Cu(II) conjugation along the transformation of a vitamin K3 derivative to a dinaphthoquinone methide radical†
Kirti D.
Badave
a,
Shalaka S.
Patil
b,
Ayesha A.
Khan
b,
Darbha
Srinivas
c,
Raymond J.
Butcher
d,
Rajesh G.
Gonnade
e,
Vedavati G.
Puranik
e,
Rahul V.
Pinjari
a,
Shridhar P.
Gejji
a and
Sandhya Y.
Rane
*a
aDepartment of Chemistry, University of Pune, Pune-411007, India. E-mail: syrane@chem.unipune.ac.in
bInstitute of Bioinformatics and Biotechnology, University of Pune, Pune-411007, India
cCatalysis Division, National Chemical Laboratory, Pune-411008, India
dChemistry Department, Howard University, D.C. 20059, USA
eCentre for Material Characterization, National Chemical Laboratory, Pune-411008, India
First published on 11th October 2013
Abstract
1,1′-Methide-bi-vitamin K3 (B) has been isolated as a dinaphthoquinone methide radical (DNQM) by the transformation of 1-imino(acetylhydrazino)-vitamin K3 (A). The transformation follows a biomimetic activation pathway mediated via Cu(II) ion catalyzed oxidative coupling. Single crystal X-ray and electron spin resonance (ESR) experiments combined with density functional calculations elucidate the “resonance structure” of the DNQM radical (B). Fluorescence investigations reveal that DNQM facilitates interaction with the cysteine residue. As compared to the parent substrate, B shows a depletion in the level of GSH, triggering apoptosis in HeLa cells.
1. Introduction
Glutathione S-transferases (GSTs) are detoxification enzymes that catalyze the conjugation reaction where glutathione (GSH) combines with different endogenous and exogenous electrophilic compounds. A variety of stimuli initiate a state of increased resistance to apoptosis, bringing about the elevation of GST levels.1–3 GSTs are known to be a promising therapeutic target. Moreover, GSTs act as an inhibitor of the mitogen-activated protein kinase (MAPK) pathway. Weydert et al.4 demonstrated that the use of 1,3-bis(2-chloroethyl)-1-nitrosourea GSH depletion chemotherapy, in combination with superoxide dismutase gene-therapy, has proved successful in the treatment of breast cancer.Any change in the intracellular GSH/GSSG ratio is critical for the activation of cell proliferation and cell death and thus a crucial consideration in treatments affecting intracellular GSH levels. The cells are more vulnerable to reactive oxygen species (ROS) attack at low intracellular GSH levels, while increased levels of ROS activate different intracellular oncogenic pathways or mutate a tumor suppressor gene pathway, thereby activating tumorigenesis.5,6 Thus changing ROS levels by GSH modulation provides a way to kill cancer cells selectively without causing significant toxicity to normal cells.7
It is known that bioactivation of the antiestrogenic drug tamoxifen forms quinone/diquinone methide intermediates that alkylate DNA and form GSH adducts.8 The data regarding the reactivity, rate of formation, and other potential biological targets of quinone methides are scanty. Quinone/diquinone methides are the intermediates which bring about the bioactivation of a selective estrogen receptor modulator in the treatment of breast cancer9 or osteoporosis.10 The transient intermediates are stabilized by the formation of metalloquinones8 or quinone methide organometallic compounds.11a,b Pursuant to this, GSH depletion12,13via the hybrid drug NO-ASA induces apoptosis, substantiating the key role of quinone methides.
Interestingly, the quinone methide as a transient organic radical structure has not yet been investigated. The present work focuses on the isolation and characterization of this radical using single crystal X-ray structure elucidation, electron spin resonance (ESR) and theoretical studies on 1,1′-methide-bi-vitamin K3 (B) as a dinaphthoquinone methide radical (DNQM), by the transformation of 1-imino(acetylhydrazino)-vitamin K3 (A)14,15 mediated via Cu(II) conjugation. To probe the chemical reactivity and toxicity of this electrophile, a reaction between DNQM and GSH has been carried out. The cellular levels of the enzyme glutathione reductase (GR) were measured, which provides an indirect method to study the GSH pool in HeLa cell lines.
2. Result and discussion
2.1 Crystal structure of compound B
Single crystal structure analysis reveals that compound B belongs to the centrosymmetric triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule in the asymmetric unit comprising two naphthoquinone rings connected by a C
with one molecule in the asymmetric unit comprising two naphthoquinone rings connected by a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (C8–C19, 1.403(2) Å) bond. Both naphthoquinone rings oriented at an angle of ∼41.2(1)° with respect to each other (Fig. 1) (C9–C8–C19–C20 torsion angle −141.3(2)°). The phenyl ring in both naphthoquinone groups shows a deviation up to ∼7(1)° from planarity and renders a slightly convex naphthoquinone moiety. The bond between C(8)–C(19), connecting the two naphthoquinone rings was observed to be 1.403(2) Å which is intermediate between C–C and CC bonds,16 points to “resonance structure” in its molecular conformation.
C (C8–C19, 1.403(2) Å) bond. Both naphthoquinone rings oriented at an angle of ∼41.2(1)° with respect to each other (Fig. 1) (C9–C8–C19–C20 torsion angle −141.3(2)°). The phenyl ring in both naphthoquinone groups shows a deviation up to ∼7(1)° from planarity and renders a slightly convex naphthoquinone moiety. The bond between C(8)–C(19), connecting the two naphthoquinone rings was observed to be 1.403(2) Å which is intermediate between C–C and CC bonds,16 points to “resonance structure” in its molecular conformation.
 | ||
| Fig. 1 ORTEP of 1,1′-methide-bi-vitamin K3 (B) showing the atom numbering scheme. Displacement ellipsoids are drawn at the 50% level. H atoms are shown as small spheres of arbitrary radii. | ||
Further, the C(9)–C(10) and C(20)–C(21) bond lengths are shorter than the rest of the C–C bonds of the ring by 0.092–0.120(2) Å. The carbonyl bond lengths [C(1)–O(1) = 1.2334(18) Å and C(12)–O(2) = 1.2278(18) Å] fall in the range of quinonoidal compounds.17 All of these structural features indicate the extended π conjugation character of the coupled rings.
Interestingly, the π⋯π interactions between the naphthoquinone moieties can be inferred (Fig. 2a and b). The carbonyl group herein paves the way for weak hydrogen bonding C–H⋯O interactions in molecular association. Further, among the aromatic π⋯π stacking interactions that exist between the molecules, the one associating the benzenoidal rings across the inversion center (symmetry code: 2 − x, 1 − y, 1 − z) seems to be relatively strong; the interplanar spacing being 3.531(1) Å. An overlap of these rings forms a convex shape dimer (Fig. 2a). This exposes the quinonoidal rings to either end of the benzenoidal π⋯π dimer without engaging in any significant interactions along the convex shape dimer. The adjacent convex shape dimers are connected to each other along the a-axis via another strong centrosymmetric π⋯π stacking interaction involving both the phenyl rings of the naphthoquinone groups, forming a concave shape dimer in this association18a (Fig. 2b).
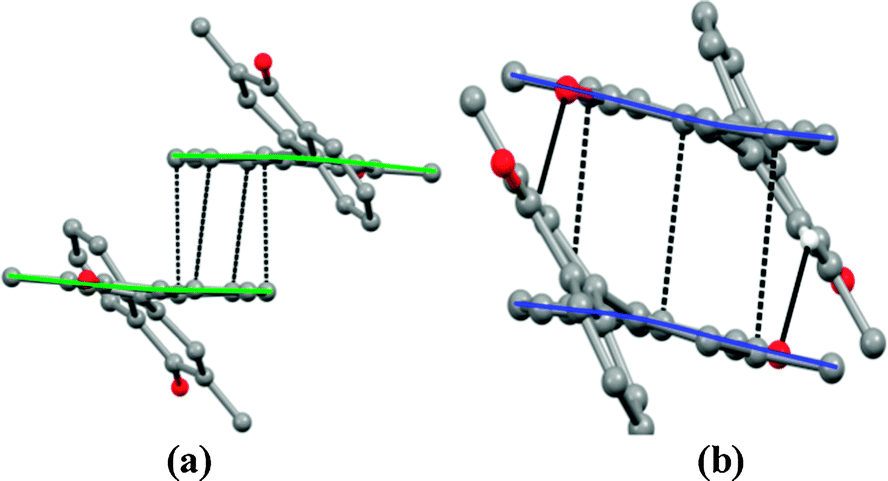 | ||
| Fig. 2 (a) Centrosymmetric π⋯π interactions mediated a convex shape dimer and (b) centrosymmetric π⋯π and C–H⋯O interactions favored a concave shape dimer. | ||
The overlap of the benzenoidal and quinonoidal rings (Cg⋯Cg = 3.623(1) Å, dihedral angle = 6.33(7)°, symmetry code: 1 − x, 1 − y, 1 − z, Cg – centroid of the benzene ring) yields a concave shape dimer. A relatively short (H20⋯O1*: 2.58 Å; symmetry code: (*) 1 − x, 1 − y, 1 − z) bond can be noticed. Nonlinear C20–H20⋯O1*, deviating up to ∼40° from linearity, suggests the presence of attractive long range interactions which supplements π⋯π stacking assembly. Such convex and concave shape dimeric assembly along the a-axis engenders layered arrangement wherein only one of the naphthoquinone moieties is involved in π⋯π stacking whereas the others protrude out from both ends of the stacking assembly (Fig. 3a). Such neighboring parallel layers are stitched together along the direction of the c-axis, engaging the protruded naphthoquinone moieties in π⋯π stacking interactions that lead to a sheet of intercalated layers (Fig. 3b).
 | ||
| Fig. 3 (a) Crystal structure showing the aromatic π⋯π and C–H⋯O interactions, (b) zipping of parallel layers through π⋯π interactions. Atom H20 is at the position x, y, z and atom O1 marked with * is at the symmetry equivalent position 1 − x, 1 − y, 1 − z. | ||
The zipping of such layers renders π⋯π interactions between the benzenoidal phenyl rings, and the Cg⋯Cg distance turns out to be 3.604(1) Å. Both phenyl rings from the naphthoquinone moieties along the zipped layer show Cg⋯Cg distance between the rings is somewhat longer at 3.904(1) Å. The interplay of such π⋯π interactions and C–H⋯O interactions from carbonyl oxygen O2 potentially govern the crystal packing. Details regarding calculating the lattice energies using the OPIX program18b are given in the ESI.†
The extended intramolecular π conjugation and the intermediate bond lengths between C(8)–C(19) indicate that the radical structure for B shown in the single crystal X-ray structure is also shown by the ESR experiment.
Formation of such a radical via the delocalization of electron density on one of the methyl groups of B is inferred from ESR spectroscopy (Fig. 4). Polycrystals [and a frozen DMSO solution Fig. S4, ESI†] of B showed a spectrum containing four hyperfine features centered at giso = 2.0049 with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3:1. This suggests that they arise from the interaction of electron spin with the nuclear spin of three protons of the methyl group of B. The relatively large hyperfine coupling constant (aH = 8 G) is attributed to the hyper-conjugation mechanism of the radical electron in the π-system.19 Due to the lack of π spin orbital overlap interactions in the angular quinonoidal rings (distance >3.9 Å)20 the species B (Fig. 2) would not exhibit antiferromagnetism. Magnetic susceptibility studies confirmed its paramagnetic nature (Fig. S5, ESI†).
:3:3:1. This suggests that they arise from the interaction of electron spin with the nuclear spin of three protons of the methyl group of B. The relatively large hyperfine coupling constant (aH = 8 G) is attributed to the hyper-conjugation mechanism of the radical electron in the π-system.19 Due to the lack of π spin orbital overlap interactions in the angular quinonoidal rings (distance >3.9 Å)20 the species B (Fig. 2) would not exhibit antiferromagnetism. Magnetic susceptibility studies confirmed its paramagnetic nature (Fig. S5, ESI†).
 | ||
| Fig. 4 X-band ESR spectrum of polycrystals of B at 300 K. | ||
2.2 Theoretical studies on compound B
The electronic structure of B in the neutral and radical states was obtained by employing the B3LYP theory. The neutral state exhibited an EPR silent nature while the radical form showed EPR with the g value being 2.0044, which agrees well with the experiment. Fig. 5 displays frontier orbitals in B. Thus, the HOMO has largely been localized near methyl protons of the quinonoidal ring. Moreover, the high electron density near the methyl proton of the radical is evident from the SOMO. | ||
| Fig. 5 HOMO, LUMO and SOMO in neutral and radical B. | ||
Methyl carbon possesses a relatively large residual negative charge (−0.714) in the molecule that further prohibits the approach of another molecule conducing dimer formation (owing to relatively strong electron repulsion interactions). The Fermi contact coupling constant for atom centers in B (radical) are displayed in Table S2 (see the ESI†). As may readily be noticed, the bridging carbon centers (C8 and C19) and the methyl substituted carbons (C10 and C21) reveal relatively large Fermi contact couplings (5.35 G and 4.92 G, respectively).
2.3 Solution studies on compound B
 | ||
| Fig. 6 Cyclic voltammogram of B in 0.1 M tetraethyl ammonium perchlorate in DMSO at 100 mV s−1 scan rate. | ||
Such a redox couple is assigned to Cu(II)/Cu(I) in the [Cu(I)(biQ)2]BF4 polymer, where the trapped Cu(I) ion is quantized after eight units.21 But, in our case the copper ion concentration is relatively very low (∼0.14%), so we do not suspect Cu(II)/Cu(I) to be a possibility and instead assign the redox couple for the one electron transfer (ΔE ∼ 0.075 V) in B as NSQ ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) NQ valence tautomers.22
NQ valence tautomers.22
 | ||
| Fig. 7 UV-Visible spectra of (A) acetonitrile and (B) DMSO solutions of (a) A, (b) A in sodium methoxide and (c) B. | ||
 | ||
| Scheme 1 The probable activation pathway for oxidative coupling in the formation of 1,1′-methide-bi-vitamin K3 (DNQM radical), with Cu(II) conjugation. | ||
B is formed in 70% yield with a concentration of Cu(II) species as low as 0.25 mol with respect to 1 mol of A. Without Cu(II) ions this transformation is not possible at all, as confirmed from experimental trials (see the ESI†). Scheme 1 displays the formation of transient species B, of the quinone methide category.8
The first step of the single electron transfer (SET) process24 which occurs has been partly attributed to the conjugation of Cu(II) ions assisted by resonance, with the probable elimination of Cu(I)–hydrazo complex ions. A biradical nucleophile is formed at the C-1 position simultaneously. An open shell nucleophile yields a carbonium ion as a resonance tautomer12 in the subsequent step, with the positive charge being delocalized over the phenyl ring. As pointed out earlier in the literature, in the resonance stabilized quinone methides the carbocation contributes significantly to the charged aromatic resonance.9,26,27 On parallel lines, the stabilization of positive charge accompanying the cleavage of the NAG–NAM substrate28 in the lysozyme enzyme has been explained. Accordingly the sporadically employed sp3–*sp2 oxidative coupling shown in Scheme 1, engenders resonance tautomers. Following this, the C–C coupled dimer undergoes proton electron transfer (PET)20 with concomitant DNQM radical (B) formation. SET or electrophilic metallation24 signifies initiation and radical formation via monodentate Cu(II) coordination. The free radical transfer via π-conjugation followed by 1,4 elimination leaves Cu(I) hydrazino complex ions in solution.
In summary, a transient QM formation in literature is speculative29 but in our case unique oxidative dimerization process with Cu(II) assistance leads to formation of a biradical nucleophile (I) coupled with the ionic enolate form (II)25 undergoes the PET process in a protic solvent to generate stabilized DNQM (B) radical in the crystal. The radical form is stable in protic and aprotic solvents (Fig. S3, ESI†). Oxidative coupling cyclization with Cu(II) assistance has been reported in the recent literature.30 In the copper catalyzed polymerization of 2,6-dimethylphenol (DMP), the radical species is shown to have a very short life time.31 It has, therefore, been conjectured that Cu(II) conjugation initiates a SET process in A that renders selective stability to the vitamin K3 radical segment of the DNQM metabolite. The biomimicry accompanying the formation of 1,1′-methide-bi-vitamin K3 as a metabolite via Cu(II) conjugation in a stable valence tautomeric radical form renders antioncogenic candidature to the vitamin K3 family,32 as follows.
2.4 Reactivity of the hybrid drug (A), DNQM (B) and vitamin K3 with GSH
A higher level of GSH is important for normal cellular functions, signal transduction and protection against certain carcinogens. However, this high level (whether induced by certain drugs or as normal response to stimulants) can slow down any effective cancer treatment that works by increasing intracellular ROS.On the other hand, when intracellular GSH levels are low due to drugs such as BSO (Buthionine sulphoximine), the cells are more vulnerable to ROS attacks. Excessive levels of ROS stress can be toxic to the cancer cells, and cells are likely to be more vulnerable to damage by further ROS induced by exogenous drugs, making them more responsive to ROS producing cancer treatments. Therefore, changing ROS levels by GSH modulation is a way to selectively kill cancer cells without causing significant toxicity to normal cells.33 The characteristic ∼2.5 times depletion of the GSH level in vivo by the isolated DNQM metabolite (B) compared to the hybrid drug A15 supports its anticancer behavior (Table 1).
| Sr. no. | Compound | Specific activity (in vitro) (μmol L−1 min−1) | Specific activity (in vivo) in HeLa cells (μmol L−1 min−1) |
|---|---|---|---|
| 1 | Vitamin K3 | 81.25 | 63.25 |
| 2 | A | 472.76 | 300.25 |
| 3 | B | 128.63 | 117.54 |
Glutathione reductase (GR) is a ubiquitous 100–120 kD dimeric flavoprotein that catalyzes the reduction of oxidized glutathione (GSSG) to reduced glutathione (GSH) using the hydrogen donor β-nicotinamide dinucleotide phosphate (NADPH). In vivo, GR activity is regulated through a redox inter-conversion mechanism mediated by GSSG regulation of the NADPH generating pathways.
Further, the GSH activity was measured for the various compounds. The GSH assay (Fig. 8) shows the comparable efficient catalytic activities of the glutathione reductase enzyme for A and B as compared to the parent drug menadione (vitamin K3). A two electron reduction of GR in the presence of NADPH produces an FAD semiquinone, sulfur radical and thiol. Purified GR tends to form aggregates in the absence of thiols and these aggregates conserve full enzymatic activity, which subsequently can be assayed by fluorescence measurements using sulfur-containing amino acids.
 | ||
| Fig. 8 Fluorescence microscopic images. The HeLa cells were able to take up the drug at 25 μM concentrations within 24 h of incubation. | ||
Menadione retains its activity in the presence of the cysteine moiety and hence no fluorescence quenching was observed, while in the case of B fluorescence quenching was observed at 460 as well as 550 nm (Fig. 9). In other words, B modulates the glutathione reductase enzyme. All of these compounds do not interact with non-sulfurated amino acids such as phenylalanine, valine or histidine.
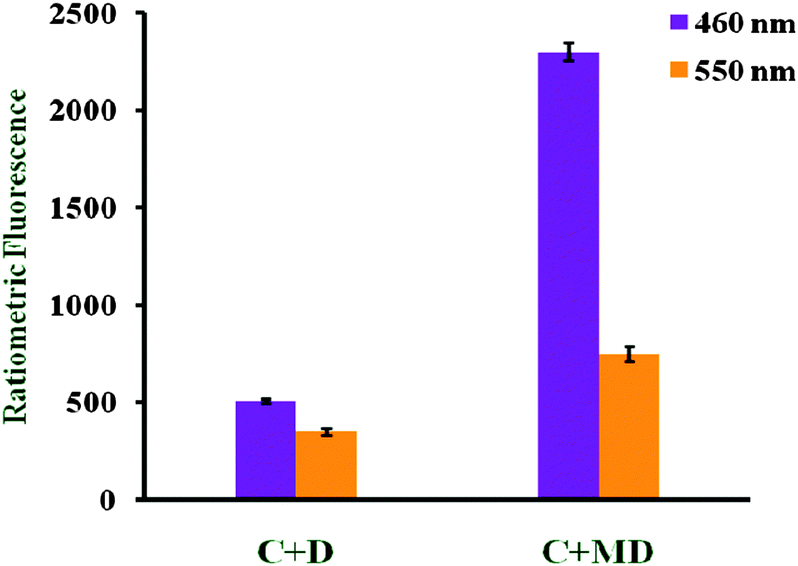 | ||
| Fig. 9 Cysteine (C) interaction with B (D) and menadione (MD). | ||
3. Experimental
3.1 General
All the reagents were of analytical grade. 3-Methyl-1,4-naphthoquinone (menadione), acetyl hydrazide and copper perchlorate hexahydrate were obtained from Sigma-Aldrich. Solvents used in synthesis and crystallization were of HPLC grade and purchased from RANKEM (India). The progress of the reaction was monitored by thin layer chromatography with F254 silica-gel precoated sheets (Merck) using hexane–acetone (60/40) as eluent; UV light was used for detection.3.2 Isolation of 1,1′-methide-bi-vitamin K3 (B)
Compound A (0.01 mol) and triethyl amine or sodium methoxide (0.01 mol) were dissolved in 30 ml of methanol while heating at refluxing temperature for 15 min. Then, a methanolic solution (5 ml) of Cu(ClO4)2·6H2O (0.01 mol) was added drop wise and the contents were refluxed for 18 h. The dark red reaction mixture so obtained was left at 298 K for crystallization. The red crystals with green fluorescence of 1,1′-methide-bi-vitamin K3 were isolated by filtration, rinsed thoroughly with methanol and dried at 298 K.3.3 Preparation for fluorescence study
A stock solution (10 mM) of the compounds in DMSO was prepared and used by dilution in aqueous DMSO solution for in vitro and in vivo fluorescence experiments. In a typical experiment, test solutions (10 mM) were prepared by adding an appropriate amount of each amino acid, and diluting the solution to 2 ml with buffered aqueous DMSO (0.10 M HEPES, pH 7.4). To conduct the fluorescence study, a 96 well flat bottom microtiter polystyrene black plate was used, to which 50 μl of compound and 150 μl of DMSO (0.10 M HEPES, pH 7.4) were added as a control. Equal amounts of AA (amino acid:compound = 1:1) were placed into the wells to check the selectivity of the compounds towards biothiols like cysteine and the GSH solution was diluted to 2 ml with (4:1) DMSO:HEPES buffer. Normally, excitation was at 420 nm. Fluorescence spectra were monitored 1 h after the addition of amino acids.
3.4 Fluorescence cell imaging of HeLa cells
For the detection of biothiols in live cells, HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 units per ml penicillin, 100 μg ml−1 streptomycin, and 10% heat-inactivated fetal bovine serum. The cells were seeded on a Ø 35 mm glass-bottomed dish at a density of 1 × 105 cells in a culture medium overnight for live-cell imaging by fluorescence microscopy on a SpectraMax M5. The HeLa cells were treated with 1 mM of compound in 2 ml of serum free medium for 0.5 h and washed 3 times with prewarmed 1 × PBS before imaging.To enhance the concentration of cellular GSH, HeLa cells were treated with α-lipoic acid (LPA, 500 μM) for 24 h, followed by the compounds for 0.5 h, and then the cellular imaging experiment was performed for the live cells. In order to reduce the concentration of GSH, HeLa cells were treated with N-ethylmaleimide (NEM, 100 μM) for 0.5 h, followed by addition of the compounds for 0.5 h, and the cellular images were taken under a fluorescence microscope by using three excitation channels (λex = 405 nm, 488 nm, and 555 nm).
After obtaining the live cell images, the mean fluorescence intensities in the blue (λex = 405 nm) and green (λex = 488 nm) channels were measured in three different fields. In order to reduce errors caused by background images outside of the cells, we also compared the intensity of the background image, but the level of the intensity is very low indicating that it did not seem to affect the mean fluorescence intensities.
3.5 Preparing samples for the GSH activity test
Dissolved samples (10 mM) were diluted to 1 mM in 1% DMSO (diluted with Tris buffer to make 100%). 100 μl of each sample was added to a 24 well plate containing trypsinized HeLa cells and incubated for 1 h. In the control well plate 1% DMSO with buffer was added to make up the volume. The samples were transferred to micro-centrifuge tubes and centrifuged in cold conditions at 1000 rpm for 5 min. These were kept overnight at 193 K for storage. Frozen samples were thawed at room temperature after 24 h of incubation and vortexed well. They were then centrifuged at 277 K and 1000 × g to obtain cell pellets. The acquired pellets were suspended in Tris buffer (200 μl). Cell samples that had been flash frozen were sonicated or freeze dried. The supernatant was drawn from the top of the tube for the assay (sample for reaction) and cell debris was discarded. The assay buffer was stored at 275–277 K. NADPH and oxidized glutathione were stored at 273 K and prepared freshly before use.3.6 GSH activity assay
The reaction mixture contained the following: T.E buffer 1700 μl, GSSG 100 μl, sample 100 μl. The contents of the cuvette were mixed thoroughly by covering with parafilm and inverting gently several times. NADPH (100 μl) was added after 5 min of incubation. The spectrophotometer was allowed to warm up for at least 15 min and the kinetic parameters were set as follows: wavelength 340 nm, lag time 40 s, rate time 60 s, total measurement time 300 s, and read intervals every 30 s. The assay was run at 298 K. The spectrophotometer was calibrated at 340 nm with deionized water. The change in A340 was recorded for 300 sec at 30 s intervals.The values obtained from the control well were subtracted from the calculations. For each mol of GSSG reduced, one mol of NADPH is oxidized with a concomitant loss of absorbance at 340 nm. One unit of GR activity is defined as the amount of enzyme that will reduce 1 μmol GSSG per minute at pH 7.6 and 25 °C.
3.7 Characterization studies
4. Conclusions
In summary, we have isolated a DNQM radical (B) form of a transient species from a hybrid drug of the vitamin K3 family viz.A as a probable metabolite catalyzed by Cu(II). The role of the DNQM radical in modulating GSH levels as compared to the parent drug vitamin K3 and hybrid drug A has been analyzed. The depletion of GSH in cancer cells inhibiting autocrine stimulated tumor growth and increasing chemotherapy-mediated apoptosis may open up new curative and palliative strategies against various carcinomas.Acknowledgements
KB is thankful to UGC for financial assistance as JRF. AK is thankful to DST for financial support. SPG acknowledges the financial support from the University Grants Commission, New Delhi [Research Grant F34-370/2008(SR)]. The authors thank Dr A. Kumbhar (P.U.) for help with CV measurements, Dr P. Garge, Chemtall-Rai India Ltd. for ICP-AES analysis and Mr S. B. Rao (NCL) for discussions. SYR and KB are thankful to Dr P. Poddar (NCL) for magnetic susceptibility measurements.Notes and references
- D. M. Townsend and K. D. Tew, Oncogene, 2003, 22, 7369–7375 CrossRef CAS PubMed.
- D. W. Voehringer, D. L. Hirschheng, J. Xiao, Q. Lu, M. Roederer, C. B. Lock, L. A. Hergenbey and L. Steinman, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2680–2685 CrossRef CAS.
- R. C. Cumming, J. Lighfoot, K. Beard, H. Youssoufien, P. J. O'Brien and M. Buchwald, Nat. Med., 2001, 7, 814–820 CrossRef CAS PubMed.
- C. J. Weydert, Y. Zhang, W. Sun, T. A. Waugh, M. L. Teoh, K. K. Andringer, N. Aykin-Burns, D. R. Spitz, B. J. Smith and L. W. Oberley, Free Radical Biol. Med., 2008, 44(5), 856–867 CrossRef CAS PubMed.
- K. Irani, Y. Xia, J. L. Zweier, S. J. Sollott, C. J. Der, E. R. Fearon, M. Sundaresan, T. Finkel and P. J. Goldschmidt-Clermont, Science, 1997, 275, 1649–1652 CrossRef CAS.
- D. Komatsu, M. Kato, J. Nakayama, S. Miyagawa and T. Kamata, Oncogene, 2008, 27(34), 4724–4732 CrossRef CAS PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8(7), 579–591 CrossRef CAS PubMed.
- A. Vigalok and D. Milstein, Acc. Chem. Res., 2001, 34, 798–807 CrossRef CAS PubMed.
- J. Liu, H. Liu, R. B. van Breemen, G. R. J. Thatcher and J. L. Bolton, Chem. Res. Toxicol., 2005, 18, 174–182 CrossRef CAS PubMed.
- L. Yu, H. Liu, W. Li, F. Zhang, C. Luckie, R. B. van Breemen, G. R. J. Thatcher and J. L. Bolton, Chem. Res. Toxicol., 2004, 17, 879–888 CrossRef CAS PubMed.
- (a) O. Rabin, A. Vigalok and D. Milstein, J. Am. Chem. Soc., 1998, 120, 7119–7120 CrossRef CAS; (b) A. D. MacIntosh, H. Yang, R. D. Pike and D. A. Sweigart, J. Organomet. Chem., 2012, 719, 14–17 CrossRef CAS PubMed.
- T. Dunlap, R. Esala, P. Chandrasena, Z. Wang, V. Sinha, Z. Wang and G. R. J. Thatcher, Chem. Res. Toxicol., 2007, 20, 1903–1912 CrossRef CAS PubMed.
- T. Dunlap, S. C. Piyankarage, G. T. Wijewikrama, S. Abdu-Hay, M. Vanni, V. Litosh, J. Luo and G. R. Thatcher, Chem. Res. Toxicol., 2012, 25, 2725–2736 CrossRef CAS PubMed.
- N. Hulsaman, J. P. Medema, C. Bos, A. Jongejan, R. Leurs, M. J. Smit, I. J. P. de Esch, D. Richel and M. Wijtmans, J. Med. Chem., 2007, 50, 2424–2431 CrossRef PubMed.
- K. Badave, Y. Patil, R. Gonnade, D. Srinivas, R. Dasgupta, A. Khan and S. Rane, J. Mol. Struct., 2011, 1006, 288–296 CrossRef CAS PubMed.
- K. Oyaizu, K. Saito and E. Tsuchida, Chem. Lett., 2000, 1318–1319 CrossRef CAS.
- H. Amouri, Y. Besace and J. L. Bras, J. Am. Chem. Soc., 1998, 120, 6171–6172 CrossRef CAS.
- (a) X.-L. Wang, C. Qin, E.-B. Wang, L. Xu, Z.-M. Su and C.-W. Hu, Angew. Chem., Int. Ed., 2004, 43, 5036–5040 CrossRef CAS PubMed; (b) A. Gavezzotti, OPIX, University of Milan, Italy, 2007 Search PubMed.
- J. E. Wertz and J. R. Bolton, Electron spin resonance elementary theory and practical application, Chapman & Hall, U.S., 1986 Search PubMed.
- K. I. Nattinen and K. Rissanen, Cryst. Growth Des., 2003, 3, 339–353 Search PubMed.
- T. V. Magdesieva, A. V. Dolganov, A. V. Yakimansky, M. Y. Goikhman and I. V. Podeshvo, Electrochim. Acta, 2009, 54, 1444–1451 CrossRef CAS PubMed.
- S. B. Zaware, S. Dagade-Waghmode, R. G. Gonnade, D. Srinivas and S. Y. Rane, J. Mol. Struct., 2009, 938, 328–335 CrossRef CAS PubMed.
- A. S. Hay, H. S. Blanchard, G. F. Endres and J. W. Eustance, J. Am. Chem. Soc., 1959, 81, 6335–6336 CrossRef CAS.
- X. Chen, G. Dobereiner, X. Hao, R. Giri, N. Maugel and J. Q. Yu, Tetrahedron, 2009, 65, 3085–3089 CrossRef CAS PubMed.
- P. Gamez, S. Gupta and J. Reedijk, C. R. Chim., 2007, 10, 295–304 CrossRef CAS PubMed.
- J. P. Richard, T. L. Ameyes, L. Bei and V. Stubblefield, Chem. Res. Toxicol., 1990, 112, 9513–9519 CAS.
- P. B. Hulbert and P. L. Grover, Biochem. Biophys. Res. Commun., 1983, 117, 129–134 CrossRef CAS.
- D. J. Vocalde, G. J. Davies, R. Laine and S. G. Withers, Nature, 2001, 412, 835–838 CrossRef PubMed.
- H. Liu, J. Liu, R. B. van Breemen, G. R. J. Thatcher and J. L. Bolton, Chem. Res. Toxicol., 2005, 18, 162–173 CrossRef CAS PubMed.
- A. Basu and G. Das, Dalton Trans., 2011, 40, 2837–2843 RSC.
- S. Tsuruya, K. Kinumi, K. Hagi and M. J. Masai, J. Mol. Catal., 1983, 22, 47–60 CrossRef CAS.
- S. Rane, K. Ahmed, S. Salunke-Gawali, S. B. Zaware, D. Shrinivas, R. Gonnade and M. Bhadbhade, J. Mol. Struct., 2008, 892, 74–83 CrossRef CAS PubMed.
- D. Trachoothan, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 801765. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj00783a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |