Functional materials from the covalent modification of reduced graphene oxide and β-cyclodextrin as a drug delivery carrier†
Guangcheng
Wei
,
Renhao
Dong
,
Dong
Wang
,
Lei
Feng
,
Shuli
Dong
,
Aixin
Song
and
Jingcheng
Hao
*
Key Laboratory of Colloid and Interface Chemistry of Ministry of Education, Shandong University, Jinan 250100, P. R. China. E-mail: jhao@sdu.edu.cn; Fax: +86-531-88564750
First published on 30th September 2013
Abstract
We report a drug delivery system based on the covalently reduced graphene oxide (rGO) with p-aminobenzoic acid (rGO-C6H4-COOH) for the loading and targeted delivery of the anticancer drug, doxorubicin (DOX). The colloidal solution of rGO-C6H4-COOH conjugated by polyethyleneimine (PEI) and Biotin was prepared. This endows the colloidal solution of rGO-C6H4-CO-NH-PEI-NH-CO-Biotin, which presents excellent water-solubility and targeting as a drug delivery system. β-Cyclodextrin (β-CD) molecules, which are host molecules for accommodating guest molecules, such as water insoluble anticancer drugs, were introduced to reduce the cytotoxicity of the drug delivery system and to improve the biocompatibility. The drug delivery of rGO-C6H4-CO-NH-PEI-NH-CO-CD-Biotin has a ∼24.64% drug (DOX) loading ratio. The drug release behavior was pH dependent at higher DOX concentrations, but salt dependent at lower DOX concentrations, which could be exploited for controlled drug release in cancer cells. The DOX loaded on rGO-C6H4-CO-NH-PEI-NH-CO-CD-Biotin could effectively induce HepG2 cancer cell apoptosis. This can be explained by the conjugation of DOX and rGO-C6H4-CO-NH-PEI-NH-CO-CD-Biotin being able to arrest the cancer cells in the G2 phase, which is the most sensitive to the anticancer drug.
1. Introduction
Biomaterials are considered a primary vehicle for targeted therapies of disease lesions because they can pass biological barriers to enter and distribute within cells by energy-dependent pathways.1,2 Graphene materials as delivery vehicles have attracted great interest in recent years due to their unique and remarkable physical and chemical properties. The drug (or biomacromolecules) delivery systems based on graphene have mainly been designed from graphene oxide (GO).3–11 As the degree of electron conjugation of GO is less than rGO-C6H4-COOH (rGO = reduced graphene oxide), rGO-C6H4-COOH as a drug delivery material has more advantages than GO, which has been verified by our previous work.12 rGO as a drug delivery material can easily produce aggregation under the complex physiological solution in the absence of a polymer dispersant or surfactant, which makes further processing difficult.13 So, one thinks that the rGO-C6H4-COOH based on graphene could be the most suitable material for drug delivery at present. The carboxylic acid active functional group of rGO-C6H4-COOH can be further modified with functionalized groups, such as –NH2. Modifying rGO-C6H4-COOH can improve water solubility and specific targeting by conjugating the carboxylic acid group with highly water-soluble polymers, such as PEI, grafting PEG star-polymers, and target molecules, such as Biotin, FA, anti CD20.3,5,14Herein, a drug delivery material based on rGO-C6H4-COOH was synthesized. The rGO-C6H4-COOH colloidal solution was firstly prepared according to our previous method12 and then the excellently water soluble PEI was engrafted to enhance the higher water-solubility of rGO-C6H4-COOH,15 and simultaneously Biotin was conjugated with PEI to enhance the targeting. β-CD was also introduced into the drug delivery material to reduce the cytotoxicity of PEI and rGO-C6H4-COOH, which forms a hydrophilic layer around the rGO-C6H4-COOH sheet. The materials increased the dispersity in water and improved the biocompatibility and performance, and greatly enhanced the half-life by delaying opsonization.16,17 On the other hand, the hydrophobic interior of β-CD can form a host–guest complex with water-insoluble drugs, which is a very important consideration as part of the drug delivery material of rGO-C6H4-COOH-NH-PEI-CD-Biotin, and it could be useful as a theranostic material for cancer therapy, without cytotoxic effects on normal cells. A schematic representation of the drug delivery material rGO-C6H4-CO-NH-PEI-NH-CO-CD-Biotin is shown in Scheme 1.
 | ||
| Scheme 1 Schematic illustration of the drug delivery materials based on rGO-C6H4-COOH and β-CD. | ||
2. Experimental methods
2.1. Synthesis of graphene oxide (GO), reduced graphene oxide (rGO), rGO-C6H4-COOH (Fig. S1, (SI†), ESI†) and the Tyndall effect (Fig. S2, SI1, ESI†)
GO, rGO, and rGO-C6H4-COOH were synthesized according to the previous method.18–21 Solid samples of GO, rGO, and rGO-C6H4-COOH were obtained through freeze-drying for the FT-IR, XPS, and TGA characterization measurements. The solutions of rGO, rGO-C6H4-COOH were put on a thin film of amorphous carbon deposited on a copper grid for TEM (JEM-1400) observation. One drop of rGO-C6H4-COOH solution was dropped onto the silicon wafer for the AFM (Nanoscope IIIA, Veeco) images.The colloidal nature of GO, rGO, and rGO-C6H4-COOH dispersion solutions was demonstrated by the Tyndall effect (Fig. S2, SI1, ESI†), which suggests excellent water dispersibility.
2.2. Synthesis of rGO-PEI-CD-Biotin and characterizations
Biotin (0.2459 g) was dissolved into 20 mL water, and EDC·HCl (0.3863 g) and NHS (0.2387 g) were added with stirring for 4 hours to activate the –COOH of Biotin. After the prescribed time, the above mixture solution was added to 20 mL PEI (M.W. 1800) aqueous solution (0.05 mol L−1) and then stirred for 24 hours to form the amide. The mixture was dialyzed against water to remove excess EDC·HCl and NHS for obtaining the PEI-Biotin solution. The rGO-C6H4-COOH (100 mL, about 0.1 g L−1) was activated with EDC·HCl (0.3811 g) and NHS (0.2227 g), and then the above PEI-biotin solution was added with stirring for 24 hours to form rGO-C6H4-CO-NH-PEI-Biotin. Dialysis was executed to remove any unnecessary molecules to obtain the rGO-C6H4-CO-NH-PEI-Biotin (rGO-PEI-Biotin) solution. β-CD-NH2-CO-C6H4-COOH (0.3868 g) was dissolved into 10 mL water, and then EDC·HCl (1.1512 g) and NHS (0.6927 g) were added with stirring for 3 hours to activate the –COOH of β-CD-NH2-CO-C6H4-COOH. The rGO-C6H4-CO-NH-PEI-Biotin solution was added with stirring for 24 hours to form the amide. Finally, the mixture was dialyzed against water to remove excess EDC·HCl and NHS for obtaining the rGO-C6H4-CO-NH-PEI-CO-C6H4-CO-NH-β-CD-Biotin (Scheme 1, rGO-PEI-CD-Biotin) aqueous solution. rGO-PEI-Biotin and rGO-PEI-CD-Biotin solid samples were obtained by freeze-drying for the FT-IR spectra. One drop of solution of rGO-PEI-CD-Biotin was dropped onto the silicon wafer for AFM determination and to the copper grid for the TFM images.According to the previous method,12,22 Cy7 was conjugated to the drug delivery materials through forming an amide (–COOH of Cy7 and –NH2 of PEI) for the cell uptake observations.
2.3. Drug loading ratio of rGO-PEI-CD-Biotin23
The rGO-PEI-CD-Biotin water solution (5 mL, 0.05 mg mL−1) was mixed with DOX DMSO solution (0.5 mL, 2.5 mmol L−1) and stirred overnight at room temperature. Excess DOX precipitates were removed by centrifugation with 2000 r min−1 for 5 min. The supernatant was dried by lyophilization to obtain the precipitates. The precipitates were re-dispersed into the 5 mL deionized water. The DOX-rGO-PEI-CD-Biotin solution was stored at 4 °C. To determine the DOX loading on rGO-PEI-CD-Biotin, UV-vis spectra of rGO-PEI-CD-Biotin, DOX-rGO-PEI-CD-Biotin, and DOX were measured by a U-4100 spectrometer.2.4. Drug releasing behavior23
The releasing behavior of the water insoluble anticancer drug on rGO-PEI-CD-Biotin was demonstrated through the choosing of the appropriate drug. DOX was chosen as the model drug. The DOX-rGO-PEI-CD-Biotin was incubated in PBS (phosphate buffer saline, pH = 7.4) and ABS (acetate buffered saline, pH = 5.5) buffer solution, respectively. After 4, 8, 12, 16, and 20 hours incubation, the released DOX was removed by centrifugation and the supernatant of DOX-rGO-PEI-CD-Biotin was re-determined for UV-vis measurements. The released percentage of DOX was calculated based on the dropping of the DOX absorption peak at 485 nm.2.5. Cell lines and cell culture
HepG2 cancer cells and primary cultured mouse central neurogliocyte cells (neurogliocyte cells) were maintained in Dulbecco's Modified Eagle's Medium (DMEM, Fanbo) supplemented with 10% (v/v) fetal bovine serum (FBS, Fanbo), penicillin–streptomycin (1.0% penicillin–streptomycin, 1.0% Glutamax, respectively, Fanbo) in an incubator supplied with 85% humidity and 5% CO2 atmosphere at 37 °C. The cell culture medium was changed every two days and the cells were split upon 80% confluence.2.6. MTT assay and FCM (flow cytometer) assay
An MTT assay of neurogliocyte cells were seeded into 96-well plates at a density of 1 × 104 per well in 200 μL of cell culture solution and grown for 24 hours. The cells were then incubated with various rGO-PEI-CD-Biotin concentrations for 48 hours. Afterwards the cells were incubated in cell culture solution containing 0.5 mg mL−1 of MTT for 4 hours, and then the cell culture solution was carefully removed. The resulting formazan violet crystals were dissolved in 200 μL of DMSO and 25 μL glycine buffer (0.1 mol L−1 glycine/0.1 mol L−1 NaCl, pH = 10.5), and the UV-vis absorbance intensity was measured at 570 nm.For the FCM assay, exponentially growing neurogliocyte cells were seeded (1 × 105 cell per well) in 12-well culture plates and pre-incubated for 24 hours, followed by co-incubation with different rGO-PEI-CD-Biotin concentrations for 48 hours. The cell culture solution was discarded and washed three times with PBS buffer solution. After trypsinization, the cells were detached through centrifugation, and re-dispersed in PBS buffer solution for the measurements. Neurogliocyte cells treated by rGO-PEI-CD-Biotin were stained with propidium iodide (PI) and Annexin V-EGFP before flowing cytometry (COULTER EPICS XL, America) analysis.
2.7. rGO-PEI-CD-Biotin/DOX induces HepG2 cell apoptosis and the cell cycle
Apoptosis and cell cycle assays were executed with the FCM. The method of the apoptosis assay is the same as the above FCM assay. The cell cycle assay was executed according to the method in our previous work.12 The HepG2 cells were cocultured with the rGO-PEI-CD-Biotin, DOX, and rGO-PEI-CD-Biotin/DOX for 48 hours, and 1 × 106 mL−1 cells were collected after 0.25% trypsin digestion, respectively. After centrifuging and removing the supernatant, the cancer cells were fixed with the 4 °C ethanol. Washing twice for the cells with the PBS and then 30 μL 1.0 mg mL−1 RNaseA was added for 30 min at 37 °C. After centrifugation and removing the supernatants, 50 μL 1.0 mg mL−1 PI was added to dye the cancer cells for the cell cycle assay with the FCM.3. Results and discussion
rGO and rGO-C6H4-COOH were synthesized according to the previous method (Fig. S1, SI1, ESI†),12 characterizations of rGO and rGO-C6H4-COOH were carried out through Fourier transform infrared (FT-IR), transmission electron microscopy (TEM), atomic force microscopy (AFM), X-ray photoelectron spectroscopy (XPS), and Thermo Gravimetric Analysis (TGA) measurements. The Tyndall effect proves the colloidal nature, and the very obvious Tyndall effect of the GO, rGO, and rGO-C6H4-COOH solutions was demonstrated, as shown in Fig. S2 (ESI†), suggesting that these solutions have excellent dispersibility in aqueous solution.The conjugation of the p-disubstituted phenyl group (–C6H4–COOH) with the planar rGO was confirmed by FT-IR measurements and AFM observations. The p-disubstituted phenyl group (–C6H4–COOH) of rGO-C6H4-COOH was confirmed by FT-IR measurements, as shown in Fig. 1a. The peak at 1015 cm−1 is the characteristic vibration of the p-disubstituted phenyl group, and the peak at 825 cm−1 is the C–H bending vibration of the p-disubstituted phenyl group.5,15 The height of the rGO-C6H4-COOH sheets was confirmed by the AFM observations, as shown in Fig. 1b, the height mostly lies in the range from 1.6–4.4 nm. According to ref. 24, the theoretical height of the single-layer rGO-C6H4-R (R = COOH, NO2, OCH3, or Br) sheet on both sides is ∼2.2 nm, and the height of the bare rGO sheets is ∼1.0 nm with the substituted aromatic groups contributing ∼0.6 nm in height. The height range from 1.6 nm to 4.4 nm suggests that single-layer and double-layer of rGO-C6H4-COOH coexist, the –C6H4–COOH groups were attached and perpendicularly standing to one side, as well as both sides of the rGO sheets.
 | ||
| Fig. 1 Characterization of rGO and rGO-C6H4-COOH. Infrared spectra of rGO and rGO-C6H4-COOH (a); AFM image of rGO-C6H4-COOH (b); TEM images of rGO (c) and rGO-C6H4-COOH (d); XPS survey curves of rGO (e) and rGO-C6H4-COOH (f). | ||
TEM observations were performed to demonstrate the morphologies of rGO and rGO-C6H4-COOH. One can see that the rGO sheets have the ripple lines between the layers, as demonstrated in Fig. 1c, suggesting high quality rGO sheets were prepared. As shown in Fig. 1d, the rGO-C6H4-COOH sheets are very seriously accumulated between the graphene layers. When the rGO-C6H4-COOH is in a solid state, the fold on the edge can also be found, which agrees with ref. 15.
XPS survey was used to analyze the change of the functionalized groups of rGO and rGO-C6H4-COOH. The percent of surface oxygen groups in rGO is about 13.64% (Fig. 1e and Fig. S3a, SI2, ESI†), but the percent of surface oxygen groups of rGO-C6H4-COOH is about 21.99% (Fig. 1f and Fig. S4a, SI2, ESI†). The introduction of –C6H4–COOH to the rGO improves the percentage of surface oxygen groups. The percent of the surface O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O– group in rGO is 0.013% (Fig. 1e and Fig. S3b, SI2, ESI†), but the surface OC–O– group in rGO-C6H4-COOH is 9.877% (Fig. 1f and Fig. S4b, SI2, ESI†). The increasing percentage of this functionalized group is only attributed to the introduction of –C6H4–COOH to rGO sheets, strongly supporting the successful conjugation of –C6H4–COOH to the rGO plane.
C–O– group in rGO is 0.013% (Fig. 1e and Fig. S3b, SI2, ESI†), but the surface OC–O– group in rGO-C6H4-COOH is 9.877% (Fig. 1f and Fig. S4b, SI2, ESI†). The increasing percentage of this functionalized group is only attributed to the introduction of –C6H4–COOH to rGO sheets, strongly supporting the successful conjugation of –C6H4–COOH to the rGO plane.
The above measurements implied that the surface of rGO was successfully functionalized. A small quantity of nitrogen content can be attributed to the formation of hydrazones (Fig. 1e and f and Fig. S3c and S4c, SI2, ESI†).18,24 The rGO-C6H4-COOH sample heated in a thermogravimetric analysis (TGA) instrument to 800 °C under argon shows some decrease in the intensity, which is consistent with the defunctionalization of rGO-C6H4-COOH upon heating (Fig. S5, SI3, ESI†).24
rGO-C6H4-CO-NH-PEI-NH-CO-C6H4-CO-NH2-CD-Biotin was synthesized according to EDC chemistry, as illustrated in Scheme 1 and Fig. S6 (SI4), ESI.† The PEI-Biotin was conjugated with rGO-C6H4-COOH to synthesize rGO-C6H4-CO-NH-PEI-NH-CO-Biotin according to EDC chemistry. The characteristic amide-carbonyl for the stretching vibration of CO vibration for the NH–CO group and the bending vibration of –C–S–C– are at ∼1650 cm−1 and ∼618 cm−1, respectively, as shown in Fig. S7a and b (SI4), ESI,† suggesting the PEI-Biotin was conjugated to the rGO-C6H4-COOH sheet. β-CD-NH2-CO-C6H4-COOH was synthesized according to the previous method (Fig. S8 and S9, SI5, ESI†),25 and then it was conjugated to rGO-C6H4-COOH-NH-PEI-Biotin via forming amide bonds between the –COOH group of β-CD-NH2-CO-C6H4-COOH and the –NH2 group of PEI in rGO-C6H4-COOH-NH-PEI-Biotin. A very obviously characteristic peak at 1155 cm−1 can be designated to be the stretching vibration of –C–O–C– for β-CD-NH2-CO-C6H4-COOH (Fig. S7c, SI4, ESI†), revealing the successfully conjugation of β-CD-NH2-CO-C6H4-COOH and rGO-C6H4-COOH-NH-PEI-Biotin.
That the particle size is small enough is an important prerequisite to ensure that the nanoparticles can step across nuclear pore complexes.26 As shown in Fig. 2a, the size of rGO-PEI-CD-Biotin was mostly less than 100 nm, which is suitable and reasonable as a drug delivery material. The rGO-PEI-CD-Biotin was very stable, and it can exist in various kinds of biological solutions without aggregation (Fig. S10, SI6, ESI†). AFM measurement reveals that the thickness of rGO-PEI-CD-Biotin is about 13.89 nm (Fig. 2b). The increase of thickness was attributed to the attachment of PEI-Biotin on both planes of the rGO-C6H4-COOH sheets and the conjugation of β-CD-NH2-CO-C6H4-COOH with PEI.
 | ||
| Fig. 2 TEM (a) and AFM (b) images of rGO-PEI-CD-Biotin. | ||
DOX3,4 was chosen as the model drug for the drug delivery tests. It is well-known that the DOX can interact with β-CD-NH2-CO-C6H4-COOH to form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion complex, the inclusion constant of DOX and β-CD-NH2-CO-C6H4-COOH was 3.16 × 104 L mol−1 (Fig. S11 and Table S1, SI7, ESI†). The inclusion ratio fits with the schematic illustration and the β-CD of rGO-PEI-Biotin-CD could be fully utilized as a host molecule to accommodate guest molecular DOX (Scheme 1).
:1 inclusion complex, the inclusion constant of DOX and β-CD-NH2-CO-C6H4-COOH was 3.16 × 104 L mol−1 (Fig. S11 and Table S1, SI7, ESI†). The inclusion ratio fits with the schematic illustration and the β-CD of rGO-PEI-Biotin-CD could be fully utilized as a host molecule to accommodate guest molecular DOX (Scheme 1).
The drug loading capacity and drug release profile are the most important performance indicators of drug delivery materials. The DOX loading capacity was executed according to the method of the previous work.12,23 The DOX loading ratio on rGO-PEI-CD-Biotin was ∼24.64% at 485 nm (Fig. S12, SI8, ESI†). It could be a synergetic result that the hydrophobic interior of β-CD and the plane of graphene sheets together load DOX. The driving force is mainly the balance of the host–guest and hydrophobic interactions, the π–π stacking, and hydrophobic interactions between DOX with the drug delivery materials.5,17In vitro release experiments were carried out at pH = 7.4 PBS (phosphate buffered saline) and pH = 5.5 ABS (acetate buffered saline) solutions, respectively (Fig. S13a and b, SI9, ESI†). We found that the release rate of DOX was faster in PBS solution than that in ABS solution during the early stages, but slower at more than 14 hours (Fig. S13c, SI9, ESI†). It can be suspected that the DOX release was pH dependent at higher DOX concentrations, but salt dependent at lower DOX concentrations. Because the physiological salt environment and the extracellular pH of the tumor tissue is significantly lower than that of normal tissues and some unique enzymes and chemicals exist, the pH and salt dependent drug release from the rGO-PEI-CD-Biotin materials should be exploited for drug delivery applications.
The HepG2 cells co-cultured with rGO-PEI-CD-Biotin-Cy7 showed quite bright red fluorescence owing to the Cy7 (rGO-PEI-CD-Biotin-Cy7) fluorescent emitting at 532 nm excitation by confocal microscopy, which suggests rGO-PEI-CD-Biotin is rapidly internalized into the cells (Fig. S14, SI10, ESI†). The rapid uptake may be the reason for the specific conjugation of Biotin of rGO-PEI-CD-Biotin-Cy7 and Biotin-specific receptors, which are often over-expressed on the HepG2 cell surface. The Biotin content in cancerous tumors is significantly higher than in normal tissue and rapid proliferations of cancer cells require extra Biotin.14,27
Neurogliocyte cells were applied for the observation of cell morphology. The cell morphology was not changed when the concentration of rGO-PEI-CD-Biotin is up to 80 mg L−1. However, 100 mg L−1 rGO-PEI-CD-Biotin is a turning point at which the cell morphology begins to be changed, and there are much more obvious changes at 120, 140, 160 and 180 mg L−1 rGO-PEI-CD-Biotin, as shown in Fig. S15 (SI11), ESI.† Some cells turned round and contractible. The spherical protrusions at the cell surface were observed and apoptotic bodies formed.12
The cytotoxicity of rGO-PEI-CD-Biotin was determined by using MTT (3-[4,5-dimethylthialzol-2-yl]-2,5-diphenyltetrazolium bromide)28 and FCM (flow cytometer) assay29 with neurogliocyte cells. In the FCM assay (Fig. 3a and Fig. S16, SI12, ESI†), the rGO-PEI-Biotin-CD has no obvious toxicity to neurogliocyte cells at the concentrations up to 50 μg mL−1, meaning that the relative cell viability is not less than 80%. Even when the concentration is increased up to 100 μg mL−1, the relative cell viability is still not less than 50%. It was found that 20 μg mL−1 is a specific concentration and the relative cell viability increases at this concentration. It can be explained due to the D-glucopyranoside units of β-CD that can enhance the cell growth and the toxicity of rGO and PEI can restrain cell growth. At low concentrations of rGO-PEI-CD-Biotin, the effect of β-CD on the cell growth is more obvious than that of the rGO and PEI, while at high concentrations of rGO-PEI-Biotin-CD, the effect is the opposite. The result of the MTT assay is very similar with that of the FCM assay, as shown in Fig. 3b. The rGO-PEI-CD-Biotin has no toxicity on neurogliocyte cells at concentrations up to 10 μg mL−1 and the relative cell viability increases a little. However, at 20 μg mL−1 rGO-PEI-CD-Biotin can slightly induce neurogliocyte cell apoptosis. The relative cell proliferation inhibition rate is 9.561%, which is a bit different from the FCM assay result and it is within a reasonable error range by different measurement methods (MTT and FCM). The relative cell viability rate decreases in sequence with the increase of concentration from 20 to 100 mg L−1, but the relative cell viability is not less than 50% at concentrations up to 100 μg mL−1, that is, the IC50 is not less than 100 μg mL−1 (Fig. 3a and b). According to the results of the FCM assay, MTT assay, and cell morphology observations, the rGO-PEI-CD-Biotin concentration of 10 μg mL−1 was used for the drug delivery in the current experiment.
 | ||
| Fig. 3 Relative cell viability: FCM assay (a), MTT assay (b), and relative HepG2 cell viability to the concentration of rGO-PEI-CD-Biotin, DOX and rGO-PEI-CD-Biotin/DOX (c), respectively. The asterisks indicate P < 0.05 versus the control group. | ||
The conjugation of rGO-PEI-CD-Biotin with DOX induces HepG2 cell apoptosis, which can be analyzed with FCM (Fig. S17, SI13, ESI†). The cell viability of the control group is 87.02%. While the 10 mg L−1 rGO-PEI-CD-Biotin group is 89.41% and the relative viability (rGO-PEI-CD-Biotin group versus control group) is 102.75% (Fig. 3c). This result fitted with the cell viability. The DOX group (1.0 mg L−1) can obviously induce apoptosis in the cancer cells, as shown in Fig. 3c, the relative cell viability was 82.14% when DOX was loaded together onto rGO-PEI-CD-Biotin. However, 1.0 mg L−1 DOX and 10 mg L−1 rGO-PEI-CD-Biotin can obviously induce apoptosis of the HepG2 cells (Fig. 3c), and the relative cell viability was 65.35%. According to Jin's formula,10 the q value is 2.22, suggesting that conjugation of DOX and rGO-PEI-CD-Biotin can effectively enhance cancer cell apoptosis. It is very likely that this is the reason why rGO-PEI-CD-Biotin loaded with DOX significantly overcomes the drug resistance of cancer cells. It is a serious challenge facing doctors and patients when drugs are used.5 This was confirmed also by the cell cycle distribution experiments.
In a cell population, the effect of internalized rGO-PEI-CD-Biotin nanomaterials and DOX on each cell varies as the cell advances through the cell cycle (Fig. S18, SI14, ESI†).30 The HepG2 cells were employed for the cell cycle assay. As shown in Fig. 4a, the HepG2 cells in the cell cycle assay tend to block in the S phase, i.e., 55.722%, while only 7.163% in G2 (G2/M) phase, and 37.115% in G1 phase (G0/G1). Compared with the control group, the DOX group (Fig. 4b) showed that the DOX can induce the HepG2 cell block mostly in the S phase (44.641%) and G1 phase (41.848%), and the arrested rate of the G1 phase increases slightly from 7.163% to 13.511%. The rGO-PEI-CD-Biotin group (Fig. 4c) induces the HepG2 cell block in the mainly G1 phase (41.152%) and S (48.657%), and only 10.19% in the G2 phase, which is similar to the control group. This suggests that the rGO-PEI-CD-Biotin is not toxic at the current concentration (10 mg L−1) to HepG2 cells. As shown in Fig. 4d for rGO-PEI-CD-Biotin/DOX group, the arresting rate of the G1 phase is 29.332% and the S phase is 48.757%, suggesting that the DNA synthesis of HepG2 cell is active after being treated with DOX and the restriction point requirements have been met,31 however, the G2 phase shows a great difference with the control groups, the arresting rate is 21.911%, suggesting that the RNA synthesis and the chromatin spiral of HepG2 cells treated with rGO-PEI-CD-Biotin/DOX are active. J. A. Kim, et al.30 found that the cells in different phases of the cell cycle have similar rates of internalizing nanomaterials. After 24 h, the concentration of nanomaterials in the cells could be ranked according to the different phases: G2/M > S > G0/G1, and the cancerous cells can pass through S or G2/M phases more often than healthy cells. The cells are the most sensitive in the late M and G2 phases and most resistant in the late S phase. The pattern of resistance and sensitivity correlates with the level of sulfhydryl compounds in the cells. Sulfhydryls are natural radioprotectors and tend to be at their highest levels in S and at their lowest near mitosis.32 As shown by rGO-PEI-CD-Biotin/DOX group in our experiments, the conjugation of rGO-PEI-CD-Biotin and DOX could effectively hinder the HepG2 cancer cells in the G2 phase and prevent the HepG2 cancer cells from entering the mitosis period (M phase).
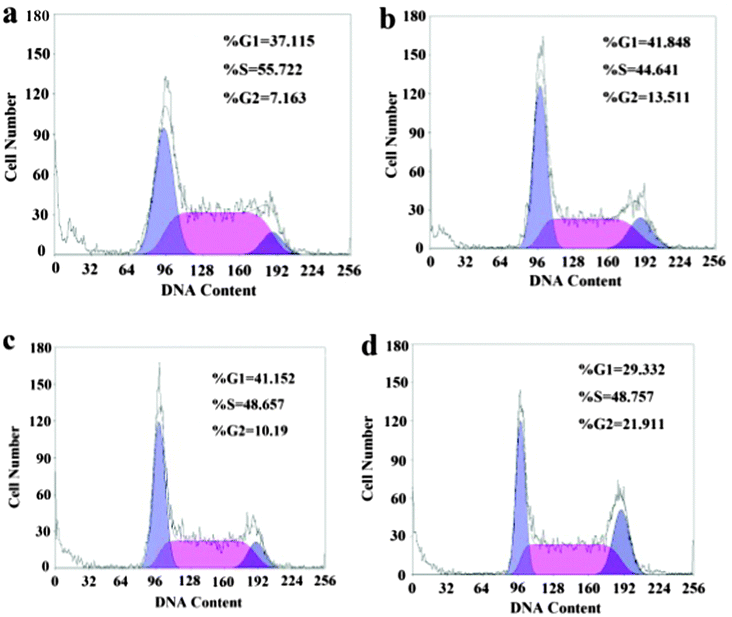 | ||
| Fig. 4 Effect of DOX, rGO-PEI-CD-Biotin, and rGO-PEI-CD-Biotin/DOX on HepG2 cancer cell cycle distribution: control group (a), 1.0 mg L−1 DOX group (b), 10 mg L−1 rGO-PEI-CD-Biotin group (c), and 1.0 mg L−1 DOX + 10 mg L−1 rGO-PEI-CD-Biotin group (d). | ||
4. Conclusions
We have illustrated an advanced concept for constructing a drug delivery system based on an rGO-C6H4-COOH dispersion solution. We synthesized rGO-C6H4-COOH branched PEI and biotin via covalent bonds to form colloidal solution for endowing the delivery materials with excellent water-solubility and targeting. β-CD molecules are host molecules to accommodate guest molecules, i.e., water insoluble anticancer drugs, and were introduced to reduce the cytotoxicity of the drug delivery system and to improve the biocompatibility. The drug (DOX) loading ratio of the drug delivery system is ∼24.64% and the drug release behavior is pH dependant at high DOX concentrations and salt dependant at low DOX concentrations, which can be exploited for the controlled drug release in cancer cells. The cytotoxicity of rGO-PEI-CD-Biotin is low and the DOX loaded on rGO-PEI-CD-Biotin can effectively induce the HepG2 cancer cell apoptosis, which can be explained because of the conjugation of DOX and rGO-PEI-CD-Biotin to arrest the HepG2 cells in the G2 phase which is the most sensitive to anticancer drugs. Even though the drug delivery system still has a long way to go to clinical application, our present work could demonstrate a novel insight for designing drug delivery systems.Acknowledgements
This work was financially supported by the NSFC (21033005 & 21273136) and the National Basic Research Program of China (973 Program, 2009CB930103). We also thank Ms Miaomiao Yan and Ms Lixia Zhang (Binzhou Medical College) for helping with the cell culture and valuable discussion.Notes and references
- J. A. Kim, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 62–68 CrossRef CAS PubMed.
- N. Nakatsuji, Biomater. Sci., 2013, 1, 9–10 RSC.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS PubMed.
- X. Yang, X. Zhang, Z. Liu, Y. Ma, Y. Huang and Y. Chen, J. Phys. Chem. C, 2008, 112, 17554–17558 CAS.
- X. Yang, X. Zhang, Y. Ma, Y. Huang, Y. Wang and Y. Chen, J. Mater. Chem., 2009, 19, 2710–2714 RSC.
- L. Zhang, Z. Lu, Q. Zhao, J. Huang, H. Shen and Z. Zhang, Small, 2011, 7, 460–464 CrossRef CAS PubMed.
- X. Yang, Y. Wang, X. Huang, Y. Ma, Y. Huang, R. Yang, H. Duan and Y. Chen, J. Mater. Chem., 2011, 21, 3448–3454 RSC.
- C. Cheng, S. Q. Nie, S. Li, H. Peng, H. Yang, L. Ma, S. D. Sun and C. S. Zhao, J. Mater. Chem. B, 2013, 1, 265–275 RSC.
- C. Cheng, S. Li, S. Q. Nie, W. F. Zhao, H. Yang, S. D. Sun and C. S. Zhao, Biomacromolecules, 2012, 13, 4236–4246 CrossRef CAS PubMed.
- G. Wei, M. Yan, R. Dong, D. Wang, X. Zhou, J. Chen and J. Hao, Chem.–Eur. J., 2012, 18, 14708–14716 CrossRef CAS PubMed.
- X. Qi, K. Pu, X. Zhou, H. Li, B. Liu, F. Boey, W. Huang and H. Zhang, Small, 2010, 6, 663–669 CrossRef CAS PubMed.
- D. N. Heo, D. H. Yang, H. J. Moon, J. B. Lee, M. S. Bae, S. C. Lee, W. J. Lee, I. C. Sun and I. K. Kwon, Biomaterials, 2012, 33, 856–866 CrossRef CAS PubMed.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- R. Ghosh, P. Zhang, A. Wang and C. Ling, Angew. Chem., Int. Ed., 2012, 51, 1548–1552 CrossRef CAS PubMed.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS.
- K. Yang, S. Zhang, G. Zhang, X. Sun, S. T. Lee and Z. Liu, Nano Lett., 2010, 10, 3318–3323 CrossRef CAS PubMed.
- Z. Liu, X. Sun, N. Nakayama-Ratchford and H. Dai, ACS Nano, 2007, 1, 50–56 CrossRef CAS PubMed.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W. F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS PubMed.
- H. Takahashi, Y. Takashima, H. Yamaguchi and A. Harada, J. Org. Chem., 2006, 71, 4878–4883 CrossRef CAS PubMed.
- L. Pan, Q. He, J. Liu, Y. Chen, M. Ma, L. Zhang and J. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS PubMed.
- W. B. Tsai and M. Wang, Biomaterials, 2005, 26, 3141–3151 CrossRef CAS PubMed.
- H. Huang, E. Pierstorff, E. Osawa and D. Ho, Nano Lett., 2007, 7, 3305–3314 CrossRef CAS PubMed.
- Y. Bae, W. D. Jang, N. Nishiyama, S. Fukushima and K. Kataoka, Mol. BioSyst., 2005, 1, 242–250 RSC.
- J. A. Kim, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 62–68 CrossRef CAS PubMed.
- M. Kõivomägi, E. Valk, R. Venta, A. Iofik, M. Lepiku, E. R. M. Balog, S. M. Rubin, D. O. Morgan and M. Loog, Nature, 2011, 480, 128–132 CrossRef PubMed.
- M. Mahmoudi, K. Azadmanesh, M. A. Shokrgozar, W. S. Journeay and S. Laurent, Chem. Rev., 2011, 111, 3407–3432 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nj00690e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |