properties. The relatively low phonon energy (about 600 cm−1) of this host enables high UC efficiency of RE ions by efficiently hindering nonradiative losses.35 Furthermore, the hollow spherical phosphors would achieve a reduction in the amount of expensive rare earth and some of the methods mentioned above are complex and it is not easy to control the morphology of the products. Hence, it is especially promising to develop a simple and green approach to prepare hollow Lu2O3 phosphors.
Herein, the as-obtained PS microspheres were utilized as hard templates to fabricate highly uniform Lu2O3 hollow microspheres using a precipitation technique followed by a subsequent calcination process. The structure, morphology, formation process and UC luminescence properties of the as-synthesized hollow microspheres were investigated in detail. The approach has been proved to be a green route without using an organic template and any etching process, where the corrosive acid or base is usually used as the etching agent. In particular, the method is suitable for high yield mass production of hollow Lu2O3:Ln3+ phosphors with controllable properties, based on its simple and environmentally friendly preparation process.
2. Experimental section
2.1 Materials
The rare-earth oxides Ln2O3 (99.99%) (Ln = La, Yb, Er, and Tm) were purchased from the Science and Technology Parent Company of Changchun Institute of Applied Chemistry, and other chemicals were purchased from Beijing Fine Chemical Company. All chemicals were analytical-grade reagents and used as purchased without further purification.
2.2 Synthesis of monodispersed polystyrene microspheres
PS fine powder consisting of microspheres was prepared by dispersion polymerization according to Paine et al.36 The PVP stabilizer (1.0 g) was dissolved in ethanol (38.2 mL) in a three-necked round bottom flask fitted with a condenser and a magnetic stirrer. The reaction vessel was then heated to 70 °C under a nitrogen blanket and purged with nitrogen for 12 h at 70 °C. A solution of AIBN (0.15 g) pre-dissolved in a styrene monomer (15 g) was added to the reaction vessel with vigorous stirring. The styrene polymerization was allowed to proceed for 12 h before cooling to room temperature. The product was purified by repeated centrifugation and washed with ethanol. A white fine powder (PS) was finally obtained after being dried in a vacuum oven at 50 °C.
2.3 Synthesis of hollow Lu2O3 microspheres
In a typical process for the synthesis of hollow Lu2O3 microspheres, 0.5 mmol of Lu2O3 was dissolved in 2 mol L−1 of HNO3 with stirring. The superfluous HNO3 was driven off by heating until the pH value of the solution reached between 2 and 3. The as-prepared Lu(NO3)3 was added to 30 mL of deionized water. Then 3.0 g of urea was dissolved in the solution under vigorous stirring to form a clear solution. The as-prepared PS microspheres (0.2 g) were then added into the above solution under ultrasonication for 1 h. Finally, the mixture was transferred into a 100 mL flask and heated at 90 °C for 4 h with vigorous stirring before the product was collected by centrifugation. The precursors were washed by deionized water and ethanol three times and dried at 60 °C in air. The final hollow Lu2O3 microspheres were obtained through a heat treatment at 800 °C in air for 4 h with a heating rate of 1 °C min−1.
2.4 Synthesis of hollow Lu2O3:Ln3+ (Ln3+ = Yb3+, Er3+, Tm3+) microspheres
First, Lu2O3, Yb2O3, Er2O3 and Tm2O3 were dissolved in dilute HNO3, respectively, forming colorless stock solutions of La(NO3)3 (0.5 M) and Ln(NO3)3 (Ln3+ = Yb3+, Er3+, Tm3+) (0.2 M), respectively. In a typical synthesis, stoichiometric Lu(NO3)3 and Ln(NO3)3 (solution) were added into deionized water, resulting in the formation of a colorless solution of [Lu(NO3)3 + Ln(NO3)3]; Ln3+/(Lu3+ + Ln3+) = 0.05–10 mol%. Then Lu2O3:Ln3+ (Ln3+ = Yb3+, Er3+, Tm3+) products can be obtained through the same preparation process of the Lu2O3 product as stated above.
2.5 Characterization
Powder X-ray diffraction (XRD) measurements were performed on a Rigaku-Dmax 2500 diffractometer with Cu Kα radiation (λ = 0.15405 nm). Fourier transform infrared (FT-IR) spectra were recorded on a Perkin-Elmer 580B infrared spectrophotometer using the KBr pellet technique. Thermogravimetric and differential thermal analyses (TG-DTA) data were recorded using a Thermal Analysis Instrument (SDT 2960, TA Instruments, New Castle, DE) with the heating rate of 10 °C min−1 in an air flow of 100 mL min−1. The morphologies and composition of the as-prepared samples were inspected under a field emission scanning electron microscope (FESEM, S4800, Hitachi) equipped with an energy-dispersive X-ray spectrum (EDX, JEOL JXA-840). Low- to high-resolution transmission electron microscopy (TEM) was performed using an FEI Tecnai G2 S-Twin with a field emission gun operating at 200 kV. Images were acquired digitally on a Gatan multiople CCD camera. The UC emission spectra were obtained using a 980 nm laser from an OPO (optical parametric oscillator, Continuum Surelite, USA) as the excitation source and detected by a photomultiplier tube (HAMAMATSU R955) from 400 to 900 nm. All measurements were performed at room temperature.
3. Results and discussion
3.1 Phase, structure and morphology
Fig. 1 shows the X-ray diffraction results of the PS spheres, as-obtained PS/Lu(OH)CO3 precursor and the final product after calcination at 800 °C for 2 h. In Fig. 1A for the PS spheres, an obviously broadened diffraction peak is seen at about 19 °C, which is a typical XRD feature of amorphous PS materials.37,38 As for the as-obtained PS/Lu(OH)CO3 precursor (Fig. 1B), the XRD pattern of the precursor shows two broad bands at 30 °C and 45 °C, which indicate that the as-formed precursor is amorphous before calcination. The component of the precursor is assigned to be Lu(OH)CO3 according to the previous literature.39–41 After the annealing process, all the diffraction peaks of the product can be indexed to the cubic phase of Lu2O3 [JCPDS No. 86-2475; space group: Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (206)] (Fig. 1C), and no other impurity peaks can be detected. The crystallite size of the sample can be estimated from Scherrer's equation:
(206)] (Fig. 1C), and no other impurity peaks can be detected. The crystallite size of the sample can be estimated from Scherrer's equation:
Dhkl = Kλ/(β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ) cosθ) |
where λ is the X-ray wavelength (0.15405 nm), β is the full width at half-maximum, θ is the diffraction angle, K is a constant (0.89), and Dhkl means the size along the (hkl) direction. The strongest crystalline plane (222) at 2θ = 29.8° was used to calculate the average crystallite sizes (D) of the sample. The estimated crystal size of Lu2O3 is calculated to be 25.3 nm. It can also be seen that the diffraction peaks of the Lu2O3 samples are very sharp and strong, revealing that the Lu2O3 product with high crystallinity can be synthesized by this method. This is important for phosphors because high crystallinity generally means fewer traps and stronger luminescence.
 |
| | Fig. 1 XRD patterns of the (A) as-prepared PS spheres, (B) uncalcined PS/Lu(OH)CO3 precursor, and (C) hollow Lu2O3 microspheres. | |
TG-DTA curves of the as-prepared PS spheres and the uncalcined core–shell structured PS/Lu(OH)CO3 precursor are shown in Fig. 2A and B, respectively. One step of the weight loss can be discovered in TG-DTA curves of the PS spheres (Fig. 2A), the sharp weight loss is attributed to the splitting burning of PS spheres at 394 °C. For the core–shell structured PS/Lu(OH)CO3 precursor (Fig. 2B), there are two major stages of sharp weight loss in the TG curve at 338 °C and 468 °C, accompanying their corresponding exothermic peaks in the DSC curve. The two weight losses can be assigned to the dehydration, burning of the PS templates, and the conversion from the Lu(OH)CO3 precursor to the Lu2O3, respectively. It is worth noting that the weight loss of the pure PS template is nearly 100%, while the residual weight percentage of the precursor is about 53.46%, suggesting a considerably high yield of the hollow phosphors prepared using this method.
 |
| | Fig. 2 TG-DTA curves of (A) the as-prepared PS spheres and (B) the uncalcined PS/Lu(OH)CO3 precursor. | |
The functional groups related to the surface information of the PS spheres, the uncalcined PS/Lu(OH)CO3 precursor, and the calcined Lu2O3 sample were examined by FT-IR, as shown in Fig. 3. The FT-IR spectrum of the PS spheres (line A) shows the characteristic adsorption bands of PS at ca. 3100–2900, 1600–1350, and 700 cm−1, which can be attributed to the stretching vibrations of aromatic C–H in-plane, stretching vibrations of aromatic C–C, and bending vibrations of aromatic C–C out-of-plane, respectively.42 Compared with the spectrum of PS, the PS/Lu(OH)CO3 uncalcined precursor (line B), not only clearly exhibits characteristic absorption bands attributed to the PS core, but also shows the IR bands at 3444, 1493, 1407, 1151, 1073, 1028, 840, 755, and 695 cm−1 corresponding to OH− (ν), CO (νas), CO (νas), CO (νs), CO (δ), OH− (δ), and CO (δ) (ν = stretch; νs = symmetric stretch; νas = asymmetric stretch; δ = deformation), which indicates that the composition of the precursor should be PS and Lu(OH)CO3.43–45 In the FTIR spectrum of the calcined Lu2O3 sample (line C), the band at about 577 cm−1 can be assigned to the Lu–O stretching adsorption of the Lu2O3,46 which also confirms the formation of the crystalline Lu2O3 sample. The typical adsorption bands of PS are not observed, which further demonstrates that the PS template is thoroughly removed from the composite microspheres by calcination. These results are consistent with those of XRD patterns and confirm the formation of the crystalline Lu2O3 sample via the urea-based precipitation and the annealing process.
 |
| | Fig. 3 FT-IR of (A) the PS spheres, (B) uncalcined PS/Lu(OH)CO3 precursor, and (C) hollow Lu2O3 microspheres. | |
The SEM and TEM images of the PS spheres show that the template consists of well-dispersed microspheres with an average size of 2.3 μm (Fig. 4A and B), and the surfaces of PS spheres are smooth. Therefore, the diameter of the final hollow spheres should also be regulated by employing PS microspheres as templates. Fig. 4C shows the SEM image of the precursor before calcination. One can see that the precursor inherits the spherical morphology, and the surfaces of precursor particles are much rougher that those of PS spheres due to the precipitation of a large amount of uniform nanoparticles. Naturally, the size of the precursor is a little larger than those of bare PS spheres due to the Lu(OH)CO3 shell. From the TEM image (Fig. 4D), it can be seen that the precursor consists of rough surface microspheres in the size range of 2.8 μm, which are similar to the SEM results. Furthermore, the core–shell structure can be clearly seen, which is due to the coated Lu(OH)CO3 layer on the surface of the PS spheres. The EDX results confirm the presence of carbon (C), oxygen (O), lutetium (Lu), and erbium (Er) elements in PS/Lu(OH)CO3:5 mol% Er3+ sample (Fig. 4E), and carbon (C), oxygen (O), lutetium (Lu), ytterbium (Yb), and thulium (Tm) elements in PS/Lu(OH)CO3:5 mol% Yb3+, 0.2 mol% Tm3+ sample (Fig. 4F), respectively (Si from the Si substrate).
 |
| | Fig. 4 (A) SEM and (B) TEM images of as-prepared PS spheres templates; (C) SEM, (D) TEM images of the uncalcined PS/Lu(OH)CO3 precursor. (E and F) EDX of the uncalcined PS/Lu(OH)CO3:5 mol% Yb3+, 0.2 mol% Tm3+, and PS/Lu(OH)CO3:5 mol% Er3+ samples. | |
Fig. 5 shows the morphology, microstructure and elemental composition of the hollow Lu2O3 microspheres after calcination at 800 °C. Fig. 5A shows the panoramic SEM image of the Lu2O3 sample, which indicates that the sample consists of a large number of well-dispersed hollow spheres. The result reveals that the PS spheres template essentially determines the morphology of the Lu2O3 products. From the enlarged SEM image, one can observe a small quantity of ruptured hollow spheres (Fig. 5B), which can provide evidence that the spheres are hollow structures. The rupture of the spheres may be caused by the release of gaseous carbon/hydrogen oxides and when the oxidation process of carbon spheres occurred in the calcination process. Moreover, the wall thickness of hollow spheres is estimated to be about 400 nm from the ruptured hollow spheres.
 |
| | Fig. 5 (A and B) SEM, (C) TEM, (D) HRTEM images of the hollow Lu2O3 microspheres. (E and F) EDX of Lu2O3:5 mol% Yb3+, 0.2 mol% Tm3+, and Lu2O3:5 mol% Er3+ samples. The inset of (D) is the corresponding SAED pattern. | |
To provide further insight into the hollow spheres, TEM measurements were also performed. The TEM image of the Lu2O3 sample exhibits the spherical morphology with good dispersion (Fig. 5C), agreeing well with the SEM images. The strong contrast between the dark edge and the pale center reveals the hollow nature of the spheres. The wall thickness of the shell is about 400 nm, which is in accordance with the SEM results (Fig. 5B). As disclosed by the corresponding HRTEM image (Fig. 5D), the interplanar distance between the adjacent lattice fringes is 0.300 nm. This plane can be indexed as the d spacing of the (222) plane of the cubic Lu2O3 phase (JCPDS no. 86-2475). The corresponding SAED pattern (inset in Fig. 5D) taken from a hollow sphere can be indexed as the (222), (400), (440), and (622) reflections of the cubic Lu2O3, which agrees well with the XRD results. The EDX results confirm the presence of oxygen (O), lutetium (Lu), and erbium (Er) elements in the La2O3:5 mol% Er3+ sample (Fig. 5E), and oxygen (O), lutetium (Lu), ytterbium (Yb), and thulium (Tm) elements in Lu2O3:5 mol% Yb3+, 0.2 mol% Tm3+ sample (Fig. 5F), respectively (Si from the Si substrate). It should be noted that the very weak carbon signal suggests that the PS core is nearly burned off by the heat treatment and Lu(OH)CO3 precursor has been converted to Lu2O3 during the calcination process, which is consistent with the XRD and FT-IR results. To evaluate the dispersion of the rare earth ions in the Lu2O3:15 mol% Yb3+/1 mol% Er3+ hollow spheres, high angle dark field (HAADF)-STEM imaging and elemental mapping were performed. From Fig. 6, we observe that the Lu, Yb, and Er rare earth ions are uniformly distributed over the entire hollow sphere.
 |
| | Fig. 6 HAADF-STEM images of (A) hollow Lu2O3:15 mol% Yb3+/1 mol% Er3+ microspheres, and the corresponding maps for (B) Lu, (C) Yb, and (D) Er. | |
In addition, it should be mentioned that the existence of other lanthanide ions in Lu2O3 host did not change the morphology of the products in our present work. The Lu2O3:RE3+ (RE3+ = 3 mol% Yb3+/0.08 mol% Tm3+, 1 mol% Er3+, 15 mol% Yb3+/1 mol% Er3+, Lu2O3:3 mol% Yb3+/0.2 mol% Tm3+/0.4 mol% Er3+) samples were prepared by a similar procedure except for using different doping lanthanide ions. The TEM images of these Lu2O3:Ln3+ samples are displayed in Fig. 7. It can be seen that all the samples have similar size and morphology, suggesting that small amounts of doping components have little influence on the final products.
 |
| | Fig. 7 TEM images of (A) Lu2O3:3 mol% Yb3+/0.08 mol% Tm3+, (B) Lu2O3:1 mol% Er3+, (C) Lu2O3:15 mol% Yb3+/1 mol% Er3+, (D) Lu2O3:3 mol% Yb3+/0.2 mol% Tm3+/0.4 mol% Er3+ samples. | |
3.2 Formation process
According to the above XRD, TG-DTA, FT-IR, SEM, and TEM analysis, the formation mechanism of the hollow Lu2O3 microspheres depends on a series of chemical and structural transformations, as shown in Scheme 1. In the initial stage, PS sphere templates were prepared by dispersion polymerization. In the second step, the core–shell structured precursor was fabricated by the precipitation of rare earth ions on the surface of spherical PS templates using urea as the precipitating agent. FT-IR confirms the presence of abundant hydroxyl groups on the PS spheres, which is beneficial to the adsorption of Lu3+, OH−, and CO32− (released from precipitator agent urea). The adsorbed components may easily precipitate on the surface of the PS spheres, then form amorphous Lu(OH)CO3 nucleus and further grow into Lu(OH)CO3 nanoparticles. Finally, the PS sphere templates were burned out and Lu(OH)CO3 particles were decomposed into Lu2O3 crystals by the heating treatment; thus uniform hollow Lu2O3 spheres were obtained. The main reaction process for the formation of hollow Lu2O3 spheres could be represented as follows.| | | CO(NH2)2 + H2O → CO2 + 2NH3 | (1) |
| | | NH3 + H2O → NH4+ + OH− | (2) |
| | | Lu3+ + 3OH− + CO2 → Lu(OH)CO3 + H2O | (3) |
| | | 2Lu(OH)CO3 → Lu2O3 + H2O + 2CO2 | (4) |
3.3 Photoluminescence properties
It is well-known that lutetium oxide (Lu2O3) is an excellent host lattice for down- and up-conversion luminescence of various optically active rare earth ions due to its favorable physical properties. The crystalline Lu2O3 belongs the cubic system with space group Ia(206) whose structure is similar to that of Y2O3. This type of structure contains two crystallographically different cation sites with C2 and S6 symmetry, respectively. The doped lanthanide ions are assumed to occupy these two types of sites in a statistical way.47,48 In the present study, we report on the design of white UC luminescence in Yb3+/Tm3+/Er3+-doped Lu2O3 samples under diode laser excitation of 980 nm.
 |
| | Scheme 1 Schematic illustration for the possible formation mechanism of the core–shell structured PS/Lu(CO)CO3 precursor, and final hollow Lu2O3 spheres. | |
Fig. 8 shows the photoluminescence (PL) spectra of Lu2O3:RE3+ samples under 980 nm laser diode (LD) excitation. Upon excitation at 980 nm, the Lu2O3:3 mol% Yb3+/0.08 mol% Tm3+ sample shows a strong blue luminescence (inset in Fig. 8A). The up-conversion emission spectrum of Lu2O3:3 mol% Yb3+/0.08 mol% Tm3+ sample is shown in Fig. 8A. In the emission spectrum, the intense blue emissions at 477 nm, corresponding to the Tm3+ 1G4 → 3H6 transition, and a much weaker emission at 654 nm, assigned to Tm3+ 1G4 → 3F4 transition, are observed and are clearly dominated by the Tm3+ 1G4 → 3H6 transition. Fig. 8B shows the bright green emission (inset in Fig. 8B) of Lu2O3:1 mol% Er3+ sample excited at 980 nm. Two primary bands in the green emission region maximized at 541 and 566 nm are assigned to the Er3+ 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions, respectively, and a weak band appearing near 665 nm is ascribed to the Er3+ 4F9/2 → 4I15/2 transition. However, the green emission of Lu2O3:1 mol% Er3+ sample changes greatly when Yb3+ was co-doped with Er3+ in Lu2O3 sample. The Lu2O3:15 mol% Yb3+/1 mol% Er3+ sample shows a strong red light emission (inset in Fig. 8C) under 980 nm LD excitation. Fig. 8C shows the emission spectrum of Lu2O3:15 mol% Yb3+/1 mol% Er3+ sample, including mainly bright red emission of Er3+ near 663 nm corresponding to Er3+ 4F9/2 → 4I15/2 transition, together with the very weak emissions near 540 and 565 nm assigned to the Er3+ 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions respectively.
 |
| | Fig. 8 Up-conversion emission spectra of Lu2O3:RE3+ samples under 980 nm LD excitation. (A) Lu2O3:3 mol% Yb3+/0.08 mol% Tm3+, (B) Lu2O3:1 mol% Er3+, and (C) Lu2O3:15 mol% Yb3+/1 mol% Er3+. Their luminescence photographs are shown as insets. | |
It is known that co-doping does not increase the efficiency of PL, but it also induces up-conversion PL between the donor and acceptor ions in some cases.49 In our present work, Yb3+ was chosen as the co-dopant with Tm3+ or Er3+, because it possesses a large absorption cross section at 980 nm, and energy transfer occurs as a result of the large spectral overlap between the Yb3+ emission 2F5/2 → 2F7/2 and the Tm3+ absorption 3H5 ← 3H6 or the Er3+ absorption 4I11/2 ← 4I15/2 bands. In addition, Yb3+ has a much longer excited-state lifetime.49 For blue-light emission of the sample of Lu2O3:3 mol% Yb3+/0.08 mol% Tm3+ sample, up-conversion from Tm3+ ions is due to energy transfer (ET) processes, because it has no ground or excited-state absorption (GSA or ESA) that matches the 980 nm photon. The population of 1G4 is accomplished by the three-step sequential ET from the excited Yb3+ to Tm3+. First, absorption of pump photons populates the long-lived 2F5/2 level in Yb3+. Nonresonant ET from Yb3+ to Tm3+ takes place upon excitation to the 3H5 level in Tm3+. This population relaxes rapidly to the 3F4 level by nonradiative multiphonon decay. A second nonresonant ET from Yb3+ to Tm3+ populates the 3F2 and 3F3 levels, and subsequently multiphonon decay occurs leading to population of 3H4 level. A third ET finally populates the 1G4 level, then the emissions at 477 and 490 nm (1G4 → 3H6) (most strong) and at 654 nm (1G4 → 3F4) emanate (Fig. 7).50 The mechanism of the up-converted green emission of the Lu2O3:1 mol% Er3+ sample has been established by others.50,51 The excitation wavelength from 980 nm LD matches the absorption transition between the ground state, 4I15/2, and the excited level 4I11/2 (GSA). After first-level excitation, the same wavelength laser pumps the excited atom from the 4I11/2 to the 4F7/2 level (ESA). Subsequent nonradiative relaxation populates the 2H11/2, 4S3/2, and 4F9/2 levels. Finally, radiant transitions from these levels yield the emissions at 540 and 565 nm (2H11/2, 4S3/2 → 4I15/2) (most strong) and at 662 nm (4F9/2 → 4I15/2), respectively (Fig. 9). For red light emission of the Lu2O3:15 mol% Yb3+/1 mol% Er3+ sample, the mechanism of up-conversion emission is predominantly due to the two-step ET from the excited Yb3+ to Er3+ and little contribution from Er3+ ground/excited-state absorption (GSA/ESA), because Yb3+ ions have a much larger absorption cross-section and ion concentration than the Er3+ ions. At first, Yb3+ ions are excited from the 2F7/2 to the 2F5/2 level by 980 nm laser, and then an excited Yb3+ transfers its energy to Er3+ (4I11/2). During the lifetime of the 4I11/2 level, a second Yb3+ ion transfers its energy again, resulting in the population of the 4F7/2 state of Er3+. Relaxation from the 4F7/2 state and some other energy-transfer processes populate the 2H11/2, 4S3/2, and the 4F9/2 states, which results in the observed emission spectra, namely, 662 nm corresponding to 4F9/2 → 4I15/2 (most strong) and 540/565 nm corresponding to 2H11/2/4S3/2 → 4I15/2, in Fig. 9.
 |
| | Fig. 9 Energy level diagrams of the Yb3+, Er3+, and Tm3+ ions and the proposed UC emission mechanism. | |
Fig. 10 shows the PL spectra of different concentrations of Yb3+ in Er3+-doped Lu2O3 samples under 980 nm LD excitation. Using a fixed Er3+ concentration (1 mol%) and upon variation of the Yb3+ concentration (from 0 to 15 mol%), it was found that the green emission intensity decreased, while the red emission intensity increased (Fig. 10). This case (the green emission decreases, while the red emission enhances) can be caused by fewer Er3+ ions holding at the green-emitting levels of 2H11/2/4S3/2 and more Er3+ ions holding at the red-emitting level of 4F9/2. One of the most likely reasons is that introduction of an increasing amount of Yb3+ dopants in the Lu2O3 host lattice would decrease the ⋯Yb3+⋯Er3+⋯ interatomic distance and thus facilitate two energy back-transfer processes 4S3/2 (Er3+) + 2F7/2 (Yb3+) → 4I13/2 (Er3+) + 2F5/2 (Yb3+) and 4F7/2 (Er3+) + 2F7/2 (Yb3+) → 4I11/2 (Er3+) + 2F5/2 (Yb3+) efficiently.52 The former energy back-transfer should subsequently suppress the population in excited levels of 4S3/2 (2H11/2), resulting in the decrease of green-light emission (2H11/2/4S3/2 → 4I15/2). At the same time, the energy back-transfer leads to the saturation of the 4I13/2 (Er3+) state and then excited Yb3+ ions transfer their energy to Er3+ ions through the energy-transfer process 4I13/2 (Er3+) + 2F5/2 (Yb3+) → 4F9/2 (Er3+) + 2F7/2 (Yb3+), which can directly populate the 4F9/2 (Er3+) level, producing the enhancement of red (4F9/2 → 4I15/2) emission. In addition, the populated 4I13/2 level might be excited to the 4F9/2 red-emitting level in Er3+ ions by the cross-relaxation process 4I13/2 + 4I11/2 → 4F9/2 + 4I15/2. The latter energy back-transfer process should depopulate the excited 4F7/2 (Er3+) level at higher Yb3+ concentrations. This results in a smaller population of the 2H11/2 and 4S3/2 green emitting levels and causes a decrease in the green emission intensity. Another possible route is the higher efficiency of the cross-relaxation in Er3+ ions, that is, 4F7/2 + 4I11/2 → 4F9/2 + 4F9/2, which can also directly populate the 4F9/2 red-emitting level and indirectly depopulate the 2H11/2 and 4S3/2 green emitting levels. Fig. 9 shows these analyses for the change in red and green emission intensities upon increasing the Yb3+ ion concentration. Fig. 11 shows the UC luminescence spectra and photographs of Lu2O3:x mol% Er3+ as a function of Er3+-doping concentration (x). At low concentrations of Er3+ (x < 1), the 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions of Er3+ ions dominate and thus show green emission. With the increase of x, the luminescence intensity of 2H11/2, 4S3/2 → 4I15/2 transitions gradually decreases while the red emission intensity of the 4F9/2 → 4I15/2 transition gradually increases. The result may be caused by the energy transfer of Er3+ → Er3+ ions (cross-relaxation effect).32 The total luminescence intensity decreases with the continued increase of Er3+ concentration due to the concentration quenching effect.53
 |
| | Fig. 10 Up-conversion PL emission spectra of different concentrations of Yb3+ in 1 mol% Er3+-doped Lu2O3 samples under 980 nm LD excitation. (A) Lu2O3:1 mol% Er3+, (B) Lu2O3:5 mol% Yb3+/1 mol% Er3+, (C) Lu2O3:10 mol% Yb3+/1 mol% Er3+, and (D) Lu2O3:15 mol% Yb3+/1 mol% Er3+. | |
 |
| | Fig. 11 Up-conversion PL emission spectra of Lu2O3:x mol% Er3+ under 980 nm LD excitation as a function of Er3+-doping concentration (x). (A)–(G) are 0.5, 1, 2, 3, 4, 5 and 6, respectively. | |
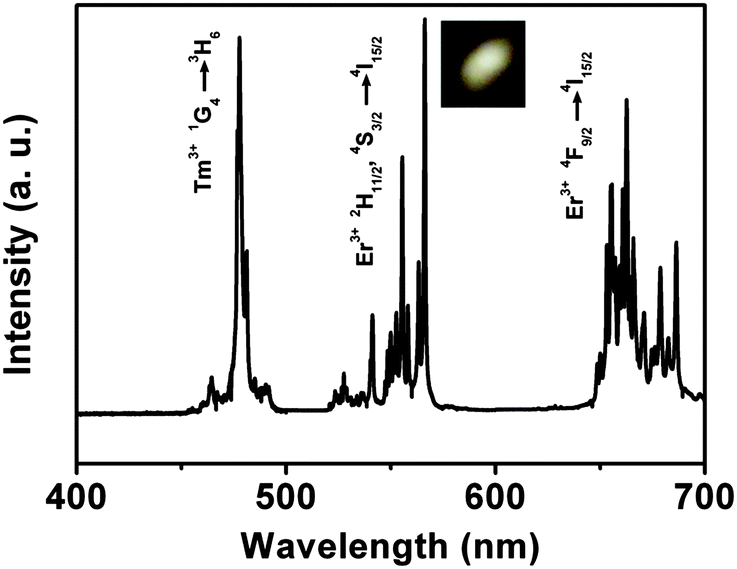
Based on the generation of red, green and blue emissions in the different doped Lu2O3:RE3+ samples, it is possible to produce the luminescence with a wide spectrum of colors, including white, by appropriate adjustment of the doping of Yb3+, Tm3+, and Er3+ in the present Lu2O3 samples. To demonstrate this idea, we prepared a Lu2O3 sample doped with 3 mol% Yb3+/0.2 mol% Tm3+/0.4 mol% Er3+. The UC emission spectrum of the sample is shown in Fig. 12. As compared with Fig. 8 and 10, the blue, green, and red bands centered at 477/490, 540/565, and 662 nm, respectively, in Fig. 12 can be easily assigned to the intra-4f electronic transitions 1G4 → 3H6 of Tm3+ ions, 2H11/2, 4S3/2 → 4I15/2, and 4F9/2 → 4I15/2 of Er3+ ions, respectively. The sample shows nearly equal intensities of blue, green, and red emissions, resulting in the production of bright white light as exhibited by the photograph in the inset of Fig. 12. The chromaticity coordinate of the 3 mol% Yb3+/0.2 mol% Tm3+/0.4 mol% Er3+ triply doped Lu2O3 sample is calculated to be about x = 0.3478 and y = 0.3143, which falls exactly within the white region of 1931 CIE diagram (Fig. 13) and is very close to the standard equal energy white light illuminate (x = 0.33 and y = 0.33), pointing to their potential use as a white light source under 980 nm LD excitation.
 |
| | Fig. 12 Up-conversion PL emission spectrum of the Lu2O3:3 mol% Yb3+/0.2 mol% Tm3+/0.4 mol% Er3+ sample under 980 nm LD excitation. The inset of the figure shows the corresponding luminescent photographs of the Lu2O3:3 mol% Yb3+/0.2 mol% Tm3+/0.4 mol% Er3+ sample. | |
 |
| | Fig. 13 Calculated CIE chromaticity coordinates for the white light luminescence in the Lu2O3:3 mol% Yb3+/0.2 mol% Tm3+/0.4 mol% Er3+ sample. | |
4. Conclusions
The well-dispersed Lu2O3 hollow spheres have been successfully fabricated via a template-directed method. The precursor was first synthesized by a precipitation technique with PS spheres as the template. Subsequently, the template was removed and the Lu2O3 hollow spheres were obtained during a calcination process. Furthermore, we have studied the Lu2O3:Yb3+/Tm3+/Er3+ samples UC PL properties under 980 nm LD excitation at room temperature. The red, green, and blue (RGB) up-conversion luminescence assigned to erbium and thulium ions both excited by means of Er3+ ground/excited-state absorption (GSA/ESA) and energy transfer from Yb3+ ions was observed. By precise control of the emission intensity balance through control of different combinations of lanthanide dopants and dopant concentration, white light was experimentally shown with the simultaneous generation of fluorescence with nearly equal intensities at the wavelengths of the three primary colors (RGB) in a single sample and by using a single near-infrared excitation source (980 nm). The calculated chromaticity coordinates (x = 0.3478, y = 0.3143) fall exactly within the white region of 1931 CIE diagram, which is very close to the standard equal energy white light illuminate (x = 0.33, y = 0.33). Because of abundant luminescent colors from RGB to white in Lu2O3:Yb3+/Er3+/Tm3+ samples under 980 nm LD excitation, they can potentially be used as fluorophores in the fields of color displays, backlights, UC lasers, photonics, and biomedicine.
References
- A. P. Alivisatos, Science, 1996, 271, 933 CAS.
-
K. J. Klabunde, Nanoscale Materials in Chemistry, Wiley-Interscience, New York, 2001 Search PubMed.
- H. S. Im, U. Jeong and Y. Xia, Nat. Mater., 2005, 4, 671 CrossRef PubMed.
- X. Feng, L. Yang, N. Zhang and Y. Liu, J. Alloys Compd., 2010, 506, 728 CrossRef CAS PubMed.
- X. Xie, H. Yang, F. Zhang, L. Li, J. Ma, H. Jiao and J. Zhang, J. Alloys Compd., 2009, 477, 90 CrossRef CAS PubMed.
- X. L. Xu and S. A. Asher, J. Am. Chem. Soc., 2004, 126, 7940 CrossRef CAS PubMed.
- Y. Li, W. Li, S. Chou and J. Chen, J. Alloys Compd., 2008, 456, 339 CrossRef CAS PubMed.
- A. N. Zelikin, Q. Li and F. Caruso, Angew. Chem., Int. Ed., 2006, 45, 7743 CrossRef CAS PubMed.
- F. Caruso and R. A. Caruso, Science, 1998, 282, 1111 CrossRef CAS.
- S. W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642 CrossRef CAS PubMed.
- H. Liang, H. Zhang, J. Hu, Y. Guo, L. Wan and C. Bai, Angew. Chem., Int. Ed., 2004, 43, 1540 CrossRef CAS PubMed.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 3827 CrossRef CAS PubMed.
- Y. G. Sun, B. T. Mayers and Y. N. Xia, Nano Lett., 2002, 2, 481 CrossRef CAS.
- J. Bao, Y. Liang, Z. Xu and L. Si, Adv. Mater., 2003, 15, 1832 CrossRef CAS.
- B. Z. Putlitz, K. Landfester, H. Fischer and M. Antonietti, Adv. Mater., 2001, 13, 500 CrossRef.
- D. Zhang, L. Qi, J. Ma and H. Cheng, Adv. Mater., 2002, 14, 1499 CrossRef CAS.
- Q. Peng, Y. Dong and Y. Li, Angew. Chem., Int. Ed., 2003, 42, 3027 CrossRef CAS PubMed.
- Z. Z. Yang, Z. W. Niu, Y. F. Lu, Z. B. Hu and C. C. Han, Angew. Chem., Int. Ed., 2003, 42, 1943 CrossRef CAS PubMed.
- P. Wang, D. Chen and F. Q. Tang, Langmuir, 2006, 22, 4832 CrossRef CAS PubMed.
- M. Chen, L. M. Wu, S. X. Zhou and B. You, Adv. Mater., 2006, 18, 801 CrossRef CAS.
- M. Agrawal, A. Pich, S. Gupta, N. E. Zafeiropoulos, P. Simon and M. Stamm, Langmuir, 2008, 24, 1013 CrossRef CAS PubMed.
- I. D. Hosein and C. M. Liddell, Langmuir, 2007, 23, 2892 CrossRef CAS PubMed.
- T. Suehiro, N. Hirosaki, R. J. Xie and T. Sato, Appl. Phys. Lett., 2009, 95, 051903 CrossRef.
- Q. Mu and Y. Wang, J. Alloys Compd., 2011, 509, 396 CrossRef CAS PubMed.
- G. Jia, H. P. You, K. Liu, Y. Zheng, N. Guo and J. Jia, Chem.–Eur. J., 2010, 16, 2930 CrossRef CAS PubMed.
- Y. Liu, P. Yang, W. Wang, H. Dong and J. Lin, CrystEngComm, 2010, 12, 3717 RSC.
- M. Yada, M. Mihara, S. Mouri, M. Kuroki and T. Kijima, Adv. Mater., 2002, 14, 309 CrossRef CAS.
- S. Polizzi, S. Bucella, A. Speghini, F. Vetrone, R. Naccache, J. C. Boyer and J. A. Capobianco, Chem. Mater., 2004, 16, 1330 CrossRef CAS.
- J. Lu, K. Takaichi, T. Uematsu, A. Shirakawa, M. Musha, K. Ueda, H. Yagi, T. Yanagitani and A. A. Kaminskii, Appl. Phys. Lett., 2002, 81, 4324 CrossRef CAS.
- O. Guillot-Noël, B. Bellamy, B. Viana and D. Vivien, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 1668 Search PubMed.
- G. Jia, Y. Zheng, K. Liu, Y. Song, H. You and H. Zhang, J. Phys. Chem. C, 2009, 113, 153 CAS.
- J. Yang, C. M. Zhang, C. Peng, C. X. Li, L. L. Wang, R. T. Chai and J. Lin, Chem.–Eur. J., 2009, 15, 4649 CrossRef CAS PubMed.
- J. Yang, C. Li, Z. Quan, C. Zhang, P. Yang, Y. Li, C. Yu and J. Lin, J. Phys. Chem. C, 2008, 112, 12777 CAS.
- R. Si, Y. W. Zhang, H. P. Zhou, L. D. Sun and C. H. Yan, Chem. Mater., 2007, 19, 18 CrossRef CAS.
- J. A. Capobianco, F. Vetrone, J. C. Boyer, A. Speghini and M. Bettinelli, Opt. Mater., 2002, 19, 259 CrossRef CAS.
- A. J. Paine and W. J. Luymes McNulty, Macromolecules, 1990, 23, 3104 CrossRef CAS.
- Y. Yang, Y. Chu, Y. Zhang, F. Yang and J. Liu, J. Solid State Chem., 2006, 179, 470 CrossRef CAS PubMed.
- Y. Chen and J. X. Lu, J. Porous Mater., 2012, 19, 289 CrossRef CAS.
- G. Jia, H. You, Y. Zheng, K. Liu, N. Guo and H. Zhang, CrystEngComm, 2010, 12, 2943 RSC.
- J. G. Li, X. D. Li, X. D. Sun, T. Ikegami and T. Ishigaki, Chem. Mater., 2008, 20, 2274 CrossRef CAS.
- J. G. Li, X. D. Li, X. D. Sun and T. Ishigaki, J. Phys. Chem. C, 2008, 112, 11707 CAS.
- C. L. Wang, J. T. Yan, X. J. Cui and H. Y. Wang, J. Colloid Interface Sci., 2011, 354, 94 CrossRef CAS PubMed.
- F. He, P. P. Yang, D. Wang, C. X. Li, N. Niu, S. L. Gai and M. L. Zhang, Langmuir, 2011, 27, 5616 CrossRef CAS PubMed.
- Z. H. Xu, P. A. Ma, C. X. Li, Z. Y. Hou, X. F. Zhai, S. S. Huang and J. Lin, Biomaterials, 2011, 32, 4161 CrossRef CAS PubMed.
- Z. H. Xu, Y. Gao, T. Liu, L. M. Wang, S. S. Bian and J. Lin, J. Mater. Chem., 2012, 22, 21695 RSC.
- P. P. Yang, S. L. Gai, Y. C. Liu, W. X. Wang, C. X. Li and J. Lin, Inorg. Chem., 2011, 50, 2182 CrossRef CAS PubMed.
- E. Zych, M. Karbowiak, K. Domagala and S. Hubert, J. Alloys Compd., 2002, 341, 381 CrossRef CAS.
- J. C. Park, H. K. Moon, D. K. Kim, S. H. Byeon, B. C. Kim and K. S. Suh, Appl. Phys. Lett., 2000, 77, 2162 CrossRef CAS.
- G. K. Das and T. Y. Tan, J. Phys. Chem. C, 2008, 112, 11211 CAS.
- D. Matsuura, Appl. Phys. Lett., 2002, 81, 4526 CrossRef CAS.
- A. Patra, C. S. Friend, R. Kapoor and P. N. Prasad, J. Phys. Chem. B, 2002, 106, 1909 CrossRef CAS.
- F. Wang and X. G. Liu, J. Am. Chem. Soc., 2008, 130, 5642 CrossRef CAS PubMed.
- Y. Zhang, J. H. Hao, C. L. Mak and X. H. Wei, Opt. Express, 2011, 19, 1824 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.