DOI:
10.1039/C3NJ00659J
(Paper)
New J. Chem., 2014,
38, 155-162
Facile synthesis of transparent and fluorescent epoxy–CdSe–CdS–ZnS core–multi shell polymer nanocomposites
Received
(in Montpellier, France)
18th June 2013
, Accepted 4th October 2013
First published on 10th October 2013
Abstract
A facile and environmentally benign approach for the synthesis of highly transparent and fluorescent CdSe–CdS–ZnS core–multi-shell polymer nanocomposites is presented. The CdSe–CdS–ZnS core–multi-shell quantum dots (QDs) were prepared via a continual precursor injection and phosphine free method in paraffin liquid and oleic acid without a protective atmosphere. The as-prepared core–multi-shell QDs were dispersed directly in an epoxy polymer matrix via a melt mixing technique. The QDs showed better dispersibility and good optical properties in the epoxy matrix. The transmission electron microscopy (TEM) images showed that the as-synthesized QDs are small, spherical and are well dispersed inside the polymer matrix without any change in morphology. It was found that the nanocomposite filled with yellow-emitting QDs had more transparency compared to the neat epoxy. The luminescence of the neat polymer shifted from the blue region to the yellow region in the nanocomposite. The fluorescent lifetime analysis of the as-prepared core–multi shell and the polymer nanocomposite showed a decrease compared to the core while the tensile measurements showed an increase in the tensile properties of the nanocomposite in comparison with the neat polymer.
1. Introduction
Semiconductor nanocrystals have generated a lot of interest among researchers in the past decades due to their wide range of applications in photonics, electronics and optoelectronics.1–4 In addition, extensive experimental and theoretical efforts have been made in the syntheses as well as tuning the size and properties of these materials. CdSe nanocrystals have been considered as an important material because of their high emission efficiency and size-tunable photoluminescence (PL) across the visible spectrum.5–9 Several studies on the syntheses of CdSe using various synthetic routes have been reported. However, most of these methods involved the use of phosphines and alkylphosphines (such as trioctylphosphine (TOP) and tributylphosphine (TBP)) as the coordinating solvent or ligand to form complex precursors with Se. Though high quality materials have been produced via these techniques, the starting materials are hazardous, expensive and unfriendly to the environment. Thus, they are good for laboratory setting but undesirable for commercial exploitation. Although some stable, inexpensive, low toxic and environmentally benign precursors or solvents have been used to synthesise core–shell quantum dots, the use of a direct, organic, phosphine-free synthetic route for CdSe based core–multi-shell structures with high optical quality is still desirable. In the quest for greener synthesis, coordinating and non-coordinating solvents such as 1-octadecene and liquid paraffin which are environmentally friendly have been used to replace the hazardous coordinating solvents like TOP and TOPO.10–14 Recently, Wang et al. have reported the first synthesis of CdSe–CdS–ZnS core multi-shell QDs using a phosphine-free one pot-injection method in paraffin liquid.15 This is an environmentally benign and simple method compared to the conventional “two step” synthetic route, which is laborious and time-consuming, and may strongly affect the properties of core QDs.
Recently the focus has been shifted from syntheses to applications of these QD materials. This includes fabrication of devices, such as solar cells, light emitting diodes (LEDs), and so on.16–21 In the fabrication of devices, the quantum dots need to be well dispersed and must be compatible with the supporting matrix while transferring it into a composite.22–25 In search of suitable matrices for device fabrication, polymers have been found to be ideal candidates. The choice of polymers as the matrix is an attractive approach because they are cheap, flexible, and can be easily processed. Moreover, some polymers can be micro- and nanopatterned by different lithographic techniques such as nanoimprint, electron beam, and ultraviolet lithography which are of significant importance for the fabrication of photonic nanostructures and more complex devices.26 One of the promising approaches for the fabrication of devices is the incorporation of nanoparticles into a suitable polymer matrix to form a nanocomposite. These multicomponent materials usually possess the combined novel properties of both the nanoparticles and the polymer matrix. Effective dispersion of the QDs inside the polymer matrices is one of the key factors which determine the optical properties of the polymer nanocomposite. Due to the organophobic surface of the QDs, they tend to agglomerate inside the polymer matrix causing light scattering inside the matrix.27,28 Various techniques have been adopted to increase the dispersion of fillers inside the polymer matrices such as modifying the QD surface, using nanoplatelets for effective dispersion, etc.29,30 In some cases these surface modifications increase the particle size which causes low transparency and lower luminescence efficiency.31 Transparent epoxy resins are mostly employed as encapsulants in LED solid-state lighting due to their high transparency, high glass transition temperature and low water absorption.32
In the present work, we synthesised oleic acid coated CdSe–CdS–ZnS core–multi shell nanocomposites using the diglycidyl ether of bisphenol A as the polymer. The core–multi shell QDs were synthesised by the continual precursor injection route while the epoxy nanocomposites were produced by the melt mixing technique. The presence of the carboxyl groups in the curing reaction of the epoxy matrix improved the stability of the QDs and their homogenous dispersion in the epoxy matrix. The resulting epoxy core–shell nanocomposites were transparent when compared to the neat epoxy matrix and showed improvement in their mechanical properties. The fluorescent lifetime analysis studies of the as-synthesized core–multi shells and their polymer nanocomposites were also conducted.
2. Experimental procedures
2.1 Materials
Cadmium oxide (CdO; 99.5%) was purchased from Lobachemie. Selenium powder (Se; 99.9%) was obtained from Sigma Aldrich, zinc acetate dihydrate (Zn(CH3COO)2·2H2O, analytical reagent), cadmium acetate dihydrate (Cd(CH3COO)2·2H2O, analytical reagent), oleic acid (OA analytical grade), sodium sulphide nonahydrate (Na2S·9H2O) and toluene were purchased from Merck. Paraffin liquid (chemical grade with boiling point higher than 300 °C) was purchased from Nice chemicals. The polymer used was diglycidyl ether of bisphenol A and the curing agent is 4,4′-diaminodiphenyl methane (DDM), both were supplied by Atul India Limited.
2.2 Preparation of CdSe–CdS–ZnS core multi shells
The CdSe–CdS–ZnS multi shell was prepared by the Wang et al. procedure with slight modification.26 In a typical synthesis, 0.2 mmol Se was dissolved in 18 mL of paraffin which served as the solvent, in a three neck flask at 220 °C under vigorous stirring. In another flask, CdO (2 mmol) was dissolved in a mixture of 6.0 mmol oleic acid (2.0 mL) and paraffin liquid (8.0 mL) at 160 °C. The oleic acid was used to dissolve CdO powder and form homogeneous Cd solution. It also acts as the capping ligand in the formation of the NCs. This cadmium precursor solution (2 mL) was quickly injected into the Se solution, and the growth temperature was kept at 220 °C. Aliquots were taken at different time intervals and immediately injected into toluene to stop any further growth. To add the CdS layer to the core, the temperature of the core CdSe solution was quickly cooled to 50 °C. Then 0.5 mmol Na2S·9H2O as the sulphur precursor was added into the solution and heated to 140 °C under vigorous stirring. The Cd precursor (2 mL) produced by dissolving cadmium acetate dihydrate in liquid paraffin was injected dropwise into the reaction to produce the CdS shell. Then the solution was cooled to 100 °C. After 30 min of reaction, another portion of Na2S·9H2O (0.5 mmol) was added into the solution, and the solution was heated to 160 °C under vigorous stirring. Then the Zn precursor (3 mL) prepared by dissolving zinc acetate dihydrate in liquid paraffin was injected dropwise into the solution and the mixture was cooled to 100 °C and kept for 90 min for the ZnS shell growth. Aliquots were taken at different reaction times and diluted with toluene for further analysis. The experimental schematic diagram is shown in Fig. 1.
 |
| | Fig. 1 Schematic representation of preparation of CdSe–CdS–ZnS core multi shell QDs. | |
2.3 Preparation of CdSe–CdS–ZnS core–shell polymer nanocomposites
As-synthesized CdSe–CdS–ZnS core shell QDs were dispersed in the epoxy polymer matrix by the melt mixing technique. A solution of 0.30 weight percentage core–multi-shell QDs was mixed with the polymer by ultra-sonication for 1 hour at room temperature. Then the curing agent DDM was added at 80 °C and stirred for 15 min. The mixture was poured into a preheated metal mould and cured at 100 °C for 5 hours in a hot air oven to produce the polymer nanocomposite. Neat epoxy resin was cured under the same reaction conditions for comparison.
2.4 Characterization
FTIR spectra were recorded using a Nicolet-Nexus 670 FTIR spectrophotometer. UV-visible absorption and photoluminescence spectra were recorded using a SHIMDTH UV2401PC spectrophotometer and SHIMDTH RF-5301PC, respectively, at room temperature. The photoluminescence quantum yield (PL QY) was obtained by comparison with standard rhodamine B in methanol and using data derived from the luminescence and the absorption spectra in the following equation: ϕ = ϕ′(I/I′)(A′/A)(n/n′)2. In this equation, I (sample) and I′ (standard) are the integrated emission peak areas, upon 400 nm excitation; A (sample) and A′ (standard) are the absorptions at 400 nm; n (sample) and n′ (standard) are the refractive indices of the solvents; and ϕ and ϕ′ are the PL QY for the sample and the standard, respectively.33 A JEOL JEM-3010 electron microscope operating at 200 kV was used for TEM and high-resolution TEM. The samples for TEM and HRTEM analyses were prepared by putting a drop of toluene solution of the core–shells onto an amorphous carbon substrate supported on a copper grid and then allowing the solvent to evaporate at room temperature. For the nanocomposite, the samples were cut into ultrathin layers using a microtone cutter. The lifetime measurements of QD and the QD–epoxy nanocomposite were performed using a Horiba Jobin Yvon IBH picosecond lifetime analyzer. The laser used was of second harmonics within the range of 350–450 nm wavelength. Tensile measurements were carried out using a Tinius Olsen H50KT.
3. Results and discussion
The synthetic method used here was a phosphine-free continual precursor injection method without any protecting atmosphere. In this reaction, the core CdSe QDs synthesized were not separated from the excess precursors for the next coating step. This is to avoid the inappropriate changes which could affect the quality of the QDs if exposed to air and moisture. The purification of the sample was performed only after the complete formation of the desired multi core shell. The addition of each layer to the core caused an increase in the size of the particles. The nanoparticles obtained were of high quality and monodispersed with highly desirable absorption and emission features. During the entire reaction process, the growth of the particles was clearly evident from both the colour change and the shift of absorption spectra to longer wavelengths as the reaction time increased.
The absorption and emission spectra of the CdSe–CdS–ZnS at different reaction times and different stages of growth are shown in Fig. 2. The absorption and emission peaks were red-shifted as the growth time and the shell coating increased indicating an increase in particle size.34 The absorption band-edges as calculated using the direct band gap method35 are shown in Table 1. The sharp absorption features are indicative of particles with narrow size distributions. The sharpness of the excitonic peak diminishes slightly as the coating increased. This broadening has been attributed to the increase in the particle size.4,36 The particle diameters as-calculated using the Yu et al. equation37 are in the range of 2.49 nm to 3.36 nm (Table 1). The emission maxima of all the as-synthesized particles (Fig. 2B) are red shifted in relation to the corresponding absorption maxima and exhibit band-edge luminescence for excitation at 400 nm with the particles emitting in the green-orange window as the coating layer increased. A red-shift in the PL and absorbance spectra observed for the CdSe–CdS QDs and the CdSe–CdS–ZnS QDs compared with CdSe core QDs has been attributed to the partial leakage of the excitons into the shell matrix and the formation of the core–shell and core–multi-shell QDs rather than the formation of alloyed QDs.26,38,39 The red-shift after the formation of the ZnS shell is much smaller than that formed after the CdS shell indicating a slower growth rate during the second coating process. In addition, the PL intensity of the CdSe–CdS core–shell QDs was obviously superior to the CdSe core QDs, and it was further improved by further coating with the ZnS shell. The fluorescence peak position remained constant as the excitation wavelength was varied indicating that the origin of the emitting state is similar in all species. This observation has also been reported by Soloviev et al.40 and Wageh et al.41 and has been attributed as strong evidence of the purity of the samples. The full width at half maximum (FWHM) and PL quantum yield of the CdSe–CdS–ZnS core–multi shell was found to be 45 nm and 70%, respectively.
 |
| | Fig. 2 The UV-vis absorption spectra (A) and photoluminescence spectra (B) of the CdSe–CdS–ZnS core–multi shell QDs at different reaction times and stages of growth. | |
Table 1 Particle size and the band edge values of the core–multi shells
| Sample code |
Absorption maxima (nm) |
Emission maxima (nm) |
Band gap (eV) |
Particle size (nm) |
| The particle diameters were calculated using the Yu et al. equation37D = (1.6122 × 10−9)λ4 − (2.6575 × 10−6)λ3 + (1.6242 × 10−3)λ2 − (0.4277)λ + (41.57). Where D (nm) is the size of the given nanocrystal sample. λ (nm) is the wavelength of the first excitonic absorption peak which comes from the UV/vis spectrum of QDs. |
| CdSe 2 min |
519 |
524 |
3.50 |
2.49 |
| CdSe 5 min |
528 |
532 |
3.30 |
2.62 |
| CdSe–CdS 10 min |
558 |
564 |
3.00 |
3.08 |
| CdSe–CdS 30 min |
562 |
564 |
2.90 |
3.20 |
| CdSe–CdS–ZnS 10 min |
564 |
569 |
2.80 |
3.29 |
| CdSe–CdS–ZnS 30 min |
566 |
571 |
2.70 |
3.36 |
Fig. 3 shows the FTIR analysis of the as-synthesized QDs, the neat polymer and the polymer core–shell nanocomposite. The core–shell spectrum (Fig. 2A) showed a peak at 3029 cm−1 assigned to the –C–H stretching vibration of paraffin. Two peaks at 727 cm−1 and 1478 cm−1 correspond to –CH2 deformation and –C–H bending vibration of the paraffin, respectively. The peak at 687 cm−1 is attributed to the –C–H out of plane bending in paraffin while the peak at 1615 cm−1 to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration. The –CH2 symmetric and asymmetric stretching of the oleic acid appeared at 2833 cm−1 and 2921 cm−1, respectively. The –CO stretching from the –COOH group in oleic acid appeared at 1751 cm−1.41 The peak at 1459 cm−1 is attributed to the in-plane stretching of the O–H group of –COOH and the peak at 1390 cm−1 corresponds to the –C–O stretching in oleic acid.42 The presence of these peaks confirmed the successful capping of the QDs by the oleic acid. In the spectrum of the neat epoxy (Fig. 3B), two characteristic absorptions of the oxirane ring are observed in the range between 4000 cm−1 and 400 cm.−1 The first one near 900 cm−1 is attributed to the –C–O deformation of the oxirane group. The second band located at approximately 3000 cm−1 is attributed to the –C–H tension of the methylene group of the epoxy ring. The broad band at 3500 cm−1 is assigned to –O–H stretching of the hydroxyl groups, revealing the presence of dimers or high molecular weight species. All the characteristic peaks observed in the neat epoxy are present in the core–shell nanocomposites but with lower intensity. This decrease in the intensity has been attributed to the coordination between the oleic acid surface of the QDs and the polymer chain.
C stretching vibration. The –CH2 symmetric and asymmetric stretching of the oleic acid appeared at 2833 cm−1 and 2921 cm−1, respectively. The –CO stretching from the –COOH group in oleic acid appeared at 1751 cm−1.41 The peak at 1459 cm−1 is attributed to the in-plane stretching of the O–H group of –COOH and the peak at 1390 cm−1 corresponds to the –C–O stretching in oleic acid.42 The presence of these peaks confirmed the successful capping of the QDs by the oleic acid. In the spectrum of the neat epoxy (Fig. 3B), two characteristic absorptions of the oxirane ring are observed in the range between 4000 cm−1 and 400 cm.−1 The first one near 900 cm−1 is attributed to the –C–O deformation of the oxirane group. The second band located at approximately 3000 cm−1 is attributed to the –C–H tension of the methylene group of the epoxy ring. The broad band at 3500 cm−1 is assigned to –O–H stretching of the hydroxyl groups, revealing the presence of dimers or high molecular weight species. All the characteristic peaks observed in the neat epoxy are present in the core–shell nanocomposites but with lower intensity. This decrease in the intensity has been attributed to the coordination between the oleic acid surface of the QDs and the polymer chain.
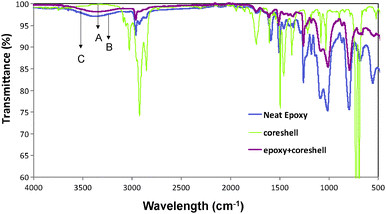 |
| | Fig. 3 The FT-IR spectra of CdSe–CdS–ZnS core–multi shell QDs (A), neat epoxy (B) and polymer core–shell nanocomposites (C). | |
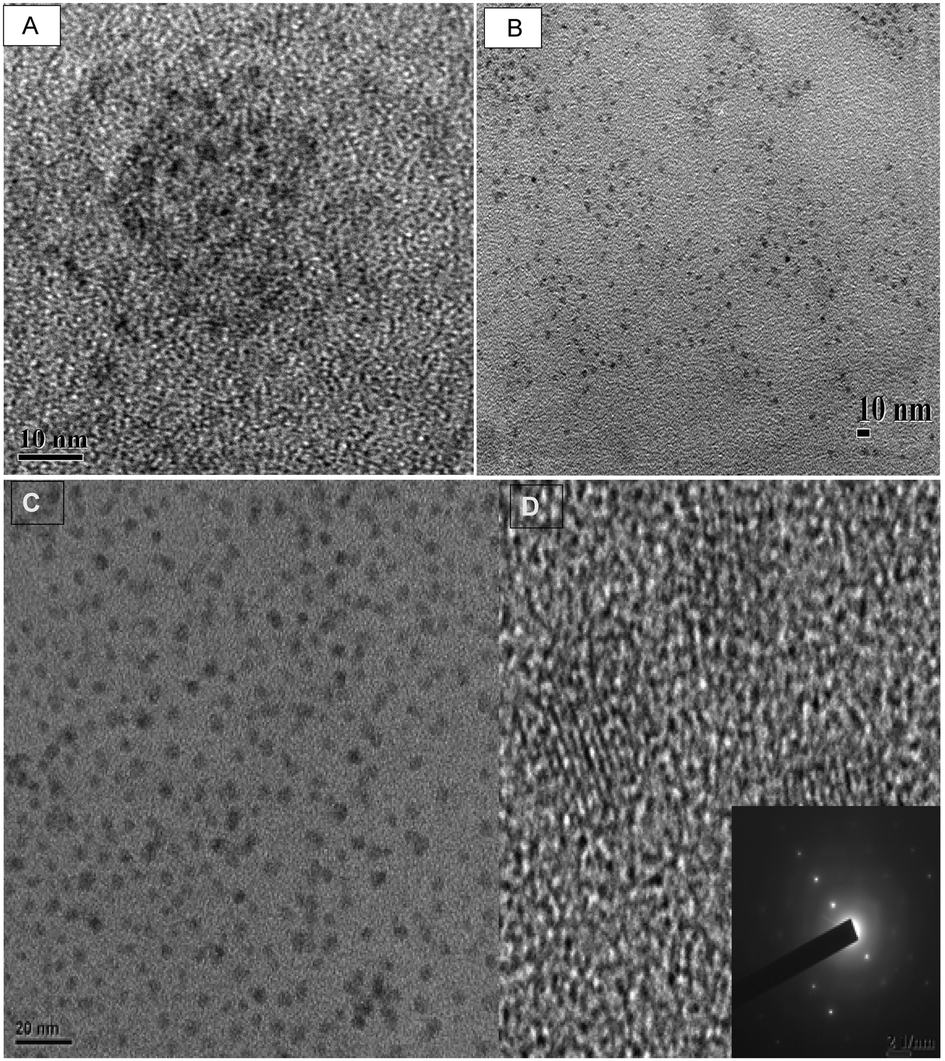
The typical transmission electron microscope (TEM) image of CdSe, CdSe–CdS, CdSe–CdS–ZnS and the high resolution TEM (HRTEM) image of CdSe–CdS–ZnS core–multi shell QDs obtained after 30 minutes are shown in Fig. 4. The images show that the as-synthesised QDs are small, spherical and well dispersed with narrow size distribution. The size distribution curve (Fig. 5A) for the CdSe–CdS–ZnS indicates that the particles are within the range of 1.0 nm to 4.5 nm with an average diameter of 2.97 ± 0.74 nm. The existence of lattice fringe in the HRTEM image (Fig. 4D) confirmed the crystallinity of the as-prepared QDs. The typical selected area electron diffraction (SAED) (Fig. 4D inset) shows lattice parameters of (111), (220), and (311), corresponding to the cubic zinc blend structure. The electron dispersion spectroscopy (EDS) measurements of the same sample (Fig. 5B) confirmed the presence of Cd, Se, Zn and S. The presence of copper is attributable to the sample grid used for the analysis which was made of copper. The dynamic light scattering (DLS) of the same sample (Fig. 6A) showed larger particles with an average particle diameter of about 10 nm. The discrepancy in particle size observed between the DLS and TEM measurements can be attributed to the formation of micelles surrounding the nanoparticles.43
 |
| | Fig. 4 The TEM images of CdSe QDs (A) and CdSe–CdS QDs (B) at 30 min reaction time. The TEM image of CdSe–CdS–ZnS QDs (C) and the corresponding HRTEM image (D), (inset SAED). | |
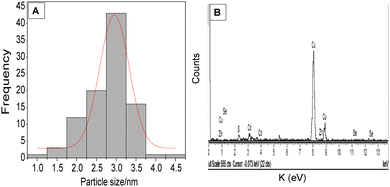 |
| | Fig. 5 The distribution curve (A) and (B) the energy dispersive spectroscopy (EDS) image of the CdSe–CdS–ZnS QDs. | |
 |
| | Fig. 6 The distribution profile of CdSe–CdS–ZnS QDs measured by DLS (A) and fitted lifetime spectrum (B) for a solution of core–multi shell QDs. | |
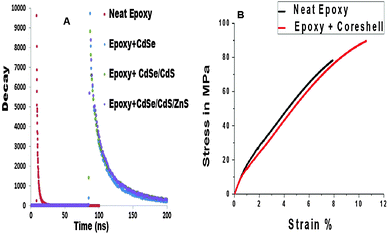
The fluorescent lifetime analysis of the samples at the different stages of the reaction is shown in Fig. 6B. Table 2 represents the PL lifetime of CdSe, CdSe–CdS and CdSe–CdS–ZnS. The delay in fluorescence after an excitation by an incident photon obeys a decaying exponential law, which depends on the lifetime of the excited state. When a fluorophore in solution or bulk is illuminated by a pulsed laser, it will emit a fluorescent signal with an exponential decaying rate, characterized by the lifetimes of the excited transitions. The correlation between the instant of the pulse and the detection of the signal investigated using fluorescence lifetime spectroscopy with characteristic decaying curves is given in Fig. 6B. All decay curves show multiexponential decay kinetics.44 The core–shells show high lifetime in the order of 10 nanoseconds indicating that the particles are highly stable. As the size of the core increases by the addition of various shells, the lifetime was found to be decreasing. This has been attributed to the decrease in the distance between the donor and the acceptor energy levels.45 As the shell grows, the distance between the valence band and the conduction band of the shell decreases and the electron–hole is confined in the core which aids the recombination process.
Table 2 PL lifetime of the CdSe–CdS–ZnS core–multi shell QDs
| Sample |
τ
1
|
τ
2
|
τ
3
|
B1 |
B2 |
B3 |
A
|
χ
2
|
| CdSe 2 min |
1.6 |
11.1 |
34.8 |
0.4476 |
0.2851 |
0.2110 |
329.832 |
1.0979 |
| CdSe–CdS 10 min |
1.58 |
7.83 |
17.81 |
0.4294 |
0.2115 |
0.2662 |
46.67 |
1.04 |
| CdSe–CdS–ZnS 30 min |
1.23 |
6.8 |
17.11 |
0.3030 |
0.2021 |
0.2299 |
33.763 |
1.062 |
The TEM micrograph of the as-synthesized CdSe–CdS–ZnS core–multi shell dispersed in the epoxy polymer matrix at a polymer to QDs ratio of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 is shown in Fig. 7A. The TEM image shows that the QDs are well dispersed in the polymer matrix without any changes in their morphology. The particles are spherical in shape thus the dispersion of the QDs in the matrix does not affect their shape. The size distribution curve in Fig. 7B indicates that the particles are in the range of 2.0 nm to 7 nm with an average diameter of 4.95 ± 1.15 nm. The slight increase in the size of the QDs nanocomposite as compared to the QDs has been attributed to the capping of polymer chains around the nanoparticles. The self-assembly of oleic acid on the surface of QDs brings the compatibility and reactive ability to QDs, and aid their dispersion in the polymers.46
:2 is shown in Fig. 7A. The TEM image shows that the QDs are well dispersed in the polymer matrix without any changes in their morphology. The particles are spherical in shape thus the dispersion of the QDs in the matrix does not affect their shape. The size distribution curve in Fig. 7B indicates that the particles are in the range of 2.0 nm to 7 nm with an average diameter of 4.95 ± 1.15 nm. The slight increase in the size of the QDs nanocomposite as compared to the QDs has been attributed to the capping of polymer chains around the nanoparticles. The self-assembly of oleic acid on the surface of QDs brings the compatibility and reactive ability to QDs, and aid their dispersion in the polymers.46
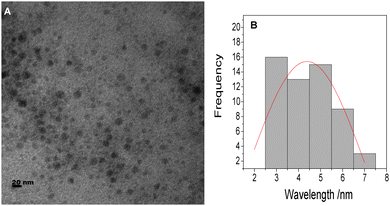 |
| | Fig. 7 The TEM image (A) and size distribution (B) of epoxy–CdSe–CdS–ZnS QD nanocomposites. | |
The emission properties of the polymer nanocomposite are presented in Fig. 8A. The maximum emission wavelength of the neat epoxy increased from 456 nm to 526 nm in the epoxy–core–multi shell nanocomposite. This indicates that the QDs have transferred their luminescence properties to the neat polymer. Considering the transparency of the composite in Fig. 8B, the core–multi shell nanocomposite is found to be more transparent than the neat epoxy. This can be attributed to the refractive index matching of the polymer and the filler, and the well dispersion of the QDs inside the polymer matrix. The lifetime measurements of the polymer nanocomposite (Fig. 9A) showed that the fluorescent lifetime of the nanocomposite core–shells decreases when compared to the pure core–multi shells. This has been attributed to the effect of polymer on the surface of the core–multi shells. The surface of the core–multi shells becomes well passivated when it is incorporated into the polymer matrix thereby reducing the number of trap states on the surface of the QDs. Thus, the distance between the energy levels of the donor and the acceptor will decrease causing the decrease in the PL lifetime.47 As far as the authors know there is no reported study regarding the PL lifetime analysis of core–multi shells inside the polymer matrix. A further detailed study regarding this phenomenon needs to be done in order to fully understand the mechanism behind this decrease in PL lifetime. The PL lifetime values of the nanocomposites are given in Table 3.
 |
| | Fig. 8 (A) Photoluminescence spectra and (B) photographs of neat epoxy and epoxy–CdSe–CdS–ZnS QD nanocomposites. | |
 |
| | Fig. 9 (A) Fitted lifetime spectra of CdSe–CdS–ZnS core–multi shell polymer nanocomposites, (B) the stress strain curves for neat epoxy and epoxy–CdSe–CdS–ZnS core–shell nanocomposites. | |
Table 3 PL lifetime of the epoxy core–multi shell QD nanocomposites
| Sample |
τ
1
|
τ
2
|
τ
3
|
B1 |
B2 |
B3 |
A
|
χ
2
|
| Neat epoxy |
86.51 |
2.61 |
9.13 |
0.6968 |
0.2104 |
0.0183 |
0.7964 |
1.295 |
| Epoxy + CdSe |
34.38 |
1.74 |
6.29 |
1.420 |
0.4179 |
0.0739 |
15.86 |
1.294 |
| Epoxy + CdSe–CdS |
50.32 |
2.6 |
0.1096 |
1.266 |
0.3298 |
0.0418 |
11.48 |
1.164 |
| Epoxy + CdSe–CdS–ZnS |
41.62 |
2.266 |
0.1029 |
1.103 |
0.3923 |
0.0464 |
12.43 |
1.294 |
The mechanical properties of the neat epoxy and the epoxy core–shell nanocomposites using the tensile testing with a speed of 1 mm min−1 and the curves are presented in Fig. 9B. The results show that the tensile modulus of the epoxy nanocomposite increases from 1594 MPa in the neat epoxy to 2430 MPa in the epoxy QD nanocomposite. This indicates that the tensile strength of the nanocomposite is higher than that of the neat polymer matrix. Fig. 10 shows the schematic diagram of the epoxy–core–multi shell polymer nanocomposite and the possible interaction between them. The oleic acid capping on the core–multi shell surface makes the surface hydrophobic. This in turn makes it compatible with the polymer matrix and hence aided the dispersion of the QDs inside the polymer matrix thereby giving a stiffening effect to the polymer chains. The tensile curve (Fig. 8B) indicates that the QD–epoxy nanocomposite shows yield behavior. The area under the tensile curve represents the absorbed energy during stretching which reflects the toughness of the material. Thus, it can be inferred that the toughness of the QD filled polymer is higher than the pure polymer matrix. Therefore, the as-prepared epoxy–QD nanocomposites can be used as encapsulating materials in the fabrication of light emitting diode devices.
 |
| | Fig. 10 The schematic representation of the interaction between the polymer and core–multi shell QDs. | |
4. Conclusions
We have successfully synthesized transparent and fluorescent epoxy CdSe–CdS–ZnS core–multi shell QD polymer nanocomposites with improved mechanical properties when compared to the neat polymer matrix. Highly fluorescent and stable CdSe–CdS–ZnS QDs were produced via a phosphine and phosphine oxide free continuous injection method. The as-synthesized QDs were successfully incorporated into the epoxy matrix by the melt mixing technique. The TEM images showed that the QDs are small, spherical and well dispersed inside the epoxy polymer matrix without any change in their morphology. The analysis of mechanical properties of the polymer QD nanocomposite showed an improvement in the tensile strength of the material. The fluorescent lifetime analysis of the core–multi shell and its polymer nanocomposite showed that the addition of shells on the core caused a decrease in the PL lifetime due to the decrease in the difference between the energy levels of the donor and acceptor. The material possesses properties that make it a good candidate for laser applications.
Acknowledgements
The authors thank the Department of Science and Technology (DST Nano mission (SR/NM/NS-54/2009)), National research foundation (NRF), South Africa under the Nanoflagship Programme (Grant no: 68706) and CV Raman Fellowship programme for financial support. The authors thank Dr IA Oluwafemi for technical assistance. Financial support from UGC-Government of India through SAP and DST-Government of India through FIST and PURSE programme is also gratefully acknowledged. This is the part of the work that OS Oluwafemi did whilst being a visiting fellow at the Centre for Nanoscience and Nanotechnology, Mahatma Gandhi University, Kerala, India, under the CV Raman Fellowship for African researchers. He thanks the DST (India) for the Fellowship and Prof. Sabu Thomas and his research group for being gracious hosts.
References
- I. Gur, N. A. Fromer, M. L. Geier and A. P. Alivisatos, Science, 2005, 310, 462 CrossRef CAS PubMed.
- P. Peng, D. J. Milliron, S. M. Hughes, J. C. Johnson, A. P. Alivisatos and R. J. Saykally, Nano Lett., 2005, 5, 1809 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS.
- O. Khani, H. R. Rajabi, M. H. Yousefi, A. A. Khosravi, M. Jannesari and M. Shamsipur, Spectrochim. Acta, Part A, 2011, 79, 361 CrossRef CAS PubMed.
- M. J. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013 CrossRef CAS.
- M. Danek, K. F. Jensen, C. B. Murray and M. G. Bawendi, Chem. Mater., 1996, 8, 173 CrossRef CAS.
- X. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019 CrossRef CAS.
- Y. A. Yang, H. M. Wu, K. R. Williams and Y. C. Cao, Angew. Chem., 2005, 117, 6870 CrossRef.
- S. Sapra, A. L. Rogach and J. J. Feldmann, J. Mater. Chem., 2006, 16, 3391 RSC.
- C. R. Bullen and P. Mulvaney, Nano Lett., 2004, 4, 2303 CrossRef CAS.
- J. Jasieniak, C. R. Bullen, J. Embden and P. Mulvaney, J. Phys. Chem. B, 2005, 109, 20665 CrossRef CAS PubMed.
- J. Y. Ouyang, M. B. Zaman, F. J. Yan, D. Johnston, G. Li, X. H. Wu, D. Leek, C. I. Ratcliffe, J. A. Ripmeester and K. Yu, J. Phys. Chem. C, 2008, 112, 13805 CAS.
- W. W. Yu and X. Peng, Angew. Chem., Int. Ed., 2002, 41, 2368 CrossRef CAS.
- Z. T. Deng, L. Cao, F. Q. Tang and B. S. Zou, J. Phys. Chem. B, 2005, 109, 16671 CrossRef CAS PubMed.
- X. Wang, W. Li and K. Sun, J. Mater. Chem., 2011, 21, 8558 RSC.
- C. W. Lee, C. H. Chou, J. H. Huang and T. P. Nguyen, Mater. Sci. Eng., B, 2008, 147, 307 CrossRef CAS PubMed.
- K. S. Leschkies, D. J. Norris and E. S. Aydil, Nano Lett., 2007, 7, 1793 CrossRef CAS PubMed.
- I. Robel, V. Subramanian and P. V. Kamat, J. Am. Chem. Soc., 2006, 128, 2385 CrossRef CAS PubMed.
- R. Koole, P. Liljeroth and D. Vanmaekelbergh, J. Phys. Chem. B, 2005, 109, 9205 CrossRef CAS PubMed.
- P. R. Yu, K. Zhu and A. J. Nozik, J. Phys. Chem. B, 2006, 110, 25451 CAS.
- Y. Q. Li, Y. Yang and S. Y. Fu, J. Phys. Chem. C, 2008, 112, 17397 CAS.
- H. J. Yang and F. Ko, Des., Manuf. Appl. Compos., 2008, 9 Search PubMed.
- Z. J. Jiang, Z. Huang, P. P. Yang and J. F. Chen, Compos. Sci. Technol., 2008, 68, 3240 CrossRef CAS PubMed.
- M. Kazes, T. Saraidarov and U. Banin, Adv. Mater., 2009, 21, 1716 CrossRef CAS.
- D. Z. Sun, H. J. Sue and N. Miyatake, J. Phys. Chem. C, 2008, 112, 16002 CAS.
- M. A. Uddin and H. P. Chan, J. Optoelectron. Adv. Mater., 2008, 10, 1 CAS.
- B. M. Novak, Adv. Mater., 1993, 5, 422 CrossRef CAS.
- Y. Q. Li, S. Y. Fu, Y. Yang and Y. W. Mai, Chem. Mater., 2008, 20, 2637 CrossRef CAS.
- Y. Q. Li, Y. Yang and S. Y. Fu, J. Phys. Chem. C, 2008, 112, 17397 CAS.
- D. Sun, H. J. Sue and N. Miyatake, J. Phys. Chem. C, 2008, 112, 16002 CAS.
- W. Zou, Z. J. Du, H. Q. Li and C. Zhang, Polym. Int., 2010, 60, 751 CrossRef.
- Y. Yang, Y. Q. Li and S. Y. Fu, J. Phys. Chem. C, 2008, 112, 10553 CAS.
- S. L. Cumberland, K. M. Hanif, A. Javier, G. A. Khitrov, G. F. Strouse, S. M. Woessner and C. S. Yun, Chem. Mater., 2002, 14, 1576 CrossRef CAS.
- H. R. Rajabi, O. Khani, M. Shamsipur and V. Vatanpour, J. Hazard. Mater., 2013, 250–251, 370 CrossRef CAS PubMed.
- J. I. Pankove, C–MRS Int. Symp. Proc., 1990, 162, 515 CAS.
- X. G. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019 CrossRef CAS.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854 CrossRef CAS.
- D. V. Talapin, I. Mekis, S. Gotzinger, A. Kornowski, O. Benson and H. Weller, J. Phys. Chem. B, 2004, 108, 18826 CrossRef CAS.
- A. R. Kortan, R. Hull, R. L. Opila, M. G. Bawendi, M. L. Steigerwald, P. J. Carroll and L. E. Brus, J. Am. Chem. Soc, 1990, 112, 1327 CrossRef CAS.
- J. Bailes, L. Vidal, D. A. Ivanov and M. Soloviev, J. Nanobiotechnol., 2009, 7, 10 CrossRef PubMed.
- S. Wageha, L. Shu-Mana, F. T. Youa and X. Xu-Ronga, J. Lumin., 2003, 102, 768 CrossRef.
- L. Zhang, R. He and H. Gu, Appl. Surf. Sci., 2006, 253, 2611 CrossRef CAS PubMed.
- M. Iwahashi, A. Umehara and K. Wakisaka, J. Phys. Chem. B, 2007, 111, 740 CrossRef CAS PubMed.
- S. J. Lee, M. Muthiah, H. J. Lee and M. Moon, Macromol. Res., 2012, 20, 188 CrossRef CAS PubMed.
- K. K. Challa, S. K. Goswami, E. Oh and E. Kim, Appl. Phys. Lett., 2011, 99, 153111 CrossRef.
- W. Zou, Z. Du, H. Li and C. Zhang, Polymer, 2011, 52, 1938 CrossRef CAS PubMed.
- X. B. Chen, Y. B. Lou, A. C. Samia and C. Burda, Nano Lett., 2003, 3, 799 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.