DOI:
10.1039/C3NJ00832K
(Paper)
New J. Chem., 2014,
38, 366-375
Synthesis, CMC determination, nucleic acid binding and cytotoxicity of a surfactant–cobalt(III) complex: effect of ionic liquid additive†
Received
(in Montpellier, France)
25th July 2013
, Accepted 7th October 2013
First published on 11th October 2013
Abstract
The surfactant–cobalt(III) complex, cis-[Co(trien)(4CNP)(DA)](ClO4)3 (trien = triethylenetetramine, 4CNP = 4-cyanopyridine and DA = dodecylamine) was synthesized and characterized by various spectroscopic and physicochemical techniques. The critical micelle concentration (CMC) value of this surfactant–cobalt(III) complex in aqueous solution was obtained from conductance measurements. The conductivity data (at 303, 308, 313, 318 and 323 K) were used for the evaluation of the temperature-dependent CMC and the thermodynamics of micellization (ΔG0m, ΔH0m and ΔS0m). Absorption, fluorescence, cyclic voltammetry, circular dichroism and viscosity experiments have been carried out to study the interaction of the surfactant–cobalt(III) complex with DNA and RNA. The results suggest that the complex can bind to nucleic acids by intercalation via the long aliphatic chain of the complex into the base pairs of DNA/RNA. In the presence of an ionic liquid additive, the binding strength of the surfactant–cobalt(III) complex to the nucleic acids increased. The complex was tested in vitro on HepG2 (human hepatocellular liver carcinoma) tumor cell lines and found to be active.
1 Introduction
During the past decade, there has been tremendous interest in studies pertaining to the interaction of various transition metal complexes with nucleic acids.1–3 Recently, applications of these transition metal complexes in the design and development of synthetic restriction enzymes, new drugs, DNA footprinting agents, and stereoselective probes of nucleic acid structure have been explored extensively.4–6 RNA is a versatile molecule that plays essential roles in many biological processes, and consequently, it is an attractive target for potential therapeutics. The structural diversity present in RNA molecules has led to specific drug recognition sites. It is currently accepted that RNA plays a key role in many biological processes involving living cells and is considered as the essential regulator of many steps of gene expression.7 Furthermore, many viruses such as HIV, HCV, influenza and flaviviruses are encoded by a unique RNA molecule that serves as the main genetic material. Therefore, metal complexes that bind to RNA and disturb RNA functions could be powerful tools for understanding and controlling gene expression.8
Recently, different ligands and metal complexes have been developed that bind to RNA.9,10 The development of reagents that can form intra-strand cross-links with nucleic acids continues to be a subject of considerable interest in the areas of microbiology and rational drug design.11 Metal complexes are usually used as catalysts for hydrolytic cleavage of RNA.12 They are also used as shape-selective probes of RNA tertiary structure,13 agents of oxidative cleavage of RNA14 and recognition of mismatches in RNA.15 However, there have been relatively few previous investigations into the binding mode and the enantioselectivity for the interactions between metal complexes and RNA. The future development of RNA-targeting drugs will rely on a deeper understanding of these binding processes. Although some experimental investigations on RNA–metal complex interactions have been carried out during the past decade, there are still some questions that need to be answered for a better understanding of the mechanism and the biological implications of the interactions, for example, how the structures of nucleic acids affect the binding behavior of metal complexes with them. Thus, a comparative study of the interactions of metal complexes with RNA and DNA, their binding mode, binding strength and enantioselectivity will be very important in understanding the mechanism of the interactions and the biological impact of metal complexes.
Ionic liquids (ILs) have currently attracted much interest for applications as novel solvents in many fields.16–18 Favorable alterations of the physicochemical properties of aqueous micellar solutions of surfactants are important due to their applications in colloidal formulation. Ionic liquids (ILs) have emerged as one of the most interesting chemicals both in fundamental and applied research during the last three decades.19–21 The utilization of ILs to alter the solution properties of surfactant solutions is an interesting and appealing concept both from environmental and applicative points of view. Armstrong et al.22 and Fletcher23 and Pandey24 have demonstrated the effectiveness of ILs in altering the key physicochemical properties of the aqueous solutions of zwitterionic, nonionic and anionic surfactants. Molecular self-assemblies formed in ILs are of great interest and may widen the applications of ILs.
Studies on the self-assembly of amphiphilic molecules in ILs have currently attracted considerable attention. This is because the behavior of amphiphilic molecules in ILs is quite interesting from an academic point of view, and from a practical point of view, the molecular aggregates are expected to solubilize inherently insoluble materials in the ILs, which would widen the application field of ILs. In addition, the self-assemblies of amphiphilic molecules in a solvent have many potential applications such as nanomaterial synthesis, drug delivery, pharmaceutical formulation, and other dispersant technologies. In this sense also, the elucidation of the self-assembling phenomena of amphiphilic molecules in ILs must be a significant research subject. Many authors have investigated the effect of ionic liquid additives on the aggregation behavior of surfactants.25,26 Rao et al.27 have reported dry micelle formation of some traditional surfactants in two ionic liquids, 1-butyl-3-methylimidazolium chloride (bmimCl) and [bmim][PF6]. They have demonstrated that changing the exact nature of the ILs can tune the size and aggregation number of the micelles. Although the aggregation behavior of surfactants in imidazolium-based ILs has been widely studied, only a few reports are available on the thermodynamics of surfactant aggregation in ILs. These investigations have clarified that the driving force to induce the self-aggregation of surfactant molecules in ILs is a solvophobic interaction between the hydrocarbon tails of the surfactant and the ILs.28 From this point of view, surfactant solutions in ILs have become a research target in the field of surfactant science. To the best of our knowledge, no studies on the interaction of surfactant metal complexes with nucleic acids in the presence of an ionic liquid additive have been reported so far. The binding behavior of such systems may be useful in the future for biological implications.
We have been interested in the synthesis and reactions involving surfactant–cobalt(III) complexes which contain one or two ligands having long aliphatic chains.29–31 It is very important in biotechnological and biomedical applications, particularly for the possibility of using such systems for in vivo gene delivery and gene transfer.32 DNA can form tight complexes with surfactants through hydrophobic or electrostatic binding.23 Among the three different kinds of surfactants, i.e., cationic, anionic and nonionic surfactants, the interaction of cationic surfactants with DNA attracts the most attention and the binding of these surfactants to DNA was shown to proceed in two stages.32 In the first stage, surfactant ions exchange with counter ions condensed on the surface of DNA driven by hydrophobic interactions, while the effective charge on the DNA does not change. In the second stage, surfactant molecules bind to DNA without exchange of condensed counter ions, thus the effective charge on the DNA changes dramatically, usually followed by phase separation at high surfactant concentration. This feature suggests that DNA–surfactant systems have some further applications, such as DNA purification and condensation.33 Meanwhile, with an increase in cationic surfactant concentration, the conformation of the DNA–surfactant complex undergoes a discrete transition from extended coils to collapsed globules, and the two states coexist below a certain critical concentration. This phenomenon has been demonstrated by a number of techniques, e.g., fluorescence microscopy and viscosity techniques.34
The present work deals with the synthesis and CMC determination of a water soluble surfactant–cobalt(III) complex, cis-[Co(trien)(4CNP)(DA)](ClO4)3 along with a comparative study on the interaction of this complex with calf thymus DNA (CT DNA) and yeast tRNA in the presence and absence of an ionic liquid additive by UV-visible absorption spectroscopy, fluorescence spectroscopy, viscometry, circular dichroism spectroscopy and cyclic voltammetric measurements. Also, the cytotoxicity of this complex has been evaluated by the MTT assay method (MTT = (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)) and some staining experiments.
2 Experimental
2.1 Materials and methods
The absorption and emission spectroscopic titrations were carried out in a buffer (tris buffer, pH = 7.1) at room temperature. Calf thymus (CT) DNA and yeast (tRNA) were purchased from Sigma-Aldrich, Germany, and used as received. Triethylenetetramine and 4-cyanopyridine were obtained from Loba Chem. Dodecylamine (DA), obtained from Sigma, was used as supplied. All the experiments involving the interaction of the surfactant–cobalt(III) complex with nucleic acids in the presence of an ionic liquid additive were carried out with twice distilled water in buffer containing 5 mM Tris-HCl–50 mm NaCl at pH 7.0.
CHN analysis of the sample was carried out at CECRI (Central ElectroChemical Research Institute), Karaikudi, India. Absorption spectra were recorded on a Shimadzu UV-1800 UV-Visible spectrophotometer using cuvettes of 1 cm path length, and emission spectra were recorded on a JASCO FP 770 spectrofluorimeter. FT-IR spectra were recorded on a FT-IR PerkinElmer spectrophotometer with samples prepared as KBr pellets. 1H-NMR spectra were recorded on a BRUKER 400 MHz Spectrometer using DMSO as solvent. Conductivity studies were done in aqueous solutions of the complex with an Elico conductivity bridge type CM 82 and a dip-type cell with a constant of 1.0.
2.2 Synthesis
2.2.1 Preparation of the precursor cobalt(III) complex.
The complex, cis-[Co(trien)Cl2]Cl was prepared as reported in the literature.35,36 Using this complex as the starting material, the complex, cis-[Co(trien)(4CNP)Cl]Cl2 was prepared by slight modification of a method reported earlier for a similar type of complex.37 About 0.01 mole of cis-[Co(trien)Cl2]Cl was mixed with 0.01 mole of 4-cyanopyridine in 60 mL of absolute ethanol. The slurries were stirred with warming over a water bath for 20–30 min until the colour changed to brown. The brown colored complex cis-[Co(trien)(4CNP)Cl]Cl2 thus obtained was filtered off, washed with alcohol (ethanol) and then with ether. It was dried at 110 °C for 24 hours.
2.2.2 Preparation of the surfactant–cobalt(III) complex, cis-[Co(trien)(4CNP)(DA)](ClO4)3.
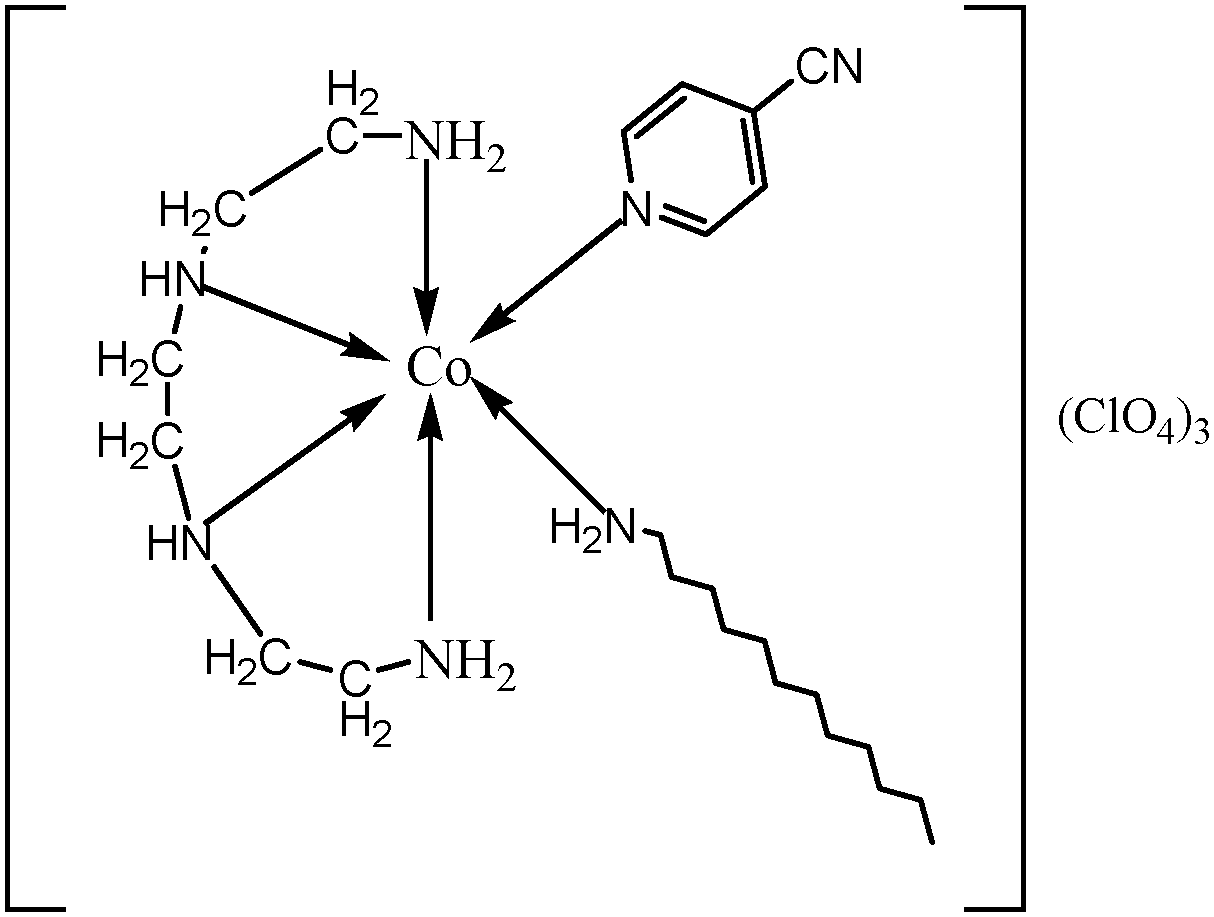
The surfactant–cobalt(III) complex of the present study was synthesized from the complex, cis-[Co(trien)(4CNP)Cl]Cl2 by a ligand substitution method. A solution containing three grams of cis-[Co(trien)(4CNP)Cl]Cl2 dissolved in 20 mL water was prepared. To this solution, slightly more than the calculated amount of dodecylamine in 3 mL ethanol was added dropwise over a period of 30 min. The brown colour of the solution gradually became red and the mixture was set aside at 40 °C for 2 days until no further change in color was observed. Afterwards, a saturated solution of sodium perchlorate was added to the solution. Slowly a pasty solid mass separated out, which was filtered off and washed with small amounts of alcohol and acetone, and then dried in air. The semi-dried solid was further dried in a drying pistol over fused calcium chloride and stored in a vacuum desiccator. The structure of the complex is shown in Fig. 1.
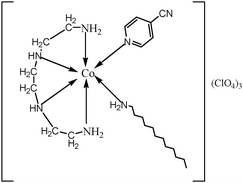 |
| | Fig. 1 The chemical structure of the complex, cis-[Co(trien)(4CNP)(DA)](ClO4)3. | |
2.3 Cobalt analysis
The cobalt content of the surfactant–cobalt(III) complex was estimated by following Kitson's method.36 A known weight of the complex was reduced with tin and concentrated hydrochloric acid. The solution volume of reduced aqueous cobalt(II) ions was made up to 10 mL in a volumetric flask using 0.1 M perchloric acid. Two millilitres of this solution and 1 mL of 50% ammonium thiocyanate solution were pipetted into a 10 mL volumetric flask and made up to the 10 mL mark with acetone. The absorbance of this solution was measured at 625 nm against a blank reagent. From the absorbance, the concentration of the cobalt was calculated.
2.4 CMC determination
The critical micelle concentration value of the surfactant–cobalt(III) complex was determined conductometrically using a conductivity meter. The conductivity cell was calibrated with KCl solutions in the appropriate concentration range. Various concentrations of the surfactant–cobalt(III) complex were used in the range of 20 × 10−5 to 20 × 10−1 mol dm−3 in aqueous solution. The conductivities of these solutions were measured at 303, 308, 313, 318 and 323 K. The temperature of the thermostat was maintained constant to within ±0.01 °C. The conductance was measured after thorough mixing and temperature equilibrating at each dilution. The establishment of equilibrium was checked by taking a series of readings after 15 min intervals until no significant change occurred.
2.5 Nucleic acid (NA) binding experiments
The nucleic acid binding experiments were performed at 25.0 ± 0.2 °C. The nucleic acid concentration was determined by electronic absorption spectroscopy using the known molar extinction coefficient values of 6600 M−1 cm−1 for DNA and 9250 M−1 cm−1 for RNA at 260 nm.38 A constant concentration of surfactant–cobalt(III) complex was maintained for UV-Visible absorption measurements to which increments of the nucleic acid stock solutions were added. The solutions were allowed to incubate for 10 min before the absorption spectra were recorded. Equal solution amounts of nucleic acid were added to both the complex solution and the reference solution to eliminate the absorbance of the nucleic acid itself. For fluorescence quenching experiments, nucleic acids were pretreated with ethidium bromide (EB) for 30 min. Solutions of the surfactant–cobalt(III) complex were added to this mixture and their effects on the emission intensity was measured. The samples were excited at 450 nm and emission was observed between 500 and 700 nm. For the cyclic voltammetry experiments, the electrode surfaces were freshly polished with alumina powder and then sonicated in ethanol and distilled water for 1 min prior to each experiment and the electrode was rinsed with doubly distilled water thoroughly between each polishing step. Finally, each electrode was cleaned thoroughly in an ultrasonic cleaner again with doubly distilled water. Cyclic voltammetric experiments were performed at 25.0 ± 0.2 °C in a single compartment cell with a three-electrode configuration, glassy carbon working electrode, platinum wire auxiliary electrode and a saturated calomel reference electrode using tris buffer. Before the experiments, all solutions were deaerated with dry nitrogen gas for 10 min to remove dissolved oxygen and kept under nitrogen atmosphere throughout the experiments. Viscosity experiments were carried out in an Ubbelohde viscometer, immersed in a thermostated water bath maintained at 30 ± 0.1 °C. DNA samples of approximately 0.5 mM were prepared by sonicating in order to minimize complexities arising from DNA flexibility. Flow time was measured with a digital stopwatch, and each sample was measured three times and an average flow time was calculated. Data were presented as (η/η0)1/3versus the concentration of the surfactant–cobalt(III) complex, where η is the viscosity of the DNA solution in the presence of the complex, and η0 is the viscosity of the solution of DNA alone. Viscosity values were calculated from the observed flow times of the DNA-containing solutions (t > 100 s) corrected for the flow time of the buffer alone (t0): η = t − t0/t0.39
2.6 Cytotoxicity assay
The cytotoxicity of the surfactant–cobalt(III) complex was measured using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay as described previously.40 The complex was first dissolved quantitatively in dimethyl sulfoxide (DMSO, Sigma, USA) to make the stock solution. Briefly, cells were seeded at a density of 5 × 104 HepG2 liver cancer cells per well into 96-well plates. After 24 h, the cells were treated with the surfactant–cobalt(III) complex at various concentrations (10, 30, 60, 90 μg mL−1) and incubated for 24 and 48 hours as indicated. At the end of the incubation, 10 μl of 3-(4,5-dimethylthiozol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) (5 mg mL−1) per well was added and incubated in the dark at 37 °C for 4 hours. The formazan crystals formed after 4 hours were dissolved in 100 μl of DMSO after aspirating the medium. The absorbance was monitored at 570 nm (measurement) and 630 nm using a 96-well plate reader (Bio-Rad, Hercules, CA, USA). The IC50 value was defined as the concentration of compound that produced a 50% reduction in cell viability.
2.6.1 Evaluation of apoptosis (acridine orange and ethidium bromide staining).
Acridine orange and ethidium bromide staining was performed as described by Baskic et al.65 Twenty-five microliters of cell suspension of each sample (both attached, and released by trypsinization and floating) containing 5 × 105 cells, was treated with AO and EB solution (one part of 100 mg mL−1 AO and one part of 100 mg mL−1 EO in PBS) and examined under a fluorescence microscope (Carl Zeiss, Germany) using an UV filter (450–490 nm). Three hundred cells per sample were counted in tetraplicates for each dose point. Cells were scored as viable, apoptotic or necrotic as judged by the staining, nuclear morphology and membrane integrity, and the percentages of apoptotic and necrotic cells were then calculated. Morphological changes were also observed and photographed.
2.6.2 Dye preparation and drug preparation.
200 μL of dye mixture (100 μL mg−1 AO and 100 μL mg−1 EB in distilled water) was mixed with 2 mL of cell suspension (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per mL) in a 6-well plate. The suspension was immediately examined and viewed under an Olympus inverted fluorescence microscope (Ti-Eclipse) at 200× and 400× magnification. We observed untreated cells as controls and cells treated with testing material at IC50 concentrations for 24 h of exposure.
000 cells per mL) in a 6-well plate. The suspension was immediately examined and viewed under an Olympus inverted fluorescence microscope (Ti-Eclipse) at 200× and 400× magnification. We observed untreated cells as controls and cells treated with testing material at IC50 concentrations for 24 h of exposure.
2.6.3 Drug treatments.
HepG2 cells were seeded in a 24-well plate (50000 cells per well). After 24 h of cells’ incubation, the medium was replaced with 100 μL of medium containing an IC50 dose of testing material. Untreated cells served as the control after the completion of 24 h, the medium was aspirated, and the cells treated with the prepared dye and observed under the fluorescence microscope.
2.6.4 Hoechst 33342 staining.
This procedure is very sensitive to cell concentration and pH of the media. Cells should number approximately 1–2 × 106 mL, in buffered media at pH 7.2. It is also helpful to include 2% fetal calf serum to maintain the cells.
The drug was added and incubated for 24 and 48 hours. Homogenously aspirated and spent media was removed and 1 mL of saline was added and centrifuged at 1500 rpm for 10 min. The cells were stained with 0.5 mL of Hoechst 33342 solution (3.5 μg mL−1 in PBS) and incubated for 30 min at 37 °C in an incubator. After 30 min, the Hoechst 33342 solution was discarded and the cells observed at 490–520 nm with a fluorescence microscope. Time is a critical factor due to the transport of the dye. Typically, 30 minutes is a minimum, but it is important to remember that the signal may begin to degrade after ∼120 minutes. It is recommended that the staining kinetics be empirically defined. Apoptosis was observed under the fluorescence microscope. Washing is not recommended.
3 Results and discussion
3.1 Spectroscopic characterization
The surfactant–cobalt(III) complex synthesized in the present study was characterized by UV-Vis, IR, and NMR techniques. The purity of the complex was checked by Co, C, N, and H analysis, and the results were found to be in good agreement with the calculated values. Elements, % found (% calculated): Co 7.52 (7.36), C 35.24 (35.09), N 12.51 (12.60) and H 6.69 (6.21). The UV-Visible absorption spectrum of this complex shows two distinctive bands at 488 and 363 nm, and exhibits intense ligand-based π–π* bands in the UV region (∼370 nm) and a broad metal to ligand charge transfer (MLCT) band in the visible region (455–550 nm). The infrared spectrum was used for the assignment of the geometrical configuration of the complex. Buckingham and Baldwin have studied the NH2 deformation mode in the 1637–1475 cm−1 region, the CH2 rocking mode in the 900–850 cm−1 region, and the Co–N stretching mode in the 610–500 cm−1 region to distinguish between the cis and trans isomers of cobalt(III) complexes,41–43 reporting that the important variations between the cis and trans isomers are found in the CH2 rocking region of 900–850 cm−1. Trans isomers always show only one peak, and cis isomers occur as shoulders. Because of the lower-symmetry cis configuration, our complex shows two bands for NH2 deformation in the 1637–1475 cm−1 region, two bands for NH2 wagging in the 1396–1103 cm−1 region, a CH2 rocking mode in the 940–830 cm−1 region, and two bands for Co–N stretching in the 718–630 cm−1 region. The medium intensity band at 2315 cm−1 confirms the free NH2 group of 4-cyanopyridine indicating that the ligand 4-cyanopyridine is coordinated to the metal ion via the pyridine nitrogen. In the 1H NMR spectrum of our complex, the amine protons of triethylenetetramine appear as a singlet at 2.1–2.3 ppm and, due to the lower symmetry, the methylene protons of triethylenetetramine show a more complex absorption around 2.1–2.9 ppm. The methylene protons of the dodecylamine moiety give rise to a multiplet, usually at 1.2–0.8 ppm. Aromatic protons around δ = 7.4 ppm are assigned to 4-cyanopyridine.
3.2 CMC determination values
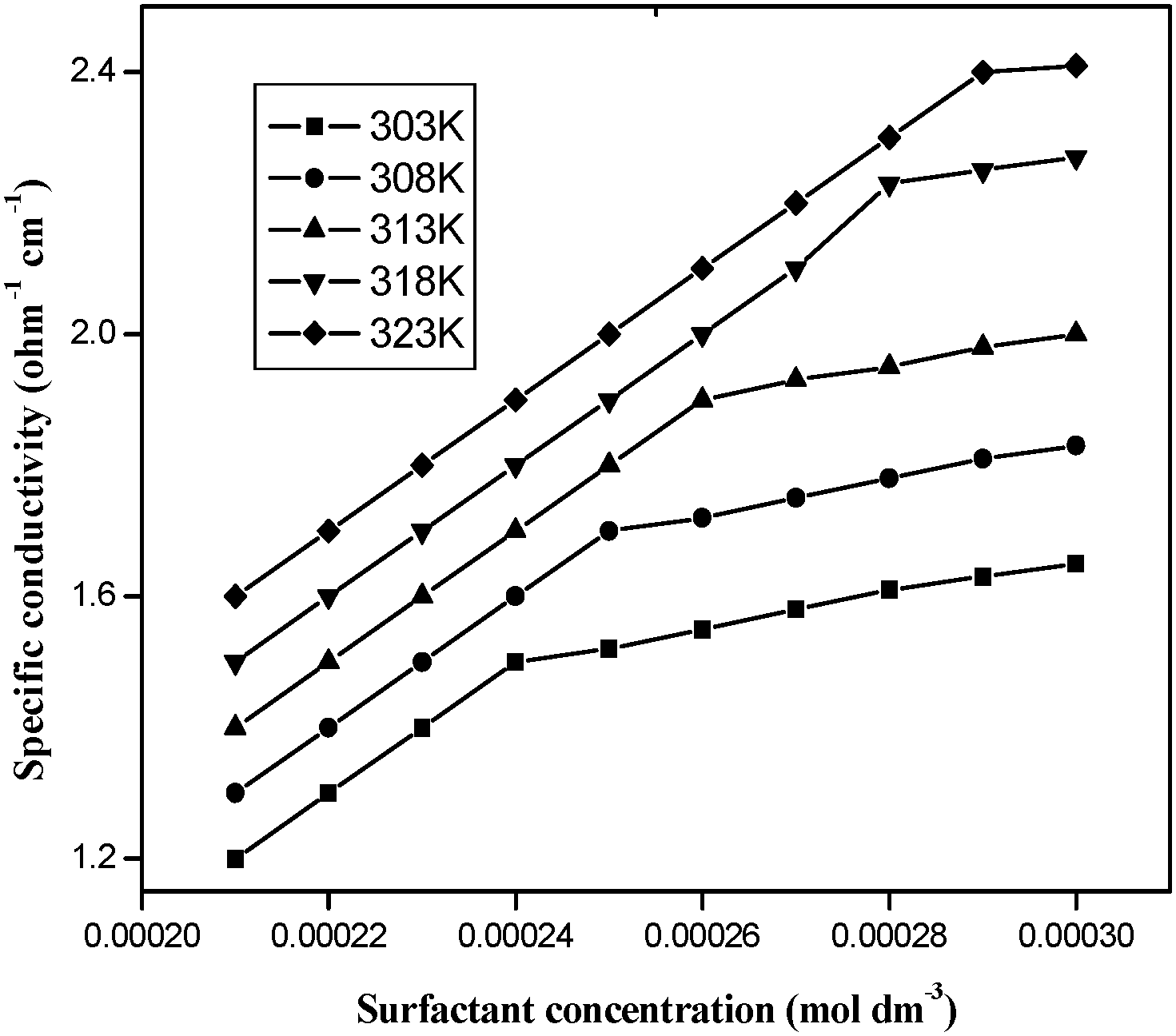
The specific conductivity of the surfactant–cobalt(III) complex increases with the complex concentration and temperature. When plots are made of [complex] versus specific conductivity, the slope is reduced after a particular value of concentration. This particular value of concentration at which the slope of the plot changes indicates micellization and this concentration is chosen as the CMC. The CMC values were determined by fitting the data points above and below the break to two equations of the form y = mx + c and solving the two equations simultaneously to obtain the point of intersection. Least-squares analysis was employed, and the correlation coefficients were greater than 99% in all the cases. The CMC values were determined at five different temperatures of 303, 308, 313, 318 and 323 K and were repeated three times and the accuracy of the CMC values (Table 1) was found to be within ±2% error. Fig. 2 illustrates the plot for the complex. It was observed that the CMC value increased with an increase in the temperature. An increase in temperature disrupts the water surrounding the hydrophobic group, and this retards micellization leading to a higher CMC value.
Table 1 CMC values of the surfactant–cobalt(III) complex cis-[Co(trien)(4CNP)(DA)](ClO4)3 in aqueous solution
| Temperature (K) |
CMC × 104 (mol dm−3) |
−ΔG0mic (kJ mol−1) |
−ΔH0mic (kJ mol−1) |
ΔS0mic (kJ mol−1) |
| 303 |
2.41 |
38.86 ± 0.2 |
14.17 ± 0.1 |
14.17 ± 0.1 |
| 308 |
2.55 |
39.36 ± 0.2 |
14.69 ± 0.2 |
14.69 ± 0.2 |
| 313 |
2.67 |
39.89 ± 0.4 |
15.21 ± 0.1 |
15.21 ± 0.1 |
| 318 |
2.83 |
40.33 ± 0.1 |
15.74 ± 0.1 |
15.74 ± 0.1 |
| 323 |
2.94 |
40.94 ± 0.3 |
16.31 ± 0.4 |
16.31 ± 0.4 |
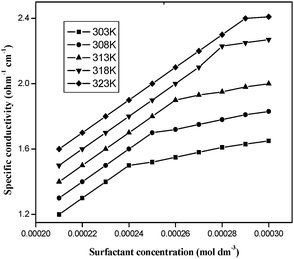 |
| | Fig. 2 Electrical conductivity vs. cobalt(III) concentration for aqueous cis-[Co(trien)(4CNP)(DA)](ClO4)3 solutions. | |
3.3 Thermodynamics of micellization
The study of CMC versus temperature is often undertaken to obtain information on hydrophobic and head group interactions. This involves deriving various thermodynamic parameters of micelle formation. The change in the CMC with temperature is generally analyzed in terms of the phase separation or the equilibrium model for micelle formation.44 According to these models, the standard Gibbs free energy of micelle formation per mole of monomer, ΔG0m, is given by| | | ΔG0m = RT(2 − αave) lnCMC, | (1) |
where R, T and αave are the gas constant, absolute temperature and average degree of micellar ionization (the micelle ionization degree at the CMC was obtained as the ratio between the slopes of the nearly linear specific conductance versus [complex] plots above and below the CMC45), respectively. The enthalpy of micelle formation can be obtained by applying the Gibbs–Helmholtz equation to eqn (1)| | | ΔH0m = −RT2(2 − αave) dlnCMC/dT. | (2) |
Once the Gibbs free energy and the enthalpy of micelle formation are obtained, obviously the entropy of micelle formation can be determined by
| | | ΔS0m = (ΔH0m − ΔG0m)/T | (3) |
The more negative Gibbs free energy of micellization observed for the surfactants with increasing head polarity could indicate more favored micellization. Since the changes of CMC with temperature are small, the values of ΔH0m and ΔS0m must be rather inaccurate and should be considered as only approximate. The negative values of enthalpy (ΔH0m) of micellization indicate the exothermic nature of the micellization process.
Nusselder and Engberts46 have suggested that the negative ΔH0m would indicate that London dispersion forces play a major role in micelle formation. The positive values of ΔS0m clearly indicate that the micellization of the studied surfactants in aqueous solution is governed mainly by hydrophobic interactions between the surfactant cations, resulting in the breakdown of the structured water surrounding the hydrophobic groups and indicating that the cationic surfactants are entropy driven. The increasing head group polarity favours the micellization process. Similar to our previous reports,29,30,47 the CMC value for the surfactant–cobalt(III) complex of the present study is very low compared to that of the simple organic surfactant, dodecylammonium chloride (CMC = 1.59 × 10−2 mol dm−3). Thus, it is suggested that this surfactant–cobalt(III) complex also has more capacity to associate itself into aggregates, when compared with common organic surfactants.
3.4 Nucleic acid binding studies
3.4.1 UV-visible absorption spectroscopy.
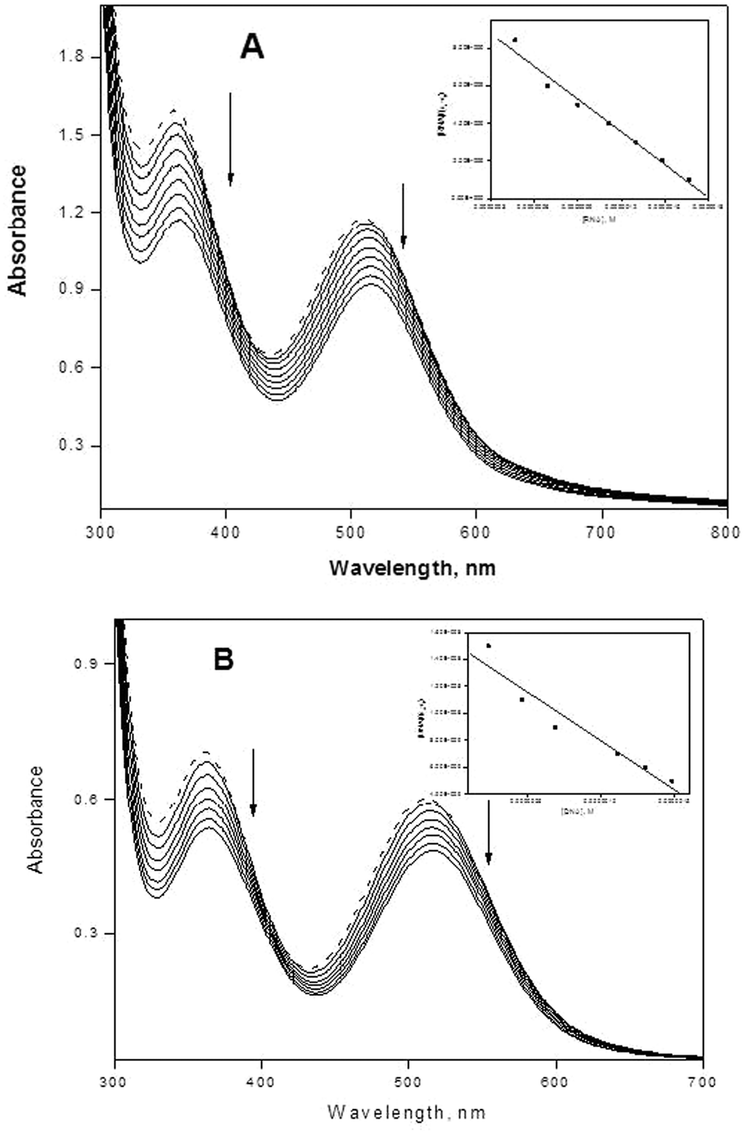
Titration with electronic spectroscopy is an effective method to examine the binding mode of DNA with metal complexes since the observed changes in the spectra may give evidence of the existing interaction mode.48 In general, hypochromism may appear in the case of the intercalative binding mode49 and is due to π → π* stacking interactions, while a red-shift (bathochromism) may be observed when the DNA duplex is stabilized.50 The surfactant–cobalt(III) complex can bind to nucleic acids in different binding modes on the basis of their structure and charge and types of ligand. The binding behavior of the present surfactant–cobalt(III) complex to nucleic acids has been followed through absorption spectral titrations. The absorption spectra of the nucleic acid bound complex in the presence and absence of an ionic liquid additive are shown in Fig. 3A and B, 4A and B. As the concentration of nucleic acid increases, the absorption band (at 515 nm for DNA and 512 nm for RNA) exhibits a decrease in intensity, which is accompanied by a slight red shift with the surfactant–cobalt(III) complex both in presence and absence of an ionic liquid additive indicating that our complex binds to nucleic acids through intercalation. This observed hypochromism is stronger in the presence of an ionic liquid additive. The ability of ionic liquid additives to change the properties of aqueous solutions of surfactants depends on the extent of the interactions between the cation–anion of the IL and the surfactants. Ionic liquids screen the electrostatic repulsion between charged head groups facilitating aggregation between surfactant molecules and thereby lowering the CMC. Through this mechanism, the ionic liquid additive facilitates aggregation of our surfactant–cobalt(III) complex. This aggregation increases the probability of interactions of each complex molecule with DNA. So the intercalative capacity is increased when an ionic liquid is added. In order to compare the strength of the nucleic acid binding of the surfactant–cobalt(III) complex, the intrinsic binding constant, Kb, has been determined using eqn (4),51,52| | | [NA]/(εa − εf) = [NA]/(ε0 − εf) + 1/Kb(ε0 − εf) | (4) |
where [NA] is the concentration of DNA or RNA expressed in base pairs, and εa, εf and ε0 are the apparent, free and fully bound cobalt(III) complex extinction coefficients. A plot of [NA]/(εa − εf) versus [NA] gives Kb as the ratio of the slope to intercept (insets in Fig. 3A and B, 4A and B). The Kb values thus obtained for our surfactant–cobalt(III) complex with nucleic acid both in the presence and absence of an ionic liquid additive are given in Table 2. As seen in Table 2, the Kb values in the presence of an ionic liquid additive are higher compared with the Kb values in the absence of an ionic liquid additive, which is consistent with the qualitative observation of a change in hypochromism. All in all, Kb values obtained for our surfactant–cobalt(III) complex are very much higher than those for similar types of ordinary complexes like [Co(bpy)3]3+ (Kb, 9.3 × 103 M−1)53 and [Co(bpy)2(imp)]3+ (Kb, 1.1 × 104 M−1)54 known in the literature. Between DNA and RNA, our surfactant–cobalt(III) complex can bind very strongly with tRNA when compared to DNA.
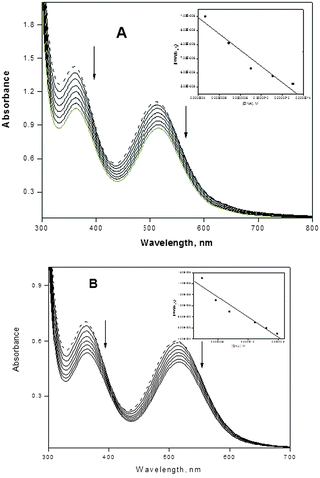 |
| | Fig. 3 (A) Absorption spectra of complex (in the presence of an ionic liquid) in the absence (dotted lines) and in the presence of increasing amounts of CT DNA (solid lines), [complex] = 7.0 × 10−4 M, [DNA] = 0–1.66 × 10−7 M. Ionic liquid = 1 × 10−3 M. Arrows show the absorbance changes upon increasing DNA concentrations. Inset: plot of [DNA]/(εa − εf) versus [DNA]. (B) Absorption spectra of complex (in the absence of an ionic liquid), in the absence (dotted lines) and in the presence of increasing amounts of CT DNA (solid lines), [complex] = 7.0 × 10−4 M, [DNA] = 0–1.66 × 10−7 M. Arrows show the absorbance changes upon increasing DNA concentrations. Inset: plot of [DNA]/(εa − εf) versus [DNA]. | |
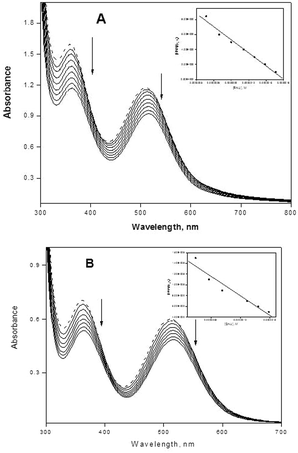 |
| | Fig. 4 (A) Absorption spectra of complex (in the presence of an ionic liquid) in the absence (dotted lines) and in the presence of increasing amounts of tRNA (solid lines), [complex] = 2.8 × 10−4 M, [tRNA] = 0–1.66 × 10−7 M. Ionic liquid = 1 × 10−3 M. Arrows show the absorbance changes upon increasing tRNA concentrations. Inset: plot of [tRNA]/(εa − εf) versus [tRNA]. (B) Absorption spectra of complex (in the absence of an ionic liquid) in the absence (dotted lines) and in the presence of increasing amounts of tRNA (solid lines), [complex] = 2.8 × 10−4 M, [tRNA] = 0–1.66 × 10−7 M. Arrows show the absorbance changes upon increasing tRNA concentrations. Inset: plot of [tRNA]/(εa − εf) versus [tRNA]. | |
Table 2 The binding constant (Kb) of cis-[Co(trien)(4CNP)(DA)]3+ with nucleic acid (in the presence and absence of an ionic liquid) using tris buffer
| Complex |
K
b (M−1) |
Hypochromism% |
| DNA |
RNA |
DNA |
RNA |
|
cis-[Co(trien)(4CNP)(DA)]3+ in the presence of IL |
4.95 × 105 |
4.46 × 106 |
22.54 |
31.04 |
|
cis-[Co(trien)(4CNP)(DA)]3+ in the absence of IL |
4.72 × 104 |
5.55 × 105 |
19.59 |
21.30 |
3.4.2 Competitive binding between (EB) and complex.
In order to examine the ability of the compound to displace EB (3,8-diamino-5-ethyl-6-phenyl-phenanthridinium bromide) from its EB–DNA complex, a competitive EB binding study has been undertaken with fluorescence experiments.55 EB is a phenanthridine fluorescent dye and is a typical indicator of intercalation, forming soluble complexes with nucleic acids and emitting intense fluorescence in the presence of CT DNA due to the intercalation of the planar phenanthridinium ring between adjacent base pairs on the double helix.56 The changes observed in the spectra of EB on its binding to CT DNA are often used for interaction studies between DNA and other compounds, such as metal complexes.57 When our surfactant–cobalt(III) complex solutions are added to nucleic acids pretreated with EB, the nucleic acid-induced emission intensity of EB decreases (ESI,† Fig. S1 and S2). Addition of a second nucleic acid binding molecule quenches the EB emission by replacing the EB from the nucleic acid. The quenching behaviour was analyzed through the canonical Stern–Volmer equation,58I0/I = 1 + Ksvr, where I0 and I are the fluorescence intensities in the absence and the presence of complex, respectively. Ksv is a linear Stern–Volmer constant which is dependent on the ratio of rbE (the ratio of the bound concentration of ethidium bromide to the concentration of NA). r is the ratio of the total concentration of complex to that of NA. When a plot of I0/I versus [complex]/[NA] is produced, Ksv is obtained from the ratio of the slope to the intercept (ESI,† Fig. S3 and S4). The Ksv values for the surfactant–cobalt(III) complex thus obtained for binding with DNA are 0.58 and 0.42 (in the presence and absence of an ionic liquid additive) and with RNA are 0.63 and 0.54 (in the presence and absence of an ionic liquid additive) are shown in Table 3. An increase in the Ksv values in the presence of an ionic liquid additive suggests that ionic liquids facilitate the interaction between surfactant complexes and nucleic acids as observed through UV-visible absorption experiments.
Table 3 Fluorescence data of cis-[Co(trien)(4CNP)(DA)]3+ of Ksv (in the presence and absence of an ionic liquid) using tris buffer
| Complex |
K
sv
|
| DNA |
RNA |
|
cis-[Co(trien)(4CNP)(DA)](ClO4)3 in the presence of IL |
0.58 |
0.63 |
|
cis-[Co(trien)(4CNP)(DA)](ClO4)3 in the absence of IL |
0.42 |
0.54 |
3.4.3 Cyclic voltammetry studies.
Electrochemical investigations of metal complex–DNA interactions can provide a useful complement to spectroscopic methods. Cyclic voltammograms of the surfactant–cobalt(III) complex were performed in Tris-HCl buffer. The voltammetric results for the surfactant–cobalt(III) complex in the absence and presence of nucleic acids in Tris-HCl buffer solutions are shown in Tables 4 and 5. As seen from these tables, the cyclic voltammogram of cis-[Co(trien)(4CNP)(DA)](ClO4)3 in the absence of nucleic acid features the reduction of Co(III) to form Co(II) at a cathodic peak potential Epc of −1.39 V versus SCE. Reoxidation of Co(II) occurs, upon scan reversal, at 1.20 V. The formal potential (E1/2) and the separation of the anodic and cathodic peak potential (ΔEp) are −0.09 mV and −2.6 V, respectively. The ratio of the oxidation peak current to reduction peak current (Ipa/Ipc) = 0.85, which indicates that the redox process was irreversible. No new peaks appeared after the addition of nucleic acid to the surfactant–cobalt(III) complex. During the course of the addition of nucleic acid (CT DNA/yeast tRNA), the anodic peak potential (Epa), cathodic peak potential (Epc) and E1/2 (calculated as the average of Epc and Epa) all showed positive shifts (E1/2, ΔEp and the ratio of the peak current are −0.07 V, −2.51 V and 0.83, respectively, for CT DNA, and −0.88 V, −0.37 V and 0.02, respectively, for yeast tRNA), indicating the intercalation of the cobalt(III) complex into the nucleic acid structure. The binding is considered principally due to the hydrophobic interactions. In the presence of an ionic liquid additive, the anodic peak potential (Epa), cathodic peak potential (Epc) and E1/2 (calculated as the average of Epc and Epa) all showed more positive shifts (Tables 6 and 7) indicating strong interaction between the surfactant–cobalt(III) complex and nucleic acid.
Table 4 Cyclic voltammetric data (mV) of cis-[Co(trien)(4CNP)(DA)]3+ (in the absence of an ionic liquid) in the absence and in the presence of CT DNA. A scan rate of 100 mV s−1 was used with tris buffer as the supporting electrolyte
| Complex (in the absence of IL) |
E
pc
|
E
pa
|
ΔEp |
E
1/2
|
I
pa/Ipc |
| [Co(trien)(4CNP)(DA)]3+ |
−1395.5 |
1204.5 |
−2600 |
−95.5 |
0.85 |
| [Co(trien)(4CNP)(DA)]3+ + CT DNA |
−1325.5 |
1184.5 |
−2510 |
−70.25 |
0.83 |
Table 5 Cyclic voltammetric data (mV) of cis-[Co(trien)(4CNP)(DA)]3+ (in the absence of an ionic liquid) in the absence and in the presence of tRNA. A scan rate of 100 mV s−1 was used with tris buffer as the supporting electrolyte
| Complex (in the absence of IL) |
E
pc
|
E
pa
|
ΔEp |
E
1/2
|
I
pa/Ipc |
| [Co(trien)(4CNP)(DA)]3+ |
−855.5 |
64.5 |
−400.5 |
910.0 |
0.03 |
| [Co(trien)(4CNP)(DA)]3+ + RNA |
−815.5 |
54.5 |
−375.5 |
880.0 |
0.02 |
Table 6 Cyclic voltammetric data (mV) of cis-[Co(trien)(4cnp)(DA)]3+ (in the presence of an ionic liquid) in the absence and in the presence of CT DNA. A scan rate of 100 mV s−1 was used with tris buffer as the supporting electrolyte
| Complex (in the presence of IL) |
E
pc
|
E
pa
|
ΔEp |
E
1/2
|
I
pa/Ipc |
| [Co(trien)(4CNP)(DA)]3+ |
−895.5 |
184.5 |
1080.0 |
−355.5 |
0.03 |
| [Co(trien)(4CNP)(DA)]3+ + CT DNA |
−840.5 |
199.5 |
1040.0 |
−320.5 |
0.01 |
Table 7 Cyclic voltammetric data (mV) of cis-[Co(trien)(4CNP)(DA)]3+ (in the presence of an ionic liquid) in the absence and in the presence of tRNA. A scan rate of 100 mV s−1 was used with tris buffer as the supporting electrolyte
| Complex (in the presence of IL) |
E
pc
|
E
pa
|
ΔEp |
E
1/2
|
I
pa/Ipc |
| [Co(trien)(4CNP)(DA)]3+ |
−835.5 |
49.5 |
895.0 |
−388.0 |
0.04 |
| [Co(trien)(4CNP)(DA)]3+ + RNA |
−800.5 |
29.5 |
850.0 |
−375.5 |
0.02 |
3.4.4 Viscosity measurements.
Optical photophysical probes generally provide necessary clues to support a binding mode, however these are not sufficiently conclusive. Hydrodynamic measurements, which are sensitive to DNA length changes, are regarded as the least ambiguous and most critical tests of a binding model in solution in the absence of crystallographic data.59 A classical intercalation model demands that the DNA helix lengthens as base pairs are separated to accommodate the binding ligand, and thus leads to an increase in the viscosity of the DNA solution. In contrast, a partial, non-classical intercalation of ligand could bend (or kink) the DNA helix, reducing its length and viscosity.60 To confirm the nature of the interactions between the title complex and nucleic acid, viscosity measurements were carried out by keeping the DNA concentration constant and varying the concentration of the complex. Plots of the relative specific viscosity (η/η0)1/3) vs. [complex]/[NA] ratio show the capability of the complex to increase the viscosity of the solution of the nucleic acid and are shown in the ESI,† Fig. S5. The increase in the concentration of the complex increases the viscosity of the solution of the nucleic acid, which is due to intercalation of our surfactant complex with nucleic acid. As the presence of an ionic liquid will itself increase the viscosity of the medium, we did not perform viscosity experiments in the presence of an ionic liquid additive.
3.5 Cytotoxicity studies
3.5.1 MTT assay.
The cytotoxicity effects of the surfactant–cobalt(III) complex (Fig. 5) on cultured HepG2 liver cancer cells were determined by exposing cells for 24 and 48 h to the medium containing the complex at 10–90 μg mL−1 concentration. In vitro antitumor activity of these compounds was determined according to the percentage of nonviable cells (%NVC), which was calculated by the following equation:
| NVC% = [number of NVC/total number of cells] × 100 |
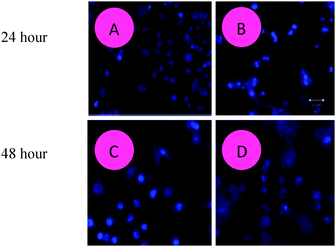 |
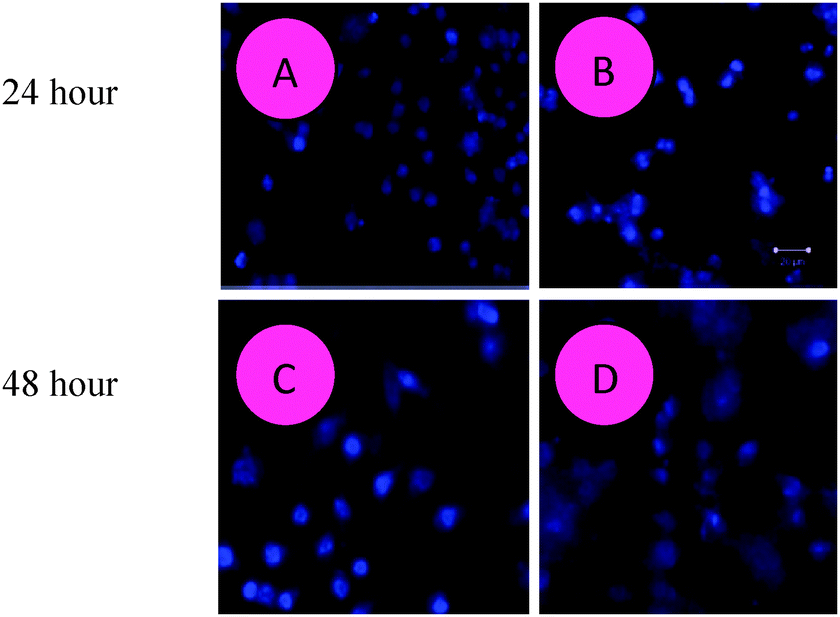
| | Fig. 5 The surfactant–cobalt(III) complex induces apoptosis in HepG2 liver cancer cells. Representative fluorescence micrographs of HepG2 liver cancer cells stained with Hoechst 33342 fluorescent dye after the compound exposure for 24 and 48 hours. (A, C) Untreated control cells; (B, D) surfactant–cobalt(III) complex treated control cells. (A, B) – 24 hours; (C, D) – 48 hours. Magnifications at 20×. | |
The results of these experiments are summarized in Table 8. As shown in the table, increasing the concentration of the surfactant–cobalt(III) complex was accompanied by a progressive decrease in the VC%. This is due to the fact that by increasing the concentration of cationic surfactant complexes, the adsorption of ions on cell membranes increases, leading to an increase in penetration and antitumor activity. The inhibition of cell viability percent showed that the surfactant–cobalt(III) complex is the most active one at a concentration of 90 μg mL−1, the VC% reaching up to 39.2%. This means that the drug at this concentration causes the death of most of the tumor cells, while at a concentration of 60 μg mL−1, the percentage reached 48.2%. For the 24 h treatment period, higher concentrations of the complex were required to kill the cells, whereas for 48 h treatment, the cell killing occurred at lower concentrations. The results of the cytotoxic activity on human tumor cell lines was determined according to the dose values of drug exposure required to reduce survival in the cell lines to 50% (IC50). The IC50 value of the complex was slightly higher for the 24 h treatment groups, i.e., in the range of 39.2−90 μg mL−1, whereas for the 48 h treatment groups, the IC50 value fell in the range of 27.4–90 μg mL−1. It should be noted that the action of the complex as an antitumor agent is found to be dependent on the type of tumor cell line tested but, as shown from the results, the surfactant–cobalt(III) complex shows excellent cytotoxic activity against tumor cell lines and, at very low concentrations, reduces the survival to 50%. This is due to the fact that cobalt complexes have the capacity to reduce the energy status in tumors, as well as to enhance tumor hypoxia, which also influences their antitumor activities. It may be also concluded that the level of cellular damage inflicted by these complexes depends on the nature of their axial ligands. It is known that phenanthroline/bipyridine-containing metal complexes possess a wide range of biological activities such as antitumor, antifungal, apoptosis61–63 and interaction with DNA inhibiting replication, transcription and other nuclear functions and arresting cancer cell proliferation so as to arrest tumor growth. From these results, surfactant–cobalt(III) complexes seem to offer promise due to the high electron affinity of the metal (which increases its ability to bind DNA) and the ready reducibility of the compounds.64
Table 8 Cell viability assay by the MTT (3-(4,5-dimethylthiozol-2-yl) 2,5-diphenyl-tetrazolium bromide) method
| Surfactant–cobalt(III) complex |
Concentration (μg mL−1) |
Cell viability (VC%) 24 hours |
Cell viability (VC%) 48 hours |
|
cis-[Co(trien)(4CNP)(DA)](ClO4)3 |
10 |
75.2 |
61.7 |
| 30 |
60.7 |
49.9 |
| 60 |
48.2 |
34.1 |
| 90 |
39.2 |
27.4 |
3.5.2 Fluorescence microscopic analysis of apoptotic cell death (AO and EB staining).
Acridine orange–ethidium bromide (AO–EB) staining adopting fluorescence microscopy also revealed apoptosis from the perspective of fluorescence, after HepG2 liver cancer cells were exposed to the various concentrations of the surfactant–cobalt(III) complex for 24 h (Fig. 6). In this study, we used the AO–EB double staining assay.65 Acridine orange is taken up by both viable and nonviable cells and emits green fluorescence if interrelated into double stranded nucleic acid (DNA) or red fluorescence if bound to single stranded nucleic acid (RNA). Ethidium bromide is taken up only by nonviable cells and emits red fluorescence by intercalation into DNA. We distinguished four types of cells according to the fluorescence emission and the morphological aspect of chromatin condensation in the stained nuclei: (1) viable cells showing light green fluorescing nuclei with a highly organized structure; (2) early apoptotic cells having bright green fluorescing nuclei with chromatin condensation and nuclear fragments; (3) late apoptotic cells having orange to red fluorescing nuclei with condensed or fragmented chromatin; and (4) necrotic cells having red fluorescing nuclei without chromatin fragmentation. Viable cells have uniform bright green nuclei with an organized structure. Apoptotic cells have orange to red nuclei with condensed or fragmented chromatin. Necrotic cells have uniformly orange to red nuclei with a condensed structure. Our results indicate that the surfactant–cobalt(III) complex induced apoptosis at the concentrations evaluated, in agreement with the cytotoxic results. The results suggest that the complex treatment caused greater cell death of HepG2 liver cancer cells.
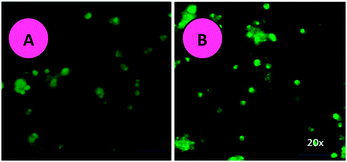 |
| | Fig. 6 Photomicrographs of the control and AO and EB stained HepG2 liver cancer cells incubated for 24 hours with the surfactant–cobalt(III) complex. (A) Untreated control cells. (B) Magnification at 20×. | |
3.5.3 Apoptosis detection with Hoechst 33342 DNA staining.
It is possible to perform apoptosis detection assays with Hoechst 33342 (Sigma B-2262), but the increase in fluorescence seen in the apoptotic cells may be less dramatic. Hoechst dyes can also be obtained from Molecular Probes®. H342 is a “vital” DNA stain that binds preferentially to A–T base-pairs. The cells require no permeabilization for labeling, but do require physiological conditions since the dye internalization is an active transport process. This condition typically varies among cell types.66 The procedure for staining and analysis of cells using Hoechst 33342 (H342): to investigate if HepG2 liver cancer cells were triggered to undergo apoptosis due to the exposure to the surfactant–cobalt(III) complex, morphological changes of apoptosis in the treated cells were determined by Hoechst 33342 staining. Apoptosis is one of the major pathways that lead to the process of cell death. After the cells were treated with IC50 concentrations of the surfactant–cobalt(III) complex (31–250 μg mL−1) for 24 and 48 h, the cells were observed for cytological changes by adopting Hoechst 33342 staining. The observations revealed that the complex brought about cytological changes such as chromatin fragmentation, binucleation, cytoplasmic vacuolation, nuclear swelling, cytoplasmic blebbing and late apoptosis indications of dot-like chromatin and condensation (Fig. 5A and C), whereas untreated cells did not show such changes (Fig. 5B and D). Data were collected by the manual counting of cells with normal and abnormal nuclear features. Both apoptotic and necrotic cells increased in a dose-dependent manner. These data clearly indicated that higher doses of the surfactant–cobalt(III) complex resulted in remarkable chromatin condensation and nuclear fragmentation in HepG2 liver cancer cells.
4 Conclusions
The synthesis and characterization of the surfactant–cobalt(III) complex, cis-[Co(trien)(4CNP)(DA)](ClO4)3 has been reported. As in our previous reports,23,24 the critical micelle concentration value of this surfactant–cobalt(III) complex is 2.41 × 10−4 M−1 and it has more capacity to associate and form aggregates, than ordinary synthetic organic surfactants. The nucleic acid binding property of the complex was investigated by electronic absorption, fluorescence, cyclic voltammetry and viscosity measurements. The experimental results indicate that the complex can bind to nucleic acids (DNA and RNA) by an intercalation mode with a binding constant Kb of 4.72 × 104 M−1 for DNA and 5.55 × 105 M−1 for RNA. The presence of an ionic liquid additive increased this intercalative capacity (Kb values are 4.95 × 105 M−1 for DNA and 4.46 × 106 M−1 for RNA) by facilitating aggregation of our surfactant–cobalt(III) complex. The surfactant–cobalt(III) complex is cytotoxic to HepG2 (human hepatocellular liver carcinoma) cells. Therefore, this complex might prove to have an application in target-based cancer therapy since the mechanism of cell death appears to be essentially apoptosis.
Acknowledgements
We are grateful to the UGC-COSIST and DST-FIST programmes of the Department of Chemistry, Bharathidasan University, and UGC-RFSMS fellowship to one of the authors, K. Nagaraj, by Bharathidasan University. Financial assistance from the CSIR (Grant No. 01(2461)/11/EMR-II), DST (Grant No. SR/S1/IC-13/2009) and UGC (Grant No. 41-223/2012(SR) sanctioned to S. Arunachalam are also gratefully acknowledged.
References
- E. L. Hegg and J. N. Burstyn, Coord. Chem. Rev., 1998, 173, 133 CrossRef CAS.
- M. Komiyama and J. Sumaoka, Curr. Opin. Chem. Biol., 1998, 2, 751 CrossRef CAS.
-
B. Norden, P. Lincoln, B. Akerman and E. Tuite, in Metal ions in biological systems, ed. A. Sigel and H. Sigel, Marcel Dekker, New York, 1996, vol. 33, p. 177 Search PubMed.
- L. F. Tan, X. L. Liang and X. H. Liu, J. Inorg. Biochem., 2009, 103, 441 CrossRef CAS PubMed.
- Y. Tor, ChemBioChem, 2003, 4, 998 CrossRef CAS PubMed.
- T. Hermann, Biopolymers, 2003, 70, 4 CrossRef CAS PubMed.
- J. Gallego and G. Varani, Acc. Chem. Res., 2001, 34, 836 CrossRef CAS PubMed.
- C. S. Chow and J. K. Barton, Biochemistry, 1992, 31, 5423 CrossRef CAS.
- N. Farrell, Met. Ions Biol. Syst., 2004, 41, 252 Search PubMed.
- X. Hong, D. Hong, Q. L. Zhang, Y. Huang, J. Z. Liu and L. N. Ji, Inorg. Chem. Commun., 2003, 6, 766 CrossRef.
- S. Aviran, C. Trapnell, J. B. Lucks, S. A. Mortimer, S. Luo, G. P. Schroth, J. A. Doudna, A. P. Arkina and L. Pachter, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11069 CrossRef CAS PubMed.
- W. K. Pogozelski and T. D. Tullius, Chem. Rev., 1998, 98, 1089 CrossRef CAS PubMed.
- T. Tran and M. D. Disney, Biochemistry, 2011, 50, 962 CrossRef CAS PubMed.
- T. D. Avery, N. F. Jenkis, M. C. Kimber, D. Lupton and D. K. Taylor, Chem. Commun., 2002, 28 RSC.
- J. G. Huddieston, H. D. Willauer, R. P. Swauoski, A. E. Visser and K. D. Rogers, Chem. Commun., 1998, 1765 RSC.
- R. Vanyúr, L. Biczók and Z. Miskolczy, Colloids Surf., A, 2007, 299, 256 CrossRef PubMed.
- R. Sharma, S. Mahajan and R. K. Mahajan, Colloids Surf., A, 2013, 427, 62 CrossRef CAS PubMed.
- H. Wang, J. Wang, S. Zhang and X. Xuan, J. Phys. Chem. B, 2008, 112, 16682 CrossRef CAS PubMed.
- M. H. Marques, N. V. Plechkova, K. R. Seddon, L. P. N. Rebelo and A. Lopes, Green Chem., 2007, 9, 481 RSC.
- T. Singh, M. Drechsler, A. H. E. Mueller, I. Mukhopadhyay and A. Kumar, Phys. Chem. Chem. Phys., 2010, 12, 11728 RSC.
- T. Singh, T. J. Trivedi and A. Kumar, Green Chem., 2010, 12, 1029 RSC.
- D. W. Armstrong, J. L. Anderson, V. Pino, E. C. Hagberg and V. V. Sheares, Chem. Commun., 2003, 2444 Search PubMed.
- K. A. Fletcher and S. Pandey, Langmuir, 2004, 20, 33 CrossRef CAS.
- K. Behera, P. Dahiya and S. Pandey, J. Colloid Interface Sci., 2007, 307, 235 CrossRef CAS PubMed.
- R. Rai, G. A. Baker, K. Behera, P. Mohanty, N. D. Kurur and S. Pandey, Langmuir, 2010, 26, 17821 CrossRef CAS PubMed.
- J. L. Anderson, V. Pino, E. C. Hagberg, V. V. Sheares and D. W. Armstrong, Chem. Commun., 2003, 2444 RSC.
- K. S. Rao, T. Singh, T. J. Trivedi and A. Kumar, J. Phys. Chem. B, 2011, 115, 13847 CrossRef PubMed.
- T. Misono, H. Sakai, K. Sakai, M. Abe and T. Inoue, J. Colloid Interface Sci., 2011, 358, 527 CrossRef CAS PubMed.
- N. Kumaraguru, S. Arunachalam, M. N. Arumugam and K. Santhakumar, Transition Met. Chem., 2006, 31, 250 CrossRef CAS PubMed.
- K. Santhakumar, N. Kumaraguru, M. N. Arumugam and S. Arunachalam, Polyhedron, 2006, 25, 1507 CrossRef CAS PubMed.
- J. Lakshmipraba, S. Arunachalam, D. A. Gandi and T. Thirunalasundari, Eur. J. Med. Chem., 2011, 46, 3013 CrossRef CAS PubMed.
- B. Dong, N. Li, L. Zheng, L. i. Yu and I. Tohru, Langmuir, 2007, 23, 4178 CrossRef CAS PubMed.
- G. Cohen and H. Eisenberg, Biopolymers, 1969, 8, 45 CrossRef CAS.
- R. M. Uda and M. Oshita, Biomacromolecules, 2012, 13, 1510 CrossRef CAS PubMed.
- C. Revathi and A. Dayalan, J. Serb. Chem. Soc., 1992, 71, 1311 Search PubMed.
- R. E. Kitson, Anal. Chem., 1950, 22, 664 CrossRef CAS.
- M. F. Reichmann, S. A. Rice and T. P. Doty, J. Am. Chem. Soc., 1954, 76, 3047 CrossRef CAS.
- G. Cohen and H. Eisenberg, Biopolymers, 1969, 8, 45 CrossRef CAS.
- R. Zana, J. Colloid Interface Sci., 1980, 78, 330 CrossRef CAS.
- V. A. Izumrudov, M. V. Zhiryakova and A. A. Goulko, Langmuir, 2002, 18, 10348 CrossRef CAS.
- T. M. Kelly, A. B. Tossi, D. J. McConnell and T. C. Strekas, Nucleic Acids Res., 1985, 13, 6017 CrossRef PubMed.
- D. A. Buckingam and D. Jones, Inorg. Chem., 1965, 4, 1387 CrossRef.
- M. E. Baldwin, J. Chem. Soc., 1960, 4369 RSC.
- M. L. Morris and D. H. Busch, J. Am. Chem. Soc., 1960, 82, 1521 CrossRef CAS.
- P. Mukerjee, J. Phys. Chem., 1962, 66, 1375 CrossRef CAS.
- J. J. H. Nusselder and J. B. F. N. Engberts, J. Colloid Interface Sci., 1992, 148, 353 CrossRef CAS.
- N. Kumaraguru, K. Santhakumar, S. Arunachalam and M. N. Arumugam, Polyhedron, 2006, 25, 3253 CrossRef CAS PubMed.
- E. C. Long and J. K. Barton, Acc. Chem. Res., 1990, 23, 271 CrossRef CAS.
- K. Nagaraj and S. Arunachalam, Int. J. Biol. Macromol., 2013, 62, 273 CrossRef PubMed.
- M. T. Carter, M. Rodriguez and A. J. Bard, J. Am. Chem. Soc., 1989, 111, 8901 CrossRef CAS.
- P. Tamil Selvi and M. Palaniandavar, Inorg. Chim. Acta, 2002, 337, 420 CrossRef.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392 CrossRef CAS.
- A. D. Samara, A. A. Pantazaki, M. Alexiou, E. Nordlander and D. P. Kessissoglou, J. Inorg. Biochem., 2008, 102, 618 CrossRef PubMed.
- S. Satyanarayana, J. C. Dabroniak and J. B. Chaires, Biochemistry, 1992, 31, 9319 CrossRef CAS.
- G. Zhao, H. Lin, S. Zhu, H. Sun and Y. Chen, J. Inorg. Biochem., 1998, 70, 219 CrossRef CAS.
- J. Olmsted and D. R. Kearns, Biochemistry, 1977, 16, 3647 CrossRef CAS.
- B. C. Baguley and M. Lebret, Biochemistry, 1984, 23, 937 CrossRef CAS.
- J. R. Lakowicz and G. Webber, Biochemistry, 1973, 12, 4161 CrossRef CAS.
- S. Satyanaryana, J. C. Daborusak and J. B. Chaires, Biochemistry, 1993, 32, 2573 CrossRef.
- S. Satyanaryana, J. C. Daborusak and J. B. Chaires, Biochemistry, 1992, 31, 9319 CrossRef.
-
B. Coyle, P. Kinsella, M. McCann, M. Devereux, R. O'Connor, M. Clynes and K. Kavanagh, International Journal Published in Association with BIBRA, 2004, 18, 63.
- B. Coyle, M. McCann, K. Kavanagh, M. Devereux and M. Geraghty, BioMetals, 2003, 16, 321 CrossRef CAS.
- S. Osinsky, I. Levitin, L. Bubnovskaya, A. Sigan and I. N. Ganusevich, Exp. Oncol., 2004, 26, 140 CAS.
- A. M. Badawi, M. Mekawias, M. Z. Mohamed and M. M. Khowdairy, J. Cancer Res. Ther., 2007, 3, 198 CrossRef CAS PubMed.
- D. Baskic, S. Popovic, P. Ristic and N. N. Arsenijevic, Cell Biol. Int., 2006, 30, 924 CrossRef CAS PubMed.
- A. Stander, S. Marais and V. Stivaktas,
et al.
, J. Ethnopharmacol., 2009, 124, 45 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nj00832k |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.