Pb4Zn2B10O21: a congruently melting lead zinc borate with a novel [B10O24] anionic group and an interesting [Pb4O12]∞ chain†
Hongwei
Yu
abcd,
Hongping
Wu
*abc,
Shilie
Pan
*abc,
Bingbing
Zhang
abcd,
Lingyun
Dong
abcd,
Shujuan
Han
abc and
Zhihua
Yang
abc
aKey Laboratory of Functional Materials and Devices for Special Environments of CAS, 40-1 South Beijing Road, Urumqi 830011, China. E-mail: slpan@ms.xjb.ac.cn; wuhp@ms.xjb.ac.cn; Fax: (+86)991-3838957; Tel: (+86)991-3674558
bXinjiang Key Laboratory of Electronic Information Materials and Devices, 40-1 South Beijing Road, Urumqi 830011, China
cXinjiang Technical Institute of Physics & Chemistry of CAS, 40-1 South Beijing Road, Urumqi 830011, China
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 14th October 2013
Abstract
A new lead zinc borate, Pb4Zn2B10O21, has been synthesized and the crystal structure was determined by single crystal X-ray diffraction. It crystallizes in the orthorhombic group Pbcn with a = 14.6062(15) Å, b = 17.4899(16) Å, c = 13.2962(15) Å, Z = 8. Interestingly, in the crystal structure, a new B–O building unit [B10O24] group is observed and the four different coordinated Pb atoms are connected with each other by sharing a corner, edge or face to form an interesting regular [Pb4O12]∞ chain. The DSC analysis proves that Pb4Zn2B10O21 melts congruently. First-principles electronic structure calculation shows that the calculated band gap of Pb4Zn2B10O21 is 3.53 eV, which is in good agreement with the experimental value measured from the UV-vis-NIR absorption spectrum, and the observed absorption peak is assigned as a charge transfer from O 2p states to Pb 6p states.
Introduction
In recent years, much work has been focused on the synthesis, characterization and physical studies of borate compounds, owing to their rich structures and interesting physical properties.1–3 Boron is a unique element, it may adopt triangular (BO3) or tetrahedral oxygen coordination (BO4). Accordingly, the atomic orbitals are hybridized to planar sp2 or three dimensional sp3 structures. Further, such structural units can connect with each other via sharing corners or edges to comprise several different BxOy groups, which are responsible for the versatile physical properties of borates.4Over the years, several borates with novel structures or important applications were reported, for example, β-BaB2O4 (BBO),5 LiB3O5 (LBO),6 CsB3O5 (CBO)7 and CsLiB6O10 (CLBO),8 which have been widely used in nonlinear optical (NLO) devices. Sodium borate halides Na11B21O36X2 (X = Cl, Br) with new graphene-like borate double layers were synthesized by the high temperature solution method by our group.9 Other borates with microporous properties have also been found, such as Zn(H2O)B2O4·xH2O, which contains one-dimensional 8-ring channels formed by ZnO4 and BO4 tetrahedra and BO3 triangles.10 In addition, some borates with edge-sharing BO4 tetrahedra, which violate Pauling's rules, have also been synthesized under high-pressure by Huppertz and von der Eltz.11 It is interesting that in Zn-containing borates, the borate with edge-sharing BO4 tetrahedra can also be synthesized under ambient pressure.12–14 Obviously the geometric flexibility of boron atoms enrich enormously the topologies of the B–O frameworks.
In this work, we will report a new lead zinc borate, Pb4Zn2B10O21. And interestingly, a new [B10O24] group and an interesting regular [Pb4O12]∞ chain are observed for the first time in its crystal structure. Also, it is a congruently melting compound, which suggests that it is easy to grow large single crystals.
Experimental section
Synthesis
Polycrystalline Pb4Zn2B10O21 was prepared by the high-temperature solid-state reaction. The reactants were PbO (Tianjin henxin Chemical Reagent Co., Ltd, 99.5%), ZnO (Tianjin henxin Chemical Reagent Co., Ltd, 99.5%), H3BO3 (Tianjin Baishi Chemical Co., Ltd, 99.5%). Initially, the stoichiometric powder mixture of PbO (8.898 g, 0.04 mol), ZnO (1.627 g, 0.02 mol) and H3BO3 (6.183 g, 0.10 mol) was prepared and packed into Pt crucibles, which were heated to 350 °C and held for 10 h to eliminate the water. Then the reaction mixture was calcined at 500 °C for 72 h with intermittent regrinding. Thus, white polycrystalline Pb4Zn2B10O21 powder was obtained.X-ray powder diffraction (XRD) analysis of Pb4Zn2B10O21 was performed at room temperature in the angular range of 2θ = 10–70° with a scan step width of 0.02° and a fixed counting time of 1 s per step using an automated Bruker D2 PHASER X-ray diffractometer equipped with a diffracted beam monochromator set for Cu Kα radiation (λ = 1.5418 Å). The experimental XRD pattern of the polycrystalline phase is in good agreement with the calculated one derived from the single-crystal data (Fig. 1).
 | ||
| Fig. 1 Experimental and calculated XRD patterns of Pb4Zn2B10O21. | ||
Single-crystal growth
Small single crystals of Pb4Zn2B10O21 were grown from a high temperature solution. The stoichiometric Pb4Zn2B10O21 was packed into a Pt crucible that was placed into a vertical programmable temperature furnace. The mixture was melted at 750 °C, held at this temperature for 15 hours, quickly cooled down to 670 °C, then a platinum wire was promptly dipped into the melt. The temperature was decreased to 620 °C at a rate of 3 °C h−1, and finally cooled to room temperature at a rate of 20 °C h−1. Thus, some clear, millimeter size, colorless crystals were discovered. Crystals of suitably high quality were selected for single-crystal XRD under an optical microscope.Structure determination
The crystal structure of Pb4Zn2B10O21 was determined by single-crystal XRD on a Bruker SMART APEX II CCD diffractometer using monochromatic Mo Kα radiation (λ = 0.71073 Å) at 296(2) K and integrated with the SAINT program.15 A colorless and transparent block of crystal with dimensions of 0.21 mm × 0.15 mm × 0.14 mm was chosen for structure determination, unit cell parameters were derived from a least-squares analysis in the range of 1.82 to 27.54°. The data were solved with SHELXS-97 by a direct method, and refined using SHELXL-97.16 The final difference Fourier synthesis map showed the maximum and minimum peaks at 2.306 (0.80 Å from Pb3) and −2.167 e Å−3 (0.80 Å from Pb3), respectively. The structures were checked for missing symmetry elements with PLATON.17 Crystal data and structure refinement information are summarized in Table 1. Final atomic coordinates and equivalent isotropic displacement parameters of the title compound are listed in Table S1 in the ESI.† Selected interatomic distances and angles are given in Table S2 in the ESI.† Inductively coupled plasma optical emission spectrometer (ICP-OES) analysis was also carried out to determine the composition of the crystal. The test was performed on a Varian Vita-Pro CCD simultaneous ICP-OES, which reveals Pb![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn:B = 4:2:9.89.
:Zn:B = 4:2:9.89.
| Empirical formula | Pb4Zn2B10O21 |
|---|---|
| a R 1 = Σ||Fo| − |Fc||/Σ|Fo| and wR2 = [Σw(Fo2 − Fc2)2/ΣwFo4]1/2 for Fo2 > 2σ(Fo2). | |
| Formula weight | 1403.60 |
| Temperature | 296(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Orthorhombic |
| Space group | Pbcn |
| Unit cell dimensions | a = 14.6062(15) Å, b = 17.4899(16) Å, c = 13.2962(15) Å |
| Volume, Z | 3396.7(6) Å3, 8 |
| Density (calculated) | 5.489 Mg m−3 |
| Absorption coefficient | 42.411 mm−1 |
| F(000) | 4848 |
| Theta range for data collection | 1.82 to 27.54° |
| Limiting indices | −18 ⩽ h ⩽ 18, −22 ⩽ k ⩽ 22, −17 ⩽ l ⩽ 16 |
| Reflections collected/unique | 52989/3918 [R(int) = 0.0848] |
| Completeness to theta = 27.54 | 99.7% |
| Refinement method | Full-matrix least-squares on F2 |
| Goodness-of-fit on F2 | 1.030 |
| Final R indices [Fo2 > 2σ(Fo2)]a | R 1 = 0.0289, wR2 = 0.0623 |
| R indices (all data)a | R 1 = 0.0488, wR2 = 0.0702 |
| Extinction coefficient | 0.000090(6) |
| Largest diff. peak and hole | 2.306 and −2.167 e Å−3 |
Infrared spectroscopy
The infrared spectrum was recorded on a Shimadzu IRAffinity-1 Fourier transform infrared spectrometer in the 400–4000 cm−1 range, the sample was mixed thoroughly with dried KBr.UV-vis-NIR diffuse reflectance spectrum
The diffuse reflectance spectrum for the Pb4Zn2B10O21 crystalline sample was measured from 190 to 2600 nm using a Shimadzu SolidSpec-3700DUV spectrophotometer equipped with an integrating sphere attachment.Differential thermal analysis
The thermal behaviors of Pb4Zn2B10O21 were carried out on a NETZSCH STA 449C simultaneous analyzer under static air. The sample was enclosed in Pt crucibles and heated from room temperature to 800 °C at a rate of 10 °C min−1 under flowing nitrogen gas.Numerical calculation details
To investigate a deep structure–property relationship, the electronic structures were obtained using density functional theory (DFT) based ab initio calculations implemented in the CASTEP package.18 The exchange–correlation potential was calculated using the Perdew–Burke–Ernzerhof (PBE) function within the Generalized Gradient Approximation (GGA) scheme.19 For the purpose of achieving energy convergence, a plane-wave basis set energy cutoff was 340.0 eV within the Norm-conserving pseudopotential. And the Monkhorst–Pack scheme was set at 1 × 1 × 1 in the Brillouin Zone (BZ) of the primitive cell for the total energy calculations. The following orbital electrons were treated as valence electrons, Pb, 5d106s26p2; Zn, 3d104s2; B, 2s22p1; O, 2s22p4. Other parameters used in the calculations were set by the default values of the CASTEP code.Results and discussion
Crystal structure
Owing to the high viscosity of melt, the Pb–Zn–B–O system is often considered as glass materials and there are few crystal structures reported, except PbZn2(BO3)2 and Pb8Zn(BO3)6.20,21 And it is also worth noting that, for PbZn2(BO3)2 and Pb8Zn(BO3)6, their basic B–O building blocks are both isolated BO3 triangles and their components are both located in the B-poor region, where the viscosity is relatively lower. However, Pb4Zn2B10O21 possesses a high B content and its component is located in the B-rich region, where the melt viscosity will be high and it is very difficult to crystallize. Therefore, in this sense, Pb4Zn2B10O21 may represent the first example which is crystallized in the B-rich region in the Pb–Zn–B–O system.Pb4Zn2B10O21 crystallizes in the space group Pbcn of the orthorhombic system with a = 14.6062(15) Å, b = 17.4899(16) Å, c = 13.2962(15) Å, and Z = 8. In the asymmetric unit, there are four unique Pb atoms, two unique Zn atoms, ten unique B atoms, and twenty-two unique O atoms. The ten unique B atoms coordinate with three or four O atoms to form BO3 triangles and BO4 tetrahedra, which condense into a new fundamental building block (FBB) [B10O24] group, which can be written as 10:2[3:(△+2T)+(1:△)+(1:T)] according to the definition given by Burns et al. (Fig. 2a).22 Other B–O groups containing ten boron atoms, have also been reported such as the [B10O24] groups in [ThB5O6(OH)6][BO(OH)2]·2.5H2O,23 the [B10O21] groups in M2M′2B10O17 (M = Cs, Tl; M′ = K, Cs)24–26 and Pb6B10O21.27 However, they all exhibit different configurations. In the [B10O24] group of [ThB5O6(OH)6][BO(OH)2]·2.5H2O, six BO4 are connected by four BO3 triangles to form a crown-like [B10O24] group, which can be written as 10:[10:6△+4T]. The [B10O21] group in M2M′2B10O17 (M = Cs, Tl; M′ = K, Cs) can be seen as two [B5O12] groups sharing one O atom and can be written as 10:2[5:3△+2T], while the [B10O21] group in Pb6B10O21 is composed of two [B4O9] groups bridged by one [B2O5] group and can be written as 10:2[4:(2T+2△)+T]. Further, the novel [B10O24] FBB is connected by sharing O atoms to form 2D [B10O21]∞ layers (Fig. 2b), which stack along the c axis and are connected by the isolated ZnO4 tetrahedra to form a [Zn2B10O21]∞ framework with Pb atoms filling in the space of the framework (Fig. 2c). And in Pb4Zn2B10O21, it should be noted that all the O atoms coordinated with Pb atoms fall within the same hemisphere around lead, which indicates that the lone-pairs on all the Pb2+ cations are stereo-active. Owing to the repulsion interactions of the lone pair, the lone pairs on the adjacent Pb2+ cations are as far apart as possible. Thus when the Pb-centered polyhedra are connected with each other to form [Pb4O12]∞ chains, the lone pairs on the adjacent Pb2+ cations are pulled to the outside of the [Pb4O12]∞ chain. Therefore, the [Pb4O12]∞ chain looks reasonably regular (Fig. 2d).
 | ||
| Fig. 2 The crystal structure of Pb4Zn2B10O21, (a) the new fundamental building block [B10O24] group; (b) the 2D [B10O21]∞ layers; (c) the 3D [Zn2B10O21]∞ framework with Pb atoms filling in the space; (d) the regular [Pb4O12]∞ chain. | ||
In addition, owing to the repulsion interactions of the lone pairs of Pb2+ cations, the coordinated environments of the Pb atoms are also complicated. When we only consider the primary coordination sphere, the Pb atoms have three kinds of obviously different coordinated environments. The Pb1 atoms are coordinated by three O atoms with Pb–O distance ranging from 2.293(6) to 2.469(6) Å, the Pb2 and Pb3 atoms are coordinated by four O atoms with Pb–O distance ranging from 2.227(6) to 2.734(6) Å, respectively, and the Pb4 atoms are coordinated by five O atoms with Pb–O distance ranging from 2.231(6) to 2.756(6) Å. The bond valence sums (BVS) of Pb2+ cations obtained by the bond valence calculations28,29 are 1.47, 1.59, 1.36, and 1.75, respectively, which is obviously smaller than the ideal oxidation state of +2. According to Davidovich,30 the coordination environments of Pb atoms are classified as the primary coordination sphere and the secondary coordination sphere. When some longer Pb–O bonds with the lengths ranging from 2.80 to 3.5 Å are considered, the BVS of the Pb atoms are 1.99, 1.94, 1.92, and 1.96, respectively, which are consistent with the oxidation state of Pb2+. The flexible coordination environments of Pb atoms are also observed in other Pb-containing borates.31,32
All the Zn atoms are coordinated by four O atoms and the Zn–O bond lengths range from 1.814(8) to 1.982(6) Å. The B atoms have two coordinated environments, BO3 triangles and BO4 tetrahedra. For the BO3 triangles, the B–O bond lengths range from 1.302(13) to 1.406(11) Å. For BO4 tetrahedra, the B–O bond lengths range from 1.430(11) to 1.517(11) Å. All the bond lengths are also consistent with those observed in other compounds.33,34
Pb4Zn2B10O21 has similar stoichiometry to Pb6B10O21, and from the formula, Pb4Zn2B10O21 seems to be the compound in which the Zn atoms substitute two Pb atoms of Pb6B10O21. However, they exhibit obviously different crystal structures. Pb6B10O21 crystallizes in space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) of the triclinic system, while Pb4Zn2B10O21 crystallizes in space group Pbcn of the orthorhombic system. And as discussed above, in Pb6B10O21 the FBBs are the isolated [B10O21] groups. However, in Pb4Zn2B10O21, the FBBs are the [B10O24] groups and they further condense into 2D [B10O21]∞ layers. The difference may be easy to understand when the coordination environments of the Pb atoms and Zn atoms are considered. Generally the Zn atoms are coordinated by four or six O atoms to form ZnO4 tetrahedra or ZnO6 octahedra. However, owing to Pb possessing the stereo-active lone pairs, the coordination environment of the Pb atoms is more complicated than the Zn atoms. For one thing, the O atoms coordinated with Pb atoms often fall within the same hemisphere around Pb atoms, leading to the Pb-centered polyhedra being extremely distorted. Also, the coordination environments of the Pb atoms are often composed of the primary coordination sphere and the secondary coordination sphere. That is, in Pb-containing compounds, it is often observed that in the coordination environment of the Pb atoms, apart from some normal Pb–O bonds, other longer Pb–O bonds with bond lengths ranging from 2.80 to 3.5 Å must be considered.20,21,31,32 The obvious differences in the coordination environments of the Pb atoms and the Zn atoms often lead to a substantial change in structure when the Zn atoms partly or totally substitute the Pb atoms.35,36
of the triclinic system, while Pb4Zn2B10O21 crystallizes in space group Pbcn of the orthorhombic system. And as discussed above, in Pb6B10O21 the FBBs are the isolated [B10O21] groups. However, in Pb4Zn2B10O21, the FBBs are the [B10O24] groups and they further condense into 2D [B10O21]∞ layers. The difference may be easy to understand when the coordination environments of the Pb atoms and Zn atoms are considered. Generally the Zn atoms are coordinated by four or six O atoms to form ZnO4 tetrahedra or ZnO6 octahedra. However, owing to Pb possessing the stereo-active lone pairs, the coordination environment of the Pb atoms is more complicated than the Zn atoms. For one thing, the O atoms coordinated with Pb atoms often fall within the same hemisphere around Pb atoms, leading to the Pb-centered polyhedra being extremely distorted. Also, the coordination environments of the Pb atoms are often composed of the primary coordination sphere and the secondary coordination sphere. That is, in Pb-containing compounds, it is often observed that in the coordination environment of the Pb atoms, apart from some normal Pb–O bonds, other longer Pb–O bonds with bond lengths ranging from 2.80 to 3.5 Å must be considered.20,21,31,32 The obvious differences in the coordination environments of the Pb atoms and the Zn atoms often lead to a substantial change in structure when the Zn atoms partly or totally substitute the Pb atoms.35,36
Spectroscopic properties
Furthermore, in order to further specify the coordination of boron in the FBBs, the infrared spectrum was measured (Fig. S1 in the ESI†). The strong peaks at 1377, 1298, 1224 and 1124 cm−1 are attributed to the asymmetric stretching of BO3 and BO4, respectively.37 The peaks at 943 and 747 cm−1 are attributed to the symmetric stretching of BO3 and BO4, respectively. The 693 and 651 cm−1 peaks belong to the out of plane bending of BO3. The peaks at 587 and 491 cm−1 are attributed to the bending of BO3 and BO4. It further confirms the existence of BO3 triangles and BO4 tetrahedra, and is consistent with the results obtained from the single-crystal X-ray structural analysis.The UV-vis-NIR diffuse-reflectance spectrum for Pb4Zn2B10O21 in the region 190–2600 nm is displayed in Fig. 3. The reflectance spectrum was converted to absorbance with the Kubelka–Munk function.38,39 It is clear that Pb4Zn2B10O21 has a wide transmission range with a UV absorption edge at 308 nm.
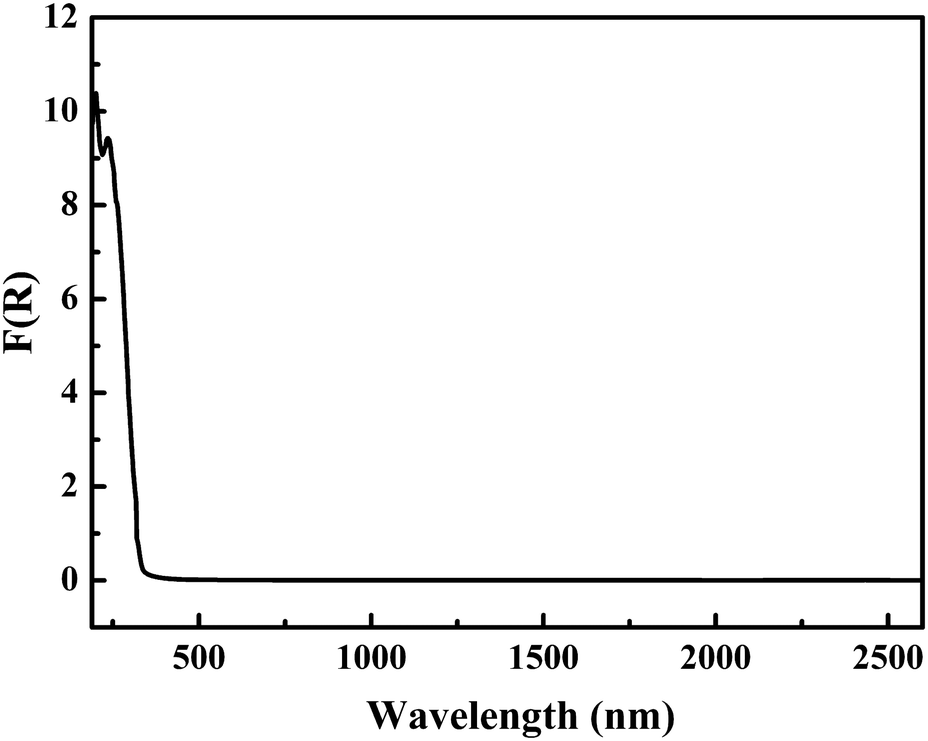 | ||
| Fig. 3 The UV-vis-NIR diffuse reflectance spectrum of Pb4Zn2B10O21. | ||
TG and DSC analysis
The thermal behavior of Pb4Zn2B10O21 is measured at a range of 50 to 800 °C. There is one endothermic peak at 658 °C on the DSC curve and there is no weight loss on the TG curve (Fig. 4), which tentatively suggests that Pb4Zn2B10O21 melts congruently at 658 °C. In order to further confirm the thermal behavior of Pb4Zn2B10O21, the solid samples of Pb4Zn2B10O21 are placed into a platinum crucible and heated to 750 °C, then slowly cooled to room temperature. Analysis of the powder XRD pattern of the solidified melt reveals that the solid product exhibits a diffraction pattern identical to that of the initial Pb4Zn2B10O21 powder (shown in Fig. 1), further demonstrating that Pb4Zn2B10O21 is a congruently melting compound, which indicates that it is easy to grow large single crystals of Pb4Zn2B10O21.40 | ||
| Fig. 4 The TG and DSC curves of Pb4Zn2B10O21. | ||
Electronic band structures
In order to examine the band structure and to better understand the relationship between electronic structures and optical properties, the electronic structure of Pb4Zn2B10O21 was calculated using density functional theory (DFT) based ab initio calculations implemented in the CASTEP package. The calculated band structures along the high symmetry lines in the unit cell are shown in Fig. 5a. It is clear that the state energies (eV) of the lowest conduction band and the highest valence band of the compound both locate at the G point, indicating that Pb4Zn2B10O21 is a direct band gap compound with a band gap of 3.53 eV. Furthermore, Fig. 5b shows the partial density of states (DOS) and total DOS of Pb4Zn2B10O21. After removing the contribution of kernel electronic orbitals, the energy band is composed of four regions. The lowest region from −21.4 to −17.1 eV mostly originates from the O 2s state, mixed with a small amount of B-2s2p states. The middle part of the valence band (−15.8 to −14.2 eV) is attributed to the isolated Pb 5d state. Around the Fermi level, the top of the valence band is mostly derived from the contributions of O 2p states with a small amount of Pb 6s, Zn 3d, B 2s2p states, and the conduction band is mainly derived from the Pb 6p state, which could contribute to the optical properties of the crystal in the visible and UV spectra. | ||
| Fig. 5 (a) Calculated band structure of Pb4Zn2B10O21; (b) the density of states of Pb4Zn2B10O21. | ||
Conclusions
In conclusion, a new borate, Pb4Zn2B10O21, has been synthesized and single crystals have been grown by the high-temperature solution method for the first time. It contains a new FBB [B10O24] group. Owing to the repulsion interactions between the lone pairs, the Pb-centered polyhedra connect with each other to form an interesting and regular [Pb4O12]∞ chain. Furthermore, the infrared spectrum confirms the existence of BO3 and BO4 units in the basic building block. The UV-vis-NIR diffuse reflectance spectrum shows that the absorption edge of Pb4Zn2B10O21 is about 308 nm and TG-DSC measurement shows that Pb4Zn2B10O21 melts congruently, which suggests that it is easy to grow large single crystals of Pb4Zn2B10O21 for applications. Furthermore, the first principles studies reveal that the title compound is a direct-gap compound with band gaps of 3.53 eV, the charge transfers from O 2p states to Pb 6p states contribute to the observed absorption edge from the UV-vis-NIR absorption spectrum.Acknowledgements
This work is supported by the National Key Basic Research Program of China (Grant No. 2012CB626803), the “National Natural Science Foundation of China” (Grant No. 21201176, U1129301, 51172277, 21101168, 11104344), Main Direction Program of Knowledge Innovation of CAS (Grant No. KJCX2-EW-H03-03), the “One Hundred Talents Project Foundation Program” of CAS, Major Program of Xinjiang Uygur Autonomous Region of China during the 12th Five-Year Plan Period (Grant No. 201130111), the “High Technology Research and Development Program” of Xinjiang Uygur Autonomous Region of China (Grant No. 201315103, 201116143), and the “Western Light Foundation” Program of CAS (Grant No. XBBS201214).Notes and references
- (a) P. Becker, Adv. Mater., 1998, 10, 979 CrossRef CAS; (b) C. T. Chen, Y. B. Wang, B. C. Wu, K. C. Wu, W. L. Zeng and L. H. Yu, Nature, 1995, 373, 322 CrossRef CAS; (c) H. P. Wu, H. W. Yu, Z. H. Yang, X. L. Hou, X. Su, S. L. Pan, K. R. Poeppelmeier and J. M. Rondinelli, J. Am. Chem. Soc., 2013, 135, 4215 CAS; (d) H. P. Wu, H. W. Yu, S. L. Pan, Z. J. Huang, Z. H. Yang, X. Su and K. R. Poeppelmeier, Angew. Chem., Int. Ed., 2013, 52, 3406 CrossRef CAS PubMed.
- (a) D. F. Xue, K. Betzler, H. Hesse and D. Lammers, Solid State Commun., 2000, 114, 21 CrossRef CAS; (b) S. C. Wang and N. Ye, J. Am. Chem. Soc., 2011, 133, 11458 CrossRef CAS PubMed; (c) S. C. Wang, N. Ye, W. Li and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8779 CAS; (d) F. Kong, H. L. Jiang, T. Hu and J. G. Mao, Inorg. Chem., 2008, 47, 10611 CrossRef CAS PubMed.
- (a) S. C. Neumair, J. S. Knyrim, O. Oeckler, R. Glaum, R. Kaindl, R. Stalder and H. Huppertz, Chem.–Eur. J., 2010, 16, 13659 CrossRef CAS PubMed; (b) R. K. Li and P. Chen, Inorg. Chem., 2010, 49, 1561 CAS; (c) H. W. Yu, H. P. Wu, S. L. Pan, Y. Wang, Z. H. Yang and X. Su, Inorg. Chem., 2013, 52, 5359 CrossRef CAS PubMed.
- (a) E. Belokoneva, Crystallogr. Rev., 2005, 11, 151 CrossRef CAS; (b) Y. Yang, S. L. Pan, X. L. Hou, C. Y. Wang, K. R. Poeppelmeier, Z. H. Chen, H. P. Wu and Z. X. Zhou, J. Mater. Chem., 2011, 21, 2890 RSC.
- C. T. Chen, B. C. Wu, A. D. Jiang and G. M. You, Sci. Sin., Ser. B, 1985, 28, 235 Search PubMed.
- C. T. Chen, Y. C. Wu, A. D. Jiang, G. M. You, R. K. Li and S. J. Lin, J. Opt. Soc. Am. B, 1989, 6, 616 CrossRef CAS.
- Y. C. Wu, T. Sasaki, S. Nakai, A. Yokotani, H. Tang and C. T. Chen, Appl. Phys. Lett., 1993, 62, 2614 CrossRef CAS.
- Y. Mori, I. Kuroda, S. Nakajima, T. Sasaki and S. Nakai, Appl. Phys. Lett., 1995, 67, 1818 CrossRef CAS.
- X. Y. Dong, H. P. Wu, Y. J. Shi, H. W. Yu, Z. H. Yang, B. B. Zhang, Z. H. Chen, Y. Yang, Z. J. Huang, S. L. Pan and Z. X. Zhou, Chem.–Eur. J., 2013, 19, 7338 CrossRef CAS PubMed.
- A. Choudhury, S. Neeray, S. Natarajan and C. N. R. Rao, J. Chem. Soc., Dalton Trans., 2002, 1535 RSC.
- H. Huppertz and B. von der Eltz, J. Am. Chem. Soc., 2002, 124, 9376 CAS.
- S. Jin, G. Cai, W. Wang, M. He, S. Wang and X. Chen, Angew. Chem., Int. Ed., 2010, 49, 4967 CrossRef CAS PubMed.
- Y. Wu, J. Y. Yao, J. X. Zhang, P. Z. Fu and Y. C. Wu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, i45 CAS.
- X. A. Chen, Y. Chen, C. Sun, X. Chang and W. Xiao, J. Alloys Compd., 2013, 568, 60 CrossRef CAS PubMed.
- SAINT, version 7.60A, Bruker Analytical X-ray Instruments, Inc., Madison, WI, 2008 Search PubMed.
- G. M. Sheldrick, SHELXTL-97 Sheldrick: Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Rrfson and M. C. Payne, Z. Kristallogr., 2005, 220, 568 Search PubMed.
- D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566 CrossRef CAS.
- X. A. Chen, Y. H. Zhao, X. A. Chang, L. Zhang and H. P. Xue, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, i11 Search PubMed.
- Z. J. Huang, S. L. Pan, Z. H. Yang, H. W. Yu, X. Y. Dong, W. W. Zhao, L. Y. Dong and X. Su, Solid State Sci., 2013, 15, 73 CrossRef CAS PubMed.
- P. C. Burns, J. D. Grice and F. C. Hawthorne, Can. Mineral., 1995, 33, 1131 CAS.
- (a) S. Wang, E. V. Alekseev, J. Diwu, W. H. Casey, B. L. Phillips, W. Depmeier and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2010, 49, 1057 CrossRef CAS PubMed; (b) S. Wang, E. V. Alekseev, W. Depmeier and T. E. Albrecht-Schmitt, Chem. Commun., 2011, 47, 10874 RSC.
- M. Touboul, N. Penin and G. Nowogrocki, Solid State Sci., 2003, 5, 1327 CrossRef CAS.
- J. M. Tu and D. A. Keszler, Inorg. Chem., 1996, 35, 463 CrossRef CAS PubMed.
- N. Penin, M. Touboul and G. Nowogrocki, J. Alloys Compd., 2004, 363, 104 CrossRef CAS.
- J. Krogh-Moe and P. S. Wold-Hansen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 2242 CrossRef CAS.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192 CrossRef.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- R. L. Davidovich, V. Stavila and K. H. Whitmire, Coord. Chem. Rev., 2010, 254, 2193 CrossRef CAS PubMed.
- (a) H. R. Tian, W. H. Wang, Y. E. Gao, T. T. Deng, J. Y. Wang, Y. L. Feng and J. W. Cheng, Inorg. Chem., 2013, 52, 6242 CrossRef CAS PubMed; (b) H. R. Tian, W. H. Wang, X. P. Zhang, Y. L. Feng and J. W. Cheng, Dalton Trans., 2013, 42, 894 RSC.
- J. L. Song, C. L. Hu, X. Xu, F. Kong and J. G. Mao, Inorg. Chem., 2013, 52, 8979 CrossRef CAS PubMed.
- H. W. Yu, S. L. Pan, H. P. Wu, J. Han, X. Dong and Z. X. Zhou, J. Solid State Chem., 2011, 184, 1644 CrossRef CAS PubMed.
- M. Zhang, S. L. Pan, J. Han, Z. H. Yang, X. Su and W. W. Zhao, J. Solid State Chem., 2012, 190, 92 CrossRef CAS PubMed.
- M. Martinez-Ripoll, S. Martinez-Carrera and S. Garcia-Blanco, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 672 CrossRef CAS.
- D. L. Corker and A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Sci., 1996, 52, 260 Search PubMed.
- (a) A. Rulmont and M. Almou, Spectrochim. Acta, Part A, 1989, 45, 603–610 CrossRef; (b) H. W. Yu, H. P. Wu, S. L. Pan, Y. Wang, Z. H. Yang and X. Su, Inorg. Chem., 2013, 52, 5359 CrossRef CAS PubMed.
- P. Kubelka and F. Z. Munk, Tech. Phys., 1931, 12, 593 Search PubMed.
- J. Tauc, Mater. Res. Bull., 1970, 5, 721 CrossRef CAS.
- G. Aka, F. Mougel, F. Auge, A. Kahn-Harari, D. Vivien, J. M. Benitez, F. Salin, D. Pelenc, F. Balembois, P. Georges, A. Brun, N. Le Nain and M. Jacquet, J. Alloys Compd., 2000, 401, 408 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The infrared spectrum of Pb4Zn2B10O21, atomic coordinates and equivalent isotropic displacement parameters, and selected bond distances and angles tables for Pb4Zn2B10O21. CCDC 954072. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj00893b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |