Benzo[a]imidazo[5,1,2-cd]indolizines – a new class of molecules displaying excited state intramolecular proton transfer†
Anton J.
Stasyuk
ab,
Marzena
Banasiewicz
c,
Barbara
Ventura
d,
Michał K.
Cyrański
*b and
Daniel T.
Gryko
*a
aWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl; Fax: +48 22 6326681; Tel: +48 22 3433063
bWarsaw University, Faculty of Chemistry, 00-273 Warsaw, Poland. Fax: +48 22 6282741; Tel: +48 22 2345801
cInstitute of Physics of the Polish Academy of Sciences, Al. Lotników 32/46, 02-668 Warsaw, Poland. Fax: +48 22 8430926; Tel: +48 22 8436601
dIstituto per la Sintesi Organica e Fotoreattivita' (ISOF), CNR, Via P. Gobetti 101, 40129 Bologna, Italy. Fax: +39 (0)51 639 98 44; Tel: +39 (0)51 639 98 12
First published on 11th October 2013
Abstract
The new class of ESIPT-capable molecules, benzo[a]imidazo[5,1,2-cd]indolizines, bearing the 2-hydroxyphenyl substituent were prepared in a straightforward manner from imidazo[1,2-a]pyridines via a tandem [8+2]cycloaddition–[2+6+2]dehydrogenation reaction. The relationship between the structure and photophysical properties was thoroughly elucidated by comparison with simple analogues i.e. 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines. Compared with parent 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines the new chromophores strongly absorb blue light and emit in the yellow part of the spectrum. In contrast to 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines, emission of π-expanded dyes in protic solvents is usually weaker than in aprotic ones, where they exhibit large Stokes shifts (5500–7000 cm−1). Unlike in solution, in the solid state luminescence for benzo[a]imidazo[5,1,2-cd]indolizines is shifted on average by 100 nm relative to the fluorescence of the corresponding imidazo[1,2-a]pyridines, which results in a relatively strong (Φfl = 0.18 to 0.27) emission of the red light. Benzo[a]imidazo[5,1,2-cd]indolizines possessing a methoxy substituent have much higher fluorescence quantum yields than analogues bearing fluorine and methyl substituents.
Introduction
Organic fluorescent molecules undergoing excited state intramolecular proton transfer (ESIPT) have been important components in the search for highly efficient organic (opto-)electronic devices.1 Their unique photophysical properties are translated into the characteristics of the bulk materials and are easily tailored by alteration of the substituents on the periphery of the π-frameworks. With multiple applications spanning laser dyes,2 fluorescent solar energy concentrators,3 chemosensors,4 photonics,5 bioimaging,6 molecular switches7 and UV-photostabilizers,8 they have attracted attention from many vantage points. ESIPT is a phototautomerization process that occurs in the electronic excited state of a molecule and it can be represented by a four-level photocycle (En → En* → Ke* → Ke → En).9 The most significant characteristic of this process is an enormous Stokes-shifted luminescence due to the emission from the keto form. Complexity of ESIPT explains why it depends on many, both internal (e.g., substituents) and external (e.g. proticity and polarity of the media), factors.In spite of a rich body of research that has been performed on ESIPT compounds, there are still many unanswered questions related to the proton transfer phenomenon. The vast majority of studies performed within last the 60 years focused on the small heterocyclic systems. This leaves an intriguing question unanswered – what is the influence of extension of the π-conjugated system on the optical properties of ESIPT dyes? A limited number of papers published on this aspect10 revealed that the π-expansion of basic ESIPT-capable chromophores can lead to quenching of excited state intermolecular proton transfer.10c Our earlier studies of this aspect led to the conclusion that in the case of π-expanded benzo[h]quinoline analogues, the energetics of ESIPT depends on the actual position of π-expansion.11 Park and co-workers1k proved that the observed behavior concerning ESIPT of π-expanded derivatives of imidazoles and related molecules can be explained by considering the nodal pattern of the wave function along with the delocalization of the electrons in the excited state (the so-called nodal-plane model developed by Nagaoka).12
Among various ESIPT-capable molecules, 2-hydroxyphenyl substituted imidazo[1,2-a]pyridines have attracted significant interest in recent years. Acuña and co-workers were first to report the photoinduced intramolecular proton transfer in imidazopyridines.13 These authors later proposed and experimentally verified the hypothesis about the switching “on–off” ESIPT phenomenon based on the involvement of molecules of the polar, protogenic solvent in this process.14 Furthermore, special attention has been given to the 2-hydroxy derivative of 2-phenylimidazo[1,2-a]pyridines as organic solid-state luminescent material.15 Araki and co-workers have found that 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine is capable of forming different polymorphs emitting at different wavelengths with different quantum yields, which was the demonstration of novel design concept for tunable organic luminescent solids. Later on derivatives of 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine were used for fabrication of colorless organic materials exhibiting white luminescence.9c It should be noted that in the solid state, 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine has a fluorescence quantum yield (Φ) of about 0.37–0.5015 (depending on the polymorph), which makes this class of compounds interesting not only from a purely theoretical point of view, but also opens a door for possible practical applications. On the other hand in solutions in non-polar solvents, the typical fluorescence quantum yield for the band corresponding to the ESIPT process is only 0.01–0.06.16
One possible way to achieve the goal of expanding the π-electron system is tandem [8+2]cycloaddition–[2+6+2]dehydrogenation reactions.17 Cossio and co-workers reported that in the case of imidazo[1,2-a]pyridines (not capable of ESIPT) final products are aromatic, blue light emitting fluorescent molecules, whose fluorescence quantum yields are enhanced with respect to the starting bicyclic molecules.
Herein we describe the synthesis of several derivatives of 2-(benzo[a]imidazo[5,1,2-cd]indolizin-1-yl)phenols, capable of undergoing the ESIPT process, based on the tandem [8+2]-cycloaddition–[2+6+2]-dehydrogenation reactions of 2-(2′-hydroxyphenyl)-imidazo[1,2-a]pyridines with 2-(trimethylsilyl)phenyl trifluoromethanesulfonate as a benzyne precursor. Also, in this paper we present comprehensive optical data for these novel functional dyes.
Results and discussion
Compounds 3a–d were prepared from appropriate amounts of acetophenones 2a–d and 2-aminopyridine (1) via an Ortoleva–King reaction followed by Chichibabin ring closure.16a,18 The corresponding imidazo[1,2-a]pyridines were subsequently reacted with a benzyne precursor in the presence of cesium fluoride and an 18-crown-6 ether (Table 1).It was shown by Aginagalde et al. that the reaction between unsubstituted imidazo[1,2-a]pyridine and 2-(trimethylsilyl)phenyl trifluoromethanesulfonate can be carried out only under microwave-promoted conditions. At the same time, the reaction performed under the classic heating conditions led to a vanishingly small amount of the corresponding benzo[a]imidazo[5,1,2-cd]indolizine. The mechanism of this reaction by quantum chemistry methods was studied in detail. It was shown that despite the fact that, in general, this process is barrierless in terms of total energy, the latter reaction (aromatization of [8+2] cycloadduct) has a significant energy barrier (about 46 kcal/mol), which in turn is likely related to the large distortion upon going from the reactant to the transition structure. Consequently, it can be argued that the aromatization of the imidazole rings of [8+2] cycloadduct is the main driving force.
Our first attempts to perform the reaction between 2-(imidazo[1,2-a]pyridin-2-yl)phenols 3a–d and the benzyne precursor were based on the fact that numerous literature sources indicate that 2-(trimethylsilyl)phenyl trifluoromethanesulfonate readily forms benzyne molecules at room temperature or gentle warming in the presence of CsF in acetonitrile. Bearing in mind the fact that starting imidazo[1,2-a]pyridines are rather thermostable, we decided to perform this reaction under classical thermal conditions. Thus, substrate 3a was mixed with 2-(trimethylsilyl)phenyl trifluoromethanesulfonate in the presence of CsF and 18-crown-6 in a sealed tube and placed in an oil bath, preheated to 140 °C (Tables 1 and 2). After overnight stirring at the stated temperature we were able to isolate the desired product, but the yield of 4a was only 11% (Table 1, entry 1). In addition we isolated another non-polar compound in comparable quantities, which apparently is a product of trimerization of benzyne.
|
|
|||||
|---|---|---|---|---|---|
| Keton | R | Imidazo[1,2-a]pyridine | Yield (%) | Benzo[a]imidazo[5,1,2-cd]indolizine | Yield (%) |
| 2a |

|
3a | 54 | 4a | 23 |
| 2b |
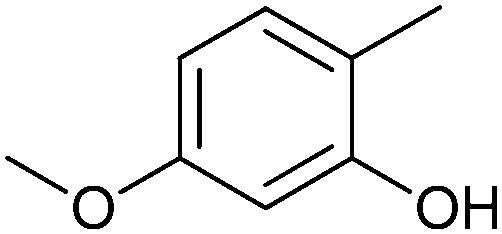
|
3b | 61 | 4b | 21 |
| 2c |
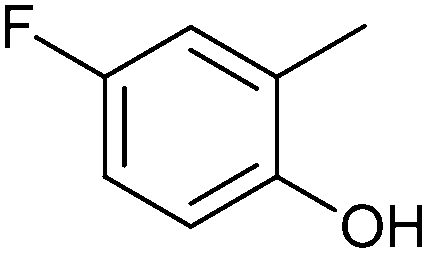
|
3c | 49 | 4c | 19 |
| 2d |

|
3d | 47 | 4d | 21 |

Indeed such trimerization is described in the literature when 2-(trimethylsilyl)phenyl trifluoromethanesulfonate is heated at high temperatures, in the presence of cesium fluoride.19 Often this reaction is carried out in the presence of a catalytic amount of palladium or copper salts with continued – about 12–15 hours – stirring or heating (depending on the used catalyst). Since the time of contact in the case of this reaction is crucial, we have decided to revert to the microwave irradiation as described by Cossio and co-workers. In the following series of experiments, the reaction between 3a and the benzyne precursor was carried out under microwave-promoted conditions. As major operating parameters, we selected temperature and exposure times as the most easily and accurately controlled (in this case the microwave oven operated in dynamically adjusted power mode to maintain the set temperature). Reactions were carried out without solvent. Thus, we have found that at temperatures above 175 °C amount of side-product substantially increases and the isolated yield of target product 4a was only several percent. On the other hand, at temperatures below 130 °C yields of compound 4a were about 10%. Eventually, we determined the temperature range of 150–160 °C as the optimal one. The reaction of 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine with 2-(trimethylsilyl)phenyl trifluoromethanesulfonate at this temperature for 25 minutes resulted in the formation of compound 4a in 23% yield. Having this procedure in hand three analogous benzoimidazopyridines 4b–d, possessing additional functionalities on phenyl substituents, were synthesized in 19–21% yield (Table 2). The choice of substituents was based on the desire to modulate the optical properties via the strength of the hydrogen bond between phenolic OH and basic nitrogen atoms.
It has been widely accepted that the driving force for the ESIPT reaction stems from the significant difference between the acidity and basicity of the involved groups;20 at the same time for most of the ESIPT molecules a strong intramolecular hydrogen bond has been detected. The presence of this hydrogen bond results in the large downfield shift (δ > 10 ppm) of the –OH protons in the 1H NMR spectrum. Thus, comparison of positions of the maxima for protons of the hydroxyl group in the NMR spectrum brings out a significant shift of these bands to a weak field side. Depending on the particular compound this shift range from 0.58 to 0.79 ppm, which in turn qualitatively indicates an increase in the strength of the hydrogen bond. The maximum value of chemical shift, as well as differences in the chemical shift values for the structures 3 and 4 was observed for the methoxy substituted compound. MeO obviously acts as an electron-donating group with a lone pair on the atom adjacent to the π-system. The methoxy group through a resonance donating effect increases the electron density on the benzene ring at the ortho- and para-positions, thereby increasing the partial positive charge on the carbon atom, which are directly connected to the hydroxyl group. That eventually leads to an increase in the acidity of the hydroxyl group. On the other hand the electron donating effect of the methoxy group via resonance may directly act on the nitrogen atom, which accepts a proton from the hydroxyl group – thus enhancing its basicity. The above processes should facilitate the ESIPT.
At the same time, one needs to keep in mind the necessary requirement of the planarity of the system, which provides the requisite spatial arrangement of the groups acting as a donor and acceptor of the proton. Earlier Araki and co-workers showed that in the 2-(imidazo[1,2-a]pyridin-2-yl)phenol deviation of the system from planarity is from 1 to 5.8 degrees depending on the polymorphic state.15a Later the same authors also showed16b that the quantum chemical calculations carried out at the RHF/6-31G(d) level of theory for the free molecules are in good agreement with the previously obtained crystallographic data. For 4-methoxyphenyl substituted benzo[a]imidazo[5,1,2-cd]indolizine, Aginagalde et al. determined that deviations from planarity were about 4 degrees for the mutual arrangement of the methoxyphenyl moiety relative to the benzo[a]imidazo[5,1,2-cd]indolizine and 5.7 degrees for the phenyl part relative to the imidazo[1,2-a]pyridine part in the benzo[a]imidazo[5,1,2-cd]indolizine core. The above evidence strongly suggests that in the case of 2-(benzo[a]imidazo[5,1,2-cd]indolizin-1-yl)phenol deviations from planarity should be comparable or smaller due to the presence of an intramolecular hydrogen bond. That in turn, should provide the possibility to induce an additional red-shift of the absorption spectra via influence of methoxy groups on the nitrogen atom by the resonance donating effect.
Subsequently, we examined the optical properties of dyes 4a–d by comparing them with the photophysics of 3a–d (Table 3, Fig. 1–3).
| Toluene | Dichloromethane | Acetonitrile | Methanol | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ abs/nm | λ em/nm | Φ | Δνss/cm−1 | λ abs/nm | λ em/nm | Φ | Δνss/ cm−1 | λ abs/nm | λ em/nm | Φ | Δνss/ cm−1 | λ abs/nm | λ em/nm | Φ | Δνss/cm−1 | |
| a Determined using quinine sulphate in 0.5 M H2SO4 as a standard. b Data taken from ref. 16a. | ||||||||||||||||
| 3a | 351 | 573 | 0.011 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
345 | 573 | 0.005 | 11500 |
328 | 389 | 0.003 | 4200 | 328 | 375 | 0.084 | 3800 |
| 577 | 0.006 | 13150 |
||||||||||||||
| 3b | 340 | 560 | 0.036 | 11600 |
334 | 572 | 0.010 | 12500 |
330 | 385 | 0.001 | 4300 | 331 | 385 | 0.124 | 4300 |
| 579 | 0.002 | 13000 |
||||||||||||||
| 3c | 337 | 582 | 0.008 | 12500 |
333 | 375 | 0.002 | 3400 | 330 | 377 | 0.006 | 3800 | 329 | 372 | 0.201 | 3500 |
| 587 | <0.001 | 13000 |
||||||||||||||
| 3d | 338 | 601 | 0.003 | 13000 |
333 | 376 | <0.001 | 3500 | 330 | 379 | 0.004 | 3900 | 329 | 375 | 0.162 | 3700 |
| 597 | <0.001 | 13000 |
||||||||||||||
| 4a | 427 | 593 | 0.060 | 6600 | 424 | 587 | 0.041 | 6600 | 420 | 435 | <0.001 | 800 | 420 | 422 | 0.017 | 110 |
| 588 | 0.003 | 6800 | ||||||||||||||
| 4b | 431 | 567 | 0.108 | 5600 | 428 | 561 | 0.084 | 5600 | 424 | 435 | 0.001 | 600 | 424 | 422 | 0.012 | 110 |
| 560 | 0.004 | 5700 | ||||||||||||||
| 4c | 428 | 604 | 0.037 | 6800 | 425 | 599 | 0.024 | 6900 | 422 | 430 | 0.002 | 450 | 421 | 422 | 0.035 | 60 |
| 595 | 0.001 | 6900 | ||||||||||||||
| 4d | 429 | 615 | 0.015 | 7100 | 426 | 612 | 0.009 | 7200 | 422 | 432 | 0.002 | 600 | 421 | 423 | 0.023 | 110 |
| 603 | <0.001 | 7100 | ||||||||||||||
 | ||
| Fig. 1 Absorption and fluorescence spectra of 3d (dashed line) and 4d (solid line) in toluene. Excitation at 330 nm (3d) and 400 nm (4d) respectively. | ||
 | ||
| Fig. 2 Absorption and fluorescence spectra of 4b in MeOH (dashed line) and in toluene (solid line). Excitation at 400 nm. | ||
 | ||
| Fig. 3 Absorption and fluorescence spectra of 4d in MeOH (dashed line) and in toluene (solid line). Excitation at 400 nm. | ||
The photophysical properties of targeted π-expanded chromophores can be compared to both 2-(2-hydroxyphenyl)imidazo[1,2-a]pyridines13–16 as well as to benzo[a]imidazo[5,1,2-cd]indolizines.17 In analogy to data revealed by Cossio and co-workers, the absorption maxima of benzo[a]imidazo[5,1,2-cd]indolizines 4a–d are bathochromically shifted by about 100 nm versus parent imidazo[1,2-a]pyridines 3a–d (Table 3, Fig. 1). This is accompanied by a strong hyperchromic effect with ε reaching 24000. The data in Table 3 show the presence of a small hypsochromic shift of absorption (7–9 nm) for the compounds of both series, with increasing polarity of the solvent. In contrast the emission maxima are shifted bathochromically only by ∼20 nm. This directly results in a decrease of Stokes shift from ∼13000 cm−1 to 5500–7000 cm−1.
The emission spectra of all of the studied compounds strongly depend on the nature of the solvent. Thus, in non-polar toluene the band shapes are structureless with a large Stokes shift. We also observed a slight increase in the value of the Stokes shift in the transition to more polar aprotic solvents. This partly results from the hypsochromic shift of the absorption band accompanying increase in the polarity of the solvent. On the other hand it is associated with the energy dissipation processes of the excited vibrational states in liquid solution.
It should be noted that the transition from non-polar to polar aprotic solvents significantly changes fluorescence spectra of all studied compounds on a qualitative level. In polar aprotic solution the emission shape consists of two bands: a “red” one which corresponds to the ESIPT luminescence (and can be observed in non-polar solvents), and a “blue” one. In contrast to toluene solution, the emission spectra in methanol for 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines show only a strong non-ESIPT emission band in the blue region accompanied by a moderate Stokes shift – 3500–4200 cm−1. The essentially different behavior of imidazo[1,2-a]pyridines in protic solution versus the non-polar, aprotic one can be explained by taking into account the hypothesis proposed by Acuña et al.13,14 about the solvent-induced stabilization of these forms, which are not able to form intramolecular hydrogen bonds, and therefore are not capable of proton transfer in the excited state.
In this case, moderate Stokes shifts are satisfactorily described in the framework of relaxation theory: explaining the shift of the emission band (energy loss) based on the reorientation of solvent dipoles, and the redistribution of electron density in the molecules of the solvent. Both the spectral evidence and structural similarity allows us to extend the applicability of that hypothesis to the class of 2-(benzo[a]imidazo[5,1,2-cd]indolizin-1-yl)phenols. In a polar aprotic solvent the value of Stokes losses is comparable to the value of losses of the vibrational energy due to relaxation processes and was on the order of 500–800 cm−1. Similar values of the Stokes shift were reported for a series of ESIPT-silent benzo[a]imidazo[5,1,2-cd]indolizine derivatives.17 It must be emphasized that for all the studied benzo[a]imidazo[5,1,2-cd]indolizine derivatives extremely small Stokes shifts (100 cm−1) were observed in methanol (Fig. 2 and 3). Fluorescence quantum yield for all the studied imidazo[1,2-a]pyridines, for the band corresponding to the ESIPT process is 0.5–4% (in toluene) while emission from the locally excited state is stronger (8–20% in methanol). These values of Φfl are in good agreement with the results of earlier reports.16b By increasing the effective π-electron system we have been able to not only shift the absorption band to the violet-blue region (∼430 nm), but also significantly increase Φfl, in non-polar solvents. Although emission in MeOH comes only from the local excited state, it is significantly weaker for dyes 4a–d than for compounds 3a–d (Table 2).
The key difference between derivatives of imidazo[1,2-a]pyridines and their novel π-expanded analogues is that in the latter case the ESIPT-emission (observed exclusively in toluene) is more intense (1.5–11% vs. 0.3–1.1%). The highest value of the fluorescence quantum yield was recorded for 2-(benzo[a]imidazo[5,1,2-cd]indolizin-1-yl)-5-methoxyphenol (4b) which amounted to 11%.
The quantum chemical study of 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines published by Araki and co-workers15b showed that the ESIPT state (S1) smoothly approached the S0–S1 conical intersection, coupled with twisting motion around the central C–C single bond connecting the phenyl and imidazo[1,2-a]pyridine rings. In other words the S0–S1 energy gap was sufficiently small at a dihedral angle of 60° for rapid radiationless decay in solution. It seems therefore that in the case of π-expanded compounds 4a–d the planar conformation is more preferred in fluid media, most probably because of limited rotation of the 2-hydroxyphenyl substituent imparted by the presence of hydrogen atoms at the fused benzene ring.
The photophysical behaviour of dyes 4a–d is different than previously reported π-expanded ESIPT-capable chromophores. 2-(2′-Hydroxyphenyl)imidazoles with extended effective conjugation length reported by Park and co-workers1k displayed properties similar to parent molecules, unless π-expansion was along the nodal plane.12 Analogues of 10-hydroxybenzo[h]quinoline bearing an additional benzene ring were characterized by moderately red-shifted absorption (∼50 nm), slightly bathochromically shifted emission (∼10 to 20 nm) and decreased Φflversus the parent molecule.11 We previously found that specific types of extending of conjugated aromatic system can result in prevention of ESIPT, in spite of the presence of strong intramolecular hydrogen bonds.11a,d,21 In contrast to all these previous examples, fusion of 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines with benzene molecule results in ESIPT-capable compounds with strongly (∼100 nm) bathochromically shifted absorption, non-shifted emission (in solution) and increased fluorescence quantum yield (from the ESIPT-state, in solution). These differences are probably associated with crucial function of free rotation and unusual mode of π-system expansion in heterocycles 4a–d.
Finally we also carried luminescence measurements for investigated compounds in the solid state (Fig. 4, Table 4). The solid samples have been gently milled with a mortar and placed inside two quartz slides. The absorption spectra of the four samples are shown in Fig. S1 (ESI†).
 | ||
| Fig. 4 Corrected emission spectra of compounds 4a–d in the solid state. Excitation at 440 nm. The spectral areas are scaled proportionally to the emission quantum yields. | ||
| Compound | λ abs /nm | λ em /nm | Φ em |
|---|---|---|---|
| a Absorption maxima from reflectance spectra analyzed using Kubelka–Munk conversion. b Emission maxima from corrected spectra. c Absolute emission quantum yields; excitation at 440 nm. d Data taken from ref. 16b. | |||
| 3a | 331 | 494 | 0.39 |
| 3b | 336 | 520 | 0.41 |
| 3c | 332 | 522 | 0.37 |
| 3d | 332 | 529 | 0.31 |
| 4a | 410, 435 | 612 | 0.267 ± 0.002 |
| 4b | 416, 443 | 568 | 0.242 ± 0.013 |
| 4c | 410, 435 | 626 | 0.184 ± 0.018 |
| 4d | 414, 439 | 626 | 0.211 ± 0.006 |
Although fluorescence is often significantly quenched in a solid state due to intermolecular interactions, Park and co-workers as well as other research groups introduced new classes of compounds displaying aggregation-induced emission enhancement (AIEE).22 AIEE was also found in the case of parent 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines, where strong fluorescence has been observed with the band corresponding to the excited state intramolecular proton transfer.9c,15a The solid state optical properties for compounds 3a–d perfectly correlate with data published earlier by Araki and co-workers (and therefore the latter ones were placed in Table 4).16b
The solid state absorption of compounds 4a–d is bathochromically shifted versus their smaller counterparts by ∼100 nm. Unlike in solution, in the solid state emission bands for benzo[a]imidazo[5,1,2-cd]indolizines 4a–d are shifted on average, by 100 nm relative to the fluorescence of the corresponding imidazo[1,2-a]pyridines. The emission measurements showed that all compounds, except 4b, have a main emission centered at 610–630 nm and a shoulder at 530 nm. Excitation spectra of the two bands revealed that the main emission is coming from a species with the absorption maximum of around 440 nm (overlapping with the absorption spectrum), whereas the shoulder comes from a species absorbing mainly in the region 490–500 nm. The emission spectrum of this second species, probably an aggregate or a form with a different crystal structure, is, in fact, well resolved upon excitation at 490 nm. The contribution of this secondary emission to the whole emission of the sample upon excitation at 440 nm can be estimated. The calculation leads to the following percentages: ca. 6% for 4a, ca. 7% for 4c and ca. 4% for 4d. Fig. S2 (ESI†) shows the contribution of the two forms and the relative excitation spectra. In the case of 4b, the emission spectral features are very different with respect to the other compounds and there is no strong evidence of emission from secondary species.
Interestingly fluorescence quantum yields in the solid state are roughly twice lower for π-expanded imidazo[1,2-a]pyridines 4a–d when compared with compounds 3a–d (Table 4). Still, they are rather high (∼20%), which suggests that AIEE also occurs in these cases. The anomalous emission of dye 4b is most probably related to the fact that an additional electron-donating MeO group acts as an additional hydrogen-bond acceptor, altering crystal packing in the solid state. The polymorphs differing in solid-state luminescence, among 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines were observed in 2008 by Araki and co-workers and later thoroughly investigated by Shigemitsu et al.15b
Conclusions
The presence of free hydroxyl groups does not interfere with tandem [8+2]cycloaddition–[2+6+2]dehydrogenation reaction in the imidazo[1,2-a]pyridine series. The fusion of imidazo[1,2-a]pyridine to a benzene ring at positions 3 and 5 leads to unusual heterocycles comprising of a four-ring core. We have demonstrated that by extension of the chromophore it is possible to easily manipulate both absorption and emission characteristics of novel yellow dyes with substantial bathochromic shifts. An expanding π-system alters the energy levels resulting in strikingly different characteristics of these novel ESIPT-capable compounds. While they still undergo ESIPT, their Φfl in non-polar solvents increases from 1–3% to 2–11% and the Stokes shift decreases to 6000 cm−1. On the other hand their emission in a protic solvent leads to radiationless deactivation rather than luminescence. Aggregation-induced emission enhancement is responsible for relatively strong red luminescence of benzo[a]imidazo[5,1,2-cd]indolizines in the solid state. These results are not only of theoretical significance in that they provide the first study of optical properties of this heterocyclic scaffold, but they may also open the door to practical applications.Experimental section
General
All chemicals were used as received unless noted otherwise. Reagent grade solvents (CH2Cl2, hexanes) were distilled prior to use. Spectrophotometric grade solvents were used without further purification. All reported 1H NMR and 13C NMR spectra were collected using 500 or 600 MHz spectrometers. Chemical shifts (δ ppm) were determined using TMS as an internal standard; J values are given in Hz. Melting points were determined using a capillary-type apparatus. Dry column vacuum chromatography (DCVC) was performed on preparative thin-layer chromatography using silica gel. Mass spectra were obtained via electron impact mass spectrometry using a double focusing sector mass analyzer (EI-MS) or using a time-of-flight spectrometer in positive electrospray ionization mode (TOF MS ES+).Absorption and fluorescence spectroscopy
The UV/Vis absorption spectra were recorded on a Perkin Elmer LAMBDA 35 spectrometer in toluene, DCM, acetonitrile and methanol. This apparatus has a 190–1100 nm wavelength range with a bandwidth of 1 nm. It is equipped with deuterium and tungsten pre-aligned sources with automatic switch-over. The optical system is a sealed double-beam scheme. Emission spectra were recorded on a fluorescence spectrophotometer Perkin Elmer 512 toluene, DCM, acetonitrile and methanol. The units are equipped with a Xe lamp (150 W), monochromator and photomultiplier tube HAMAMATSU R446. It has a 200–900 nm wavelength range with 3 nm resolution. Spectra are presented in photon units and have been corrected for instrumental factors.Solid-state determination made use of powder samples gently milled with a mortar and placed inside two quartz slides. Reflectance spectra were acquired with a Perkin–Elmer Lambda 9 UV/Vis/NIR spectrophotometer equipped with a 60 mm integrating sphere. Absolute photoluminescence quantum yields were measured on a Edinburgh FLS920 fluorimeter equipped with a 6 inches Labsphere integrating sphere, according to the method of deMello.23 Each measurement was repeated from three to five times. The limit of detection of the system is 2%.
General procedure for the preparation of imidazo[1,2-a]pyridines starting from 2-aminopyridine
A sealed tube was charged with the appropriate amounts of ketone (1.20 mmol), 2-aminopyridine (2.76 mmol, 2.3 eq.) and iodine (1.44 mmol, 1.2 eq.). The reaction mixture was stirred at 110 °C. After 4 h, the mixture was cooled to 70 °C and stirred overnight. The residue was diluted with 5 mL of distilled water, and an excess of aqueous sodium hydroxide (45%) was added. The reaction mixture was stirred at 100 °C for 1 h. After cooling to room temperature, the reaction mixture was diluted with 25 mL of CH2Cl2. 10% aqueous HCl was added to the water–organic mixture until a neutral pH was obtained. The mixture was then extracted with suitable solvents. The organic layer was washed with water, dried over Na2SO4, and concentrated under reduced pressure. The solid was isolated by column chromatography. Imidazo[1,2-a]pyridines 3a and 3b were prepared as described previously.General procedure for the preparation of 2-(benzo[a]imidazo[5,1,2-cd]indolizin-1-yl)phenol
2-(Trimethylsilyl)phenyl trifluoromethanesulfonate (120 mg, 0.40 mmol) was added to a mixture of appropriate amounts of imidazo[1,2-a]pyridine (1.20 mmol, 3 eq.), 18-crown-6 (211 mg, 0.80 mmol) and CsF (122 mg, 0.80 mmol) in a glass reactor. The reaction mixture was irradiated in a monomode microwave reactor at 160 °C for 25 min. The resulting mixture was dissolved in ethyl acetate and evaporated. The product was isolated by DCVC chromatography on silica gel and obtained as a solid.4-Fluoro-2-(imidazo[1,2-a]pyridine-2-yl)phenol (3c)
The compound was prepared as described above using CH2Cl2 for the extraction. The pure product was obtained as a yellow solid (180 mg, 49%). Data: mp 155–156 °C (lit.16b mp 161–162 °C); 1H NMR (500 MHz, CDCl3, δ) 6.86 (t, 1H, J = 6.8), 6.93–6.98 (m, 2H), 7.23–7.25 (m, 2H), 7.82 (s, 1H), 8.14 (d, 1H, J = 6.8), 12.48 (broad s, 1H); 13C NMR (125 MHz, CDCl3, δ) 107.1, 111.3, 111.5, 113.4, 116.2, 116.4, 116.9, 118.4, 118.5, 125.5, 153.4, 155.0, 156.8; HR – TOF MS(ES+) obsd 229.0776, calcd exact mass 229.0777 C13H10FN2O – [M–H]+.2-(Imidazo[1,2-a]pyridine-2-yl)-4-methylphenol (3d)
The compound was prepared as described above using CH2Cl2 for the extraction. The product was isolated by DCVC chromatography (silica, CH2Cl2/hexane 1:2 → CH2Cl2). The pure product was obtained as a yellow solid (175 mg, 47%). Data: mp 148–149 °C (lit.16b mp 150.9–151.2 °C); 1H NMR (500 MHz, CDCl3, δ) 6.75 (t, 1H, J = 6.8 Hz), 6.86 (d, 1H, J = 8.3 Hz), 6.95 (dd, 1H, J1 = 1.6 Hz, J2 = 8.2 Hz), 7.12 (t, 1H, J = 7.9 Hz), 7.31 (s, 1H), 7.49 (d, 1H, J = 9.1 Hz), 7.76 (s, 1H), 8.05 (d, 1H, J = 6.6 Hz) 12.41 (broad s, 1H); 13C NMR (125 MHz, CDCl3, δ) 20.7, 106.7, 113.2, 115.9, 116.9, 117.6, 125.2, 125.5, 126.1, 128.0, 130.6, 143.6, 145.6, 155.3; HR – TOF MS(ES+) obsd 225.1029, calcd exact mass 225.1028 C14H13N2O – [M–H]+.
2-(Benzo[a]imidazo[5,1,2-cd]indolizin-1-yl)phenol (4a)
The compound was prepared as described above. The pure product was obtained as a yellow solid (26 mg, 23%). Data: mp 177–178 °C; 1H NMR (500 MHz, CDCl3, δ) 7.12 (t, 1H, J = 7.5 Hz), 7.18 (dd, 1H, J1 = 0.9 Hz, J2 = 8.2 Hz), 7.38 (ddd, 1H, J1 = 1.6 Hz, J2 = 7.2 Hz, J3 = 7.7 Hz), 7.61 (ddd, 1H, J1 = 0.9 Hz, J2 = 7.4 Hz, J3 = 7.6 Hz), 7.79 (ddd, 1H, J1 = 1.1 Hz, J2 = 7.4 Hz, J3 = 7.8 Hz), 7.94–7.99 (m, 2H), 8.04 (dd, 1H, J1 = 1.7 Hz, J2 = 7.9 Hz), 8.28 (dd, 1H, J1 = 1.6 Hz, J2 = 7.7 Hz), 8.36 (d, 1H, J = 7.9 Hz), 8.46 (d, 1H, J = 8.2) 13.40 (broad s, 1H); 13C NMR (125 MHz, CDCl3, δ) 109.05, 111.94, 117.20, 118.03, 119.15, 119.83, 121.19, 123.15, 125.00, 126.93, 128.63, 128.81, 129.42, 130.11, 130.89, 131.13, 137.23, 146.38, 158.59; EI-HR obsd 284.0948, calcd exact mass 284.0950 (C19H12N2O).2-(Benzo[a]imidazo[5,1,2-cd]indolizin-1-yl)-5-methoxyphenol (4b)
The compound was prepared as described above. The pure product was obtained as a yellow solid (27 mg, 21%). Data: mp 192–194 °C; 1H NMR (500 MHz, CDCl3, δ) 3.91 (s, 3H), 6.70–6.74 (m, 2H), 7.59 (ddd, 1H, J1 = 0.9 Hz, J2 = 7.3 Hz, J3 = 7.6 Hz), 7.77 (ddd, 1H, J1 = 1.1 Hz, J2 = 7.2 Hz, J3 = 7.7 Hz), 7.93–7.94 (m, 1H), 8.02 (dd, 1H, J1 = 2.4 Hz, J2 = 5.5 Hz), 8.19 (d, 1H, J = 8.4 Hz), 8.36 (d, 1H, J = 8.0 Hz), 8.42 (d, 1H, J = 8.3 Hz), 13.64 (broad s, 1H); 13C NMR (125 MHz, CDCl3, δ) 55.42, 101.97, 106.89, 108.73, 110.44, 111.29, 120.93, 123.11, 124.63, 126.61, 128.71, 129.24, 129.66, 129.79, 130.09, 130.84, 137.31, 146.89, 160.53, 162.04; EI-HR obsd 314.1056, calcd exact mass 314.1055 (C20H14N2O2).2-(Benzo[a]imidazo[5,1,2-cd]indolizin-1-yl)-4-fluorophenol (4c)
The compound was prepared as described above. The pure product was obtained as a yellow solid (23 mg, 19%). Data: mp 233–234 °C; 1H NMR (500 MHz, CDCl3, δ) 7.11–7.12 (m, 2H), 7.65 (ddd, 1H, J1 = 0.9 Hz, J2 = 7.4 Hz, J3 = 7.7 Hz), 7.83 (dd, 1H, J1 = 1.1 Hz, J2 = 8.3 Hz), 7.94 (dd, 1H, J1 = 2.5 Hz, J2 = 9.3 Hz), 7.99–8.01 (m, 2H), 8.08 (dd, 1H, J1 = 2.8 Hz, J2 = 5.0 Hz), 8.40 (d, 1H, J = 7.8 Hz), 8.46 (d, 1H, J = 8.2 Hz), 13.12 (broad s, 1H); 13C NMR (125 MHz, CDCl3, δ) 109.30, 112.28, 113.93, 114.12, 117.67, 118.91, 120.06, 121.19, 123.27, 125.33, 127.26, 128.61, 129.70, 130.33, 131.30, 137.23, 145.11, 154.68, 156.82; EI-HR obsd 302.0851, calcd exact mass 302.0855 (C19H11N2OF).2-(Benzo[a]imidazo[5,1,2-cd]indolizin-1-yl)-4-methylphenol (4d)
The compound was prepared as described above. The pure product was obtained as a yellow solid (25 mg, 21%). Data: mp 174–175 °C; 1H NMR (500 MHz, CDCl3, δ) 2.51 (s, 3H), 7.07 (d, 1H, J = 8.3 Hz), 7.18 (d, 1H, J = 8.2 Hz), 7.57 (t, 1H, J = 7.5 Hz), 7.76 (t, 1H, J = 7.6 Hz), 7.92–7.94 (m, 2H), 7.99–8.01 (m, 1H), 8.04 (s, 1H), 8.32 (d, 1H, J = 8.0 Hz), 8.38 (d, 1H, J = 8.2 Hz), 13.09 (broad s, 1H); 13C NMR (125 MHz, CDCl3, δ) 20.86, 108.90, 111.84, 116.75, 117.72, 119.68, 121.01, 123.10, 124.85, 126.76, 128.02, 128.75, 128.81, 129.35, 129.98, 131.08, 131.68, 137.28, 146.64, 156.38; EI-HR obsd 298.1097, calcd exact mass 298.1106 (C20H14N2O).Acknowledgements
A. J. S., M. K. C. and D. T. G. gratefully acknowledge the Foundation for Polish Science for supporting this work under the MPD/2010/4 project “Towards Advanced Functional Materials and Novel Devices – Joint UW and WUT International PhD Programme”.Notes and references
- (a) S. Ameer-Beg, S. M. Ormson, R. G. Brown, P. Matousek, M. Towrie, E. T. J. Nibbering, P. Foggi and F. V. R. Neuwahl, J. Phys. Chem. A, 2001, 105, 3709–3718 CrossRef CAS; (b) J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed; (c) S.-J. Lim, J. Seo and S. Y. Park, J. Am. Chem. Soc., 2006, 128, 14542–14547 CrossRef CAS PubMed; (d) S. J. Formosinho and L. G. Arnaut, J. Photochem. Photobiol., A, 1993, 75, 21–48 CrossRef CAS; (e) D. McMorrow and M. Kasha, J. Phys. Chem., 1984, 88, 2235–2243 CrossRef CAS; (f) A. S. Klymchenko and A. P. Demchenko, Phys. Chem. Chem. Phys., 2003, 5, 461–468 RSC; (g) K. Das, N. Sarkar, A. K. Ghosh, D. Majumdar, D. N. Nath and K. Bhattacharyya, J. Phys. Chem., 1994, 98, 9126–9132 CrossRef CAS; (h) S. Lochbrunner, A. J. Wurzer and E. Riedle, J. Phys. Chem. A, 2003, 107, 10580–10590 CrossRef CAS; (i) P.-T. Chou, Y.-C. Chen, W.-S. Yu, Y.-H. Chou, C.-Y. Wei and Y.-M. Cheng, J. Phys. Chem. A, 2001, 105, 1731–1740 CrossRef CAS; (j) J. Waluk, Acc. Chem. Rev., 2003, 36, 832–838 CrossRef CAS PubMed; (k) S. Park, J.-E. Kwon and S. Y. Park, Phys. Chem. Chem. Phys., 2012, 14, 8878–8884 RSC.
- (a) P.-T. Chou, M. L. Martinez and J. H. Clements, Chem. Phys. Lett., 1993, 204, 395–399 CrossRef CAS; (b) K. Sakai, T. Tsuzuki, Y. Itoh, M. Ichikawa and Y. Taniguchi, Appl. Phys. Lett., 2005, 86, 081103 CrossRef; (c) V. A. Corbellini, M. L. Scroferneker, M. Carissimi, F. S. Rodembusch and V. Stefani, J. Photochem. Photobiol., B, 2010, 99, 126–132 CrossRef CAS PubMed; (d) P.-T. Chou, D. McMorrow, T. J. Aartsna and M. Kasha, J. Phys. Chem., 1984, 88, 4596–4599 CrossRef CAS.
- (a) T. Saraidarov, V. Levchenko, A. Grabowska, P. Borowicz and R. Reisfeld, Chem. Phys. Lett., 2010, 492, 60–62 CrossRef CAS PubMed; (b) O. F. Mohammed, D. Pines, E. T. J. Nibbering and E. Pines, Angew. Chem., Int. Ed., 2007, 46, 1458–1461 CrossRef CAS PubMed; (c) F. Vollmer and W. Rettig, J. Photochem. Photobiol., A, 1996, 95, 143–155 CrossRef CAS.
- (a) K.-C. Tang, M.-J. Chang, T.-Y. Lin, H.-A. Pan, T.-C. Fang, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Am. Chem. Soc., 2011, 133, 17738–17745 CrossRef CAS PubMed; (b) S. Mintova, V. De Waele, U. Schmidhammer, E. Riedle and T. Bein, Angew. Chem., Int. Ed., 2003, 42, 1611–1614 CrossRef CAS PubMed; (c) V. V. Shynkar, A. S. Klymchenko, C. Kunzelmann, G. Duportail, C. D. Muller, A. P. Demchenko, J.-M. Freyssinet and Y. Mely, J. Am. Chem. Soc., 2007, 129, 2187–2193 CrossRef CAS PubMed; (d) A. S. Klymchenko and A. P. Demchenko, J. Am. Chem. Soc., 2002, 124, 12372–12379 CrossRef CAS PubMed; (e) B. Liu, H. Wang, T. Wang, Y. Bao, F. Du, J. Tian, Q. Li and R. Bai, Chem. Commun., 2012, 48, 2867–2869 RSC.
- (a) A. R. Morales, K. J. Schafer-Hales, C. O. Yanez, M. V. Bondar, O. V. Przhonska, A. I. Marcus and K. D. Belfield, ChemPhysChem, 2009, 10, 2073–2081 CrossRef CAS PubMed; (b) S. Kim, J. Seo and S. Y. Park, J. Photochem. Photobiol., A, 2007, 191, 19–24 CrossRef CAS PubMed; (c) J. Seo, S. Kim, Y.-S. Lee, O.-H. Kwon, K. H. Park, S. Y. Choi, Y. K. Chung, D.-J. Jang and S. Y. Park, J. Photochem. Photobiol., A, 2007, 191, 51–58 CrossRef CAS PubMed.
- (a) F. Wang, J. Wu, X. Zhuang, W. Zhang, W. Liu, P. Wang and S. Wu, Sens. Actuators, B, 2010, 146, 260–265 CrossRef CAS PubMed; (b) M. Tasior, V. Hugues, M. Blanchard-Desce and D. T. Gryko, Chem.–Asian J., 2012, 7, 2656–2661 CrossRef CAS PubMed.
- (a) C. Benesch, M. F. Rode, M. Čížek, R. Härtle, O. Rubio-Pons, M. Thoss and A. L. Sobolewski, J. Phys. Chem. C, 2009, 113, 10315–10318 CrossRef CAS; (b) J. Herbich, C. Y. Hung, R. P. Thummel and J. Waluk, J. Am. Chem. Soc., 1996, 118, 3508–3518 CrossRef CAS; (c) V. S. Patil, V. S. Padalkar, K. R. Phatangare, V. D. Gupta, P. G. Umape and N. Sekar, J. Phys. Chem. A, 2012, 116, 536–545 CrossRef CAS PubMed.
- (a) D. A. Parthenopoulos, D. P. McMorrow and M. Kasha, J. Phys. Chem., 1991, 95, 2668–2674 CrossRef CAS; (b) A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2006, 8, 3410–3417 RSC.
- (a) A. V. Gaenko, A. Devarajan, I. V. Tselinskii and U. Ryde, J. Phys. Chem. A, 2006, 110, 7935–7942 CrossRef CAS PubMed; (b) S. H. Kim, S. Park, J. E. Kwon and S. Y. Park, Adv. Funct. Mater., 2011, 21, 644–651 CrossRef CAS; (c) H. Shono, T. Ohkawa, H. Tomoda, T. Mutai and K. Araki, ACS Appl. Mater. Interfaces, 2011, 3, 654–657 CrossRef CAS PubMed; (d) S. Park, S. Kim, J. Seo and S. Y. Park, Macromol. Res., 2008, 16, 385–395 CrossRef CAS; (e) A. I. Ciuciu, L. Flamigni, K. Skonieczny and D. T. Gryko, Phys. Chem. Chem. Phys., 2013, 15, 16907–16916 RSC; (f) D. Yao, S. Zhao, J. Guo, Z. Zhang, H. Zhang, Y. Liu and Y. Wang, J. Mater. Chem., 2011, 21, 3568–3570 RSC.
- (a) J. Seo, S. Kim and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 11154–11155 CrossRef CAS PubMed; (b) S. Park, J. E. Kwon, S. H. Kim, J. Seo, K. Chung, S.-Y. Park, D.-J. Jang, B. M. Medina, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 14043–14049 CrossRef CAS PubMed; (c) P. Yang, J. Zhao, W. Wu, X. Yu and Y. Liu, J. Org. Chem., 2012, 77, 6166–6178 CrossRef CAS PubMed; (d) J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC; (e) J. Ma, J. Zhao, P. Yang, D. Huang, C. Zhang and Q. Li, Chem. Commun., 2012, 48, 9720–9722 RSC; (f) J. Zhou, H. Liu, B. Jin, X. Liu, H. Fu and D. Shangguan, J. Mater. Chem. C, 2013, 1, 4427–4436 RSC.
- (a) D. T. Gryko, J. Piechowska and M. Gałęzowski, J. Org. Chem., 2010, 75, 1297–1300 CrossRef CAS PubMed; (b) J. Piechowska and D. T. Gryko, J. Org. Chem., 2011, 76, 10220–10228 CrossRef CAS PubMed; (c) J. Piechowska, K. Huttunen, Z. Wróbel, H. Lemmetyinen, N. V. Tkachenko and D. T. Gryko, J. Phys. Chem. A, 2012, 116, 9614–9620 CrossRef CAS PubMed; (d) I. Deperasińska, D. T. Gryko, E. Karpiuk, B. Kozankiewicz, A. Makarewicz and J. Piechowska, J. Phys. Chem. A, 2012, 116, 2109–2116 CrossRef PubMed; (e) I. Deperasińska, D. T. Gryko, E. Karpiuk, B. Kozankiewicz, A. Makarewicz and J. Piechowska, J. Phys. Chem. A, 2012, 116, 12049–12055 CrossRef PubMed.
- (a) S.-i. Nagaoka, U. Nagashima, N. Ohta, M. Fujita and T. Takemura, J. Phys. Chem., 1988, 92, 166–171 CrossRef CAS; (b) S.-i. Nagaoka, H. Teramae and U. Nagashima, Bull. Chem. Soc. Jpn., 2009, 82, 570–573 CrossRef CAS; (c) S.-i. Nagaoka, A. Nakamura and U. Nagashima, J. Photochem. Photobiol., A, 2002, 154, 23–32 CrossRef CAS.
- A. Douhal, F. Amat-Guerri and A. U. Acuña, J. Phys. Chem., 1995, 99, 76–80 CrossRef CAS.
- A. Douhal, F. Amat-Guerri and A. U. Acuña, Angew. Chem., Int. Ed. Engl., 1997, 36, 1514–1516 CrossRef CAS.
- (a) T. Mutai, H. Tomoda, T. Ohkawa, Y. Yabe and K. Araki, Angew. Chem., Int. Ed., 2008, 47, 9522–9524 CrossRef CAS PubMed; (b) Y. Shigemitsu, T. Mutai, H. Houjou and K. Araki, J. Phys. Chem. A, 2012, 116, 12041–12048 CrossRef CAS PubMed.
- (a) A. J. Stasyuk, M. Banasiewicz, M. K. Cyrański and D. T. Gryko, J. Org. Chem., 2012, 77, 5552–5558 CrossRef CAS PubMed; (b) T. Mutai, H. Sawatani, T. Shida, H. Shono and K. Araki, J. Org. Chem., 2013, 78, 2482–2489 CrossRef CAS PubMed; (c) D. Firmansyah, A. I. Ciuciu, V. Hugues, M. Blanchard-Desce, L. Flamigni and D. T. Gryko, Chem.–Asian J., 2013, 8, 1279–1294 CrossRef CAS PubMed.
- M. Aginagalde, Y. Vara, A. Arrieta, R. Zangi, V. L. Cebolla, A. Delgado-Camón and F. P. Cossío, J. Org. Chem., 2010, 75, 2776–2784 CrossRef CAS PubMed.
- A. E. Tschitschibabin, Chem. Ber., 1925, 58, 1704–1706 CrossRef.
- (a) D. Peña, S. Escudero, D. Pérez, E. Guitián and L. Castedo, Angew. Chem., 1998, 110, 2804–2806 CrossRef; (b) C. Xie, L. Liu, Y. Zhang and P. Xu, Org. Lett., 2008, 10, 2393–2396 CrossRef CAS PubMed.
- (a) P. F. Barbara, P. K. Walsh and L. E. Brus, J. Phys. Chem., 1989, 93, 29–34 CrossRef CAS; (b) J. L. Herek, S. Pedersen, L. Bañares and A. H. Zewail, J. Chem. Phys., 1992, 97, 9046–9061 CrossRef CAS; (c) A. Douhal, F. Lahmani and A. H. Zewail, Chem. Phys., 1996, 207, 477–498 CrossRef CAS.
- K. Skonieczny, A. I. Ciuciu, E. M. Nichols, V. Hugues, M. Blanchard-Desce, L. Flamigni and D. T. Gryko, J. Mater. Chem., 2012, 22, 20649–20664 RSC.
- (a) B. Z. Tang, X. Zhan, G. Yu, P. P. S. Lee, L. Liu and D. Zhu, J. Mater. Chem., 2001, 11, 2974–2978 RSC; (b) B.-K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed; (c) G. Huang, G. Zhang, Y. Wu, Q. Liao, H. Fu and D. Zhang, Asian J. Org. Chem., 2012, 1, 352–358 CrossRef CAS.
- A. Barbieri and G. Accorsi, EPA Newsl., 2006, 26–35 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of compounds 3c, 3d and 4a–d. See DOI: 10.1039/c3nj00842h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |