Diamondoids: functionalization and subsequent applications of perfectly defined molecular cage hydrocarbons
Maria A.
Gunawan
ab,
Jean-Cyrille
Hierso
*ac,
Didier
Poinsot
a,
Andrey A.
Fokin
bd,
Natalie A.
Fokina
b,
Boryslav A.
Tkachenko
b and
Peter R.
Schreiner
*b
aUniversité de Bourgogne, Institut de Chimie Moléculaire de l'Université de Bourgogne (ICMUB), UMR CNRS 6302, 9 avenue Alain Savary, 21078 cedex, Dijon, France. E-mail: hiersojc@u-bourgogne.fr; Fax: +33 (0)38039 3682; Tel: +33 (0)38039 6107
bInstitut für Organische Chemie, Justus-Liebig-Universität, Heinrich-Buff-Ring 58, 35392 Giessen, Germany. E-mail: prs@uni-giessen.de; Fax: +49 641 99 34309; Tel: +49 641 99 34300
cInstitut Universitaire de France (IUF), France
dDepartment of Organic Chemistry, Kiev Polytechnic Institute, pr. Pobedy 37, 03056, Kiev, Ukraine
First published on 20th September 2013
Abstract
The term “diamondoid” describes cage hydrocarbon molecules that are superimposable on the diamond lattice. Diamondoids that are formally built by face-fusing of adamantane units, namely diamantane, triamantane, tetramantane, etc., have fascinated chemists since the beginning of the last century. The functionalization of these perfectly defined (C,H)-molecules is described here. Thus, diamondoid halides and diamondoid alcohols are first rank precursors for amino and phosphine-substituted diamondoids that have proved to be highly useful in therapeutic applications and metal catalysis, respectively. The extent of functionalization and polyfunctionalization achieved for adamantane and diamantane, and the synthesis and applications of the resulting organohybrids are illustrated, revealing their high potential in fields such as organocatalysis, polymers, molecular electronics and mechanics.
 Maria A. Gunawan | Maria A. Gunawan was born in Malang (Indonesia). She studied chemistry at Clermont-Ferrand's Superior National School of Chemistry and achieved in 2010 both an engineering degree and a master’s degree from Blaise Pascal University in Clermont-Ferrand. She is now an international PhD student under the joint supervision of Prof. Jean-Cyrille Hierso (Dijon, France) and Prof. Peter R. Schreiner (Giessen, Germany) on the topic of diamondoids functionalization and metallization. |
 Jean-Cyrille Hierso | Jean-Cyrille Hierso was born in Toulouse (France) in 1971. He is Full Professor of Chemistry, heading the group of “Organometallic Chemistry and Catalysis” in the Institute of Molecular Chemistry at the University of Burgundy (Dijon, France). He is interested in metals and ligands chemistry, catalysis, and nanomaterials. In 2011 he was awarded the Prize for Coordination Chemistry from the French Chemical Society (SCF). In 2012 he received the EurJIC Young Researcher Award and he was appointed Member of the Institut Universitaire de France (IUF). He shares with his friend Peter R. Schreiner a passion for nanodiamonds (less expensive for girls…). |
Didier Poinsot was born in Strasbourg (France) in 1972. He studied physical chemistry at the University Louis Pasteur of Strasbourg (master’s degree in thin film materials, 1998). He became assistant engineer in CNRS, at the University of Burgundy (1999), and is in charge of the “Catalytic tests under pressure laboratory” in the Institute of Molecular Chemistry. He joined with enthusiasm the French-German cooperation team built in 2011 around “nanodiamonds for chemical vapour deposition”. |
 Andrey A. Fokin | Andrey A. Fokin is a Head of the Department of organic chemistry at the National Technical University of Ukraine “Kiev Polytechnic Institute”. He obtained his Candidate (1985) and Doctor of Chemical Sciences (1995) degrees from this institution. He was a DAAD (1996) and Alexander von Humboldt (1997–1998) research fellow in the group of Professor P. v. R. Schleyer at the University of Erlangen-Nürnberg (Germany). He is the author of over 150 publications on the chemistry of cage compounds, pesticide chemistry, alkane activation mechanisms, computational chemistry of radicals and radical ions, and the generation and transformations of hydrocarbon radical cations. |
 Natalie A. Fokina | Natalie A. Fokina was born in Kiev (Ukraine). She studied chemistry at the National Technical University of Ukraine “Kiev Polytechnic Institute”, and after obtaining her MS (1983) she worked as a production designer at Scientific Technological Institute of Domestic Chemistry (Kiev). She obtained a PhD (2002) in Bioorganic Chemistry at the Institute of Bioorganic Chemistry and Petrochemistry NAS of Ukraine and there served as Deputy Head of the Department of Fine Organic Synthesis. Since 2004 she has been working in the group of P. R. Schreiner as a research associate. She is the author of 25 publications on the chemistry of cage compounds and chemo-enzymatic approaches to the synthesis of fluorinated amino acids. |
Boryslav A. Tkachenko was born in Kyiv (Ukraine) in 1976. He studied chemistry at the National Technical University of Ukraine “Kyiv Polytechnic Institute” (Ukraine) where he received his MS (1999) and Candidate of Chemical Sciences (2003, PhD) degrees in Organic Chemistry. Currently he is a research associate in the group of P. R. Schreiner and a member of the diamondoid research team. |
 Peter R. Schreiner | Peter R. Schreiner (b. 1965) studied and received his Dr. rer. nat. (1994) in organic chemistry at the University of Erlangen-Nürnberg (Germany) and a PhD (1995) in Computational Chemistry from the University of Georgia (UGA), USA. He was assistant professor in Göttingen and associate professor at UGA before accepting the chair of organic chemistry at the Justus-Liebig University Giessen (Germany) in 2002, where he serves also as Vice President for Research. P. R. Schreiner received the Dirac Medal (2003) and the ADUC-Prize (1999). He was bestowed with honorary lifetime memberships of the Israel (2009) and the Polish (2013) Chemical Society. In 2013 he became a member of the German National Academy of Science (Leopoldina). In the little spare time he has, he enjoys playing fierce tennis matches and loud rock music. |
1. Introduction
Nanometre-sized particles possessing a perfect diamond cubic lattice are fascinating molecules that have recently been employed for applications that span the fields of organic and inorganic chemistry, physics, materials science, bioengineering, medicine and beyond.1–3 The term “diamondoid” has been used to name cage hydrocarbon molecules of a size larger than adamantane (C10H16) that are totally or mostly superimposable on the cubic diamond lattice.4 In this context most diamondoids are unique hydrocarbon nanostructures that can be described as fully hydrogen-terminated nanometre-sized diamonds (Scheme 1), and they have rightly been termed nanodiamonds (or molecular nanodiamonds), as opposed to detonation nanodiamond2,3 for which a clearly defined surface functionalization is particularly challenging. Thus, among diamondoids the family of polymantanes includes molecules that can only be formed by the face-fusing of adamantane units, sharing six carbon atoms per pair of adamantane units. The polymantanes form a series of which the smallest molecules are called lower diamondoids. For adamantane through triamantane the general formula (C4n+6H4n+12) does not comprise isomers whereas the next higher member of this series (C22H28) corresponds to four tetramantane stereoisomers (Scheme 1), including one pair of enantiomers. Diamondoids are very stable and possess carbon atoms of different but precisely defined “environments” (as exemplified in Scheme 1 with diamantane). As a consequence, their surface functionalization is expected to be conducted accurately by chemical means.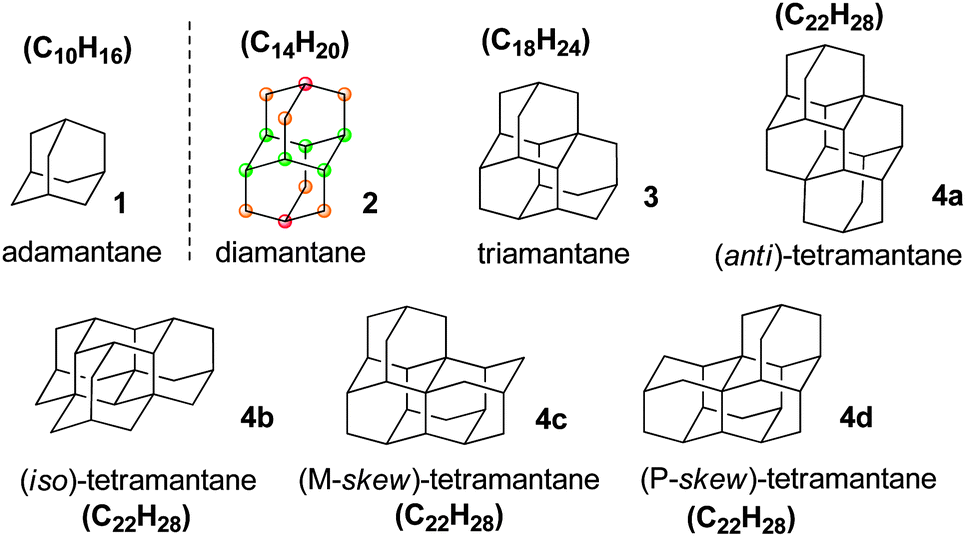 | ||
| Scheme 1 Lower diamondoids: polymantanes (2–4d) are built by face-fusing of adamantane (1) units. | ||
The Baeyer system is used for IUPAC naming of polycyclic hydrocarbons, including diamondoids and polymantanes.5 For naming polymantanes, which are regularly repeating fused adamantane units, the Baeyer system can be advantageously substituted by a nomenclature introduced by Balaban and Schleyer.4 Indeed, for instance, diamantane is named pentacyclo[7.3.1.1.4,12.0.2,706,11]tetradecane in the Baeyer system. In contrast, Balaban and Schleyer nomenclature system is based on graph theory and thus considers a step by step growth of adamantane cages. With this convenient convention the first lower diamondoids, after adamantane, are named [1]diamantane, [12]triamantane and [121]tetramantane (anti-tetramantane), and the higher diamondoids are easily pictured following the vectors indicated in brackets for adding further cages; this was nicely illustrated recently by Filik.6
From a historical perspective, adamantane was identified in 1933 from a sample of petroleum collected in Czechoslovakia.7 It was a real breakthrough for alkane chemistry when Landa showed that adamantane reacts with neat bromine at room temperature to give the tertiary monobromo derivative almost quantitatively.8 Efficient synthetic preparation of lower diamondoids has been reported for adamantane9 and for diamantane;10,11 the latter hydrocarbon was also isolated from petroleum.12 In 1966 the challenging synthesis of triamantane was also reported.13 The first higher diamondoid was synthesized in 1976: anti-tetramantane {= [121]tetramantane} received this name from its structural similarity to butane in its anti-conformation.14 Schleyer reported a perspective on this period of synthetic success, which has ended with the synthetic attempts of higher diamondoids.15 Synthetic efforts for higher diamondoids preparation via carbocation rearrangements similar to those successfully devised to prepare the lower diamondoids failed,16 apparently blocked by the large number of possible intermediates and the complex reaction pathways involved in the processes. Nevertheless, higher diamondoids up to hexamantane (C30H36) have been shown to exist in petroleum on the basis of gas chromatography/mass spectrometry analysis.17 It was suggested later that diamondoids may occur largely in all petroleum sources and thus may be used as decisive indicators of natural oil cracking.18 The breakthrough evidence of diamondoids' ubiquity in oils and fuels was provided in 2003 through the isolation and identification of twenty-one different higher polymantanes by HPLC techniques.19 Additionally, there is a method akin to chemical vapour deposition that yields higher diamondoids, albeit in low yields.20 Lower diamondoids are low-strained, and kinetically as well as thermodynamically very stable. These characteristics are accompanied by high melting points in comparison to other hydrocarbons; for adamantane, diamantane and tetramantane, melting points are estimated between 220 and 270 °C (they fairly easily sublime at room temperature under normal or low pressure). Solubility limits of adamantane and diamantane in liquid organic solvents at 25 °C (alkanes, benzene, etc.) have been reported.21 Commonly used spectroscopic methods to analyse diamondoids are NMR, FT-IR and Raman spectroscopy;6 Raman spectra of lower diamondoids have been reported.22 The vibrational spectra of diamondoids have been compared with macroscopic diamond entities such as nanocrystalline and bulk diamond.23 A significant difference is found in the CCC deformation vibration corresponding to “cage-breathing” modes, which produce the highest intensity Raman signals in the spectral region below 800 cm−1.
Stimulated by the perspective of a transposition of the properties of diamond in the nanoscale range, a large body of work has been devoted to the development of methods to selectively and efficiently functionalize diamondoids. These efforts have generated a great number of molecules having high potential for applications in biology, polymer chemistry, molecular electronics, molecular mechanics, synthesis and catalysis. We review herein the most practical functionalizations of lower diamondoids together with the related applications of the resulting compounds in the transdisciplinary fields mentioned above.
2. Direct functionalization of diamondoids
2.1 Halogenation
The functionalization of adamantane (1) is more selective than for polymantanes because there are only two different types of C–H bonds (Scheme 1). Monofunctionalized adamantane derivatives at a bridgehead tertiary carbon with all possible halides have been reported.24–26 Bromination and dibromination of 1 with Br2 in the absence or presence of aluminum Lewis catalyst leads directly to 1-bromoadamantane (1-1) and 1,3-dibromoadamantane, respectively.27 The bromination of diamantane (2) is a more intricate process (Scheme 2). When using Lewis acid catalysts the conditions have to be tuned to improve the selectivity for one of the three isomers obtained: 2-1, 2-2 and 2-3. Neat Br2 selectively gives 1-bromodiamantane (2-1) in 80% yield,28,29 while the apical isomer 4-bromodiamantane (2-3) forms in the presence of traces of AlBr3 with tert-butyl bromide in 58% yield albeit in a mixture with 2-1.29 Radical bromination of diamantane under phase transfer catalytic (PTC) conditions with CBr4 and NaOH/n-Bu4NBr predominantly gives 2-1 (56%), some 2-3 (23%) and traces of the secondary-C functionalized 3-bromodiamantane (2-2) (11%).30 The latter can be prepared in 47% overall yield in two steps from 3-diamantanone.31 A mixture of 1-chlorodiamantane (2-4) and 4-chlorodiamantane (2-5) ensues in moderate yield (25%) from the reaction of 2 with CrO2Cl2 in CCl4, with 1-hydroxydiamantane as the side product.32 The synthesis of secondary-C functionalized 3-chlorodiamantane (2-6) is possible in three steps from 2via 3-diamantanol formation followed by chlorination with SOCl2 (yield 49%).33 1-Iododiamantane (2-7), 4-iododiamantane (2-8) and 3-iododiamantane (2-9) form in the direct iodination of 2 under PTC conditions with CHI3 and NaOH in CH2Cl2.30 Compound 2-7 is the major halide formed in 39% yield. Fluorodiamantanes 2-10 and 2-11 are obtained in excellent yields from the corresponding bromides 2-1 and 2-3, respectively; AgF is used as the fluorinating agent in cyclohexane, and alternatively the corresponding hydroxylated diamantanes can also be used as precursors for reactions with diethylaminosulfur trifluoride (DAST).34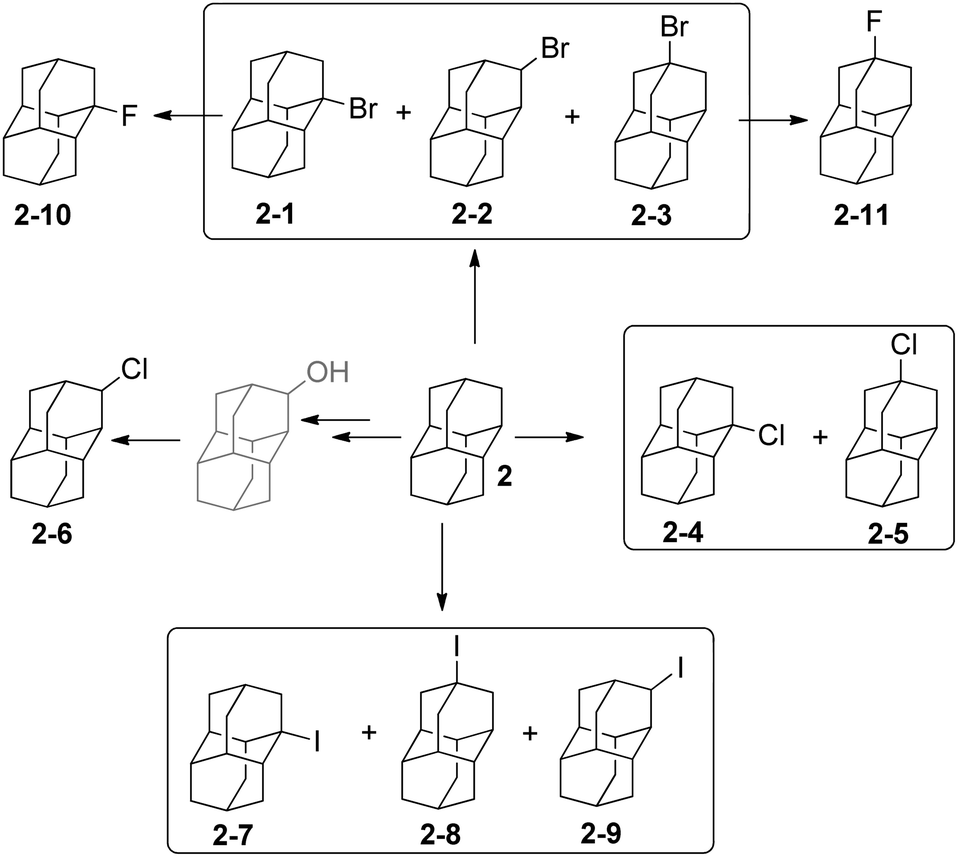 | ||
| Scheme 2 Diamantylhalides 2-1–2-11. | ||
Triamantane, tetramantane, and pentamantane bromides and fluorides have been also synthesized.35 Fluorotriamantanes are obtained by treatment of hydroxylated analogues36 with DAST.34 From anti-tetramantane treated with Br2 the access to dibrominated analogue is possible in a modest 17% yield.36 [1(2,3)4]-Pentamantane is the higher diamondoid which has been efficiently functionalized with halides and surprisingly reacts more selectively with electrophiles than triamantane and anti-tetramantane. This pentamantane gives in multistep reactions overall yields of 25 to 75% of mono- or dibrominated products.37
2.2 Hydroxylation
The hydroxy derivatives of 1 can be obtained either by hydrolysis of the corresponding halides or by direct functionalization of 1; mono-, di- and tetrahydroxylated adamantanes have been made.38–40Scheme 3 summarizes the main methods for the preparation of hydroxylated diamantanes. 1-Hydroxydiamantane (2-13) forms in 23% to 82% yield upon treatment of 2 with various oxidants (m-chloroperbenzoic acid, Pb(OAc)4, dimethyldioxirane, nitric acid), among which HNO3 is the most efficient.32,36 Concomitant formation of 4-hydroxydiamantane (2-12) and 4,9-dihydroxydiamantane (2-14) in low yields (5–13% and 6–11%, respectively) generally occurs; these can be separated by silica gel chromatography. 1,4-Dihydroxydiamantane (2-15) can be synthesized via treatment of 2 with HNO3via 1,4-dinitroxydiamantane as an isolatable intermediate (overall yield of 2-15: 30%).36 Other hydroxy derivatives can be obtained such as the species functionalized on a secondary-C2-16 and 2-17.31,33 2-Methyldiamantan-2-ol (2-17) is accessible in 93% yield from a Grignard reaction of 2-diamantanone; the latter can alternatively be reduced by LiAlH4 for the formation of 2-16 (73%). The oxidation of triamantane (3) with HNO3 is also appropriate to produce the mono- or dihydroxylated analogues.36 Conditions to yield the monohydroxylated35,41 and dihydroxylated [121]tetramantanes have been reported via the hydrolysis of bromide or nitroxy precursors.37,42 Similarly, the mono- and dihydroxylated [1(2,3)4]pentamantanes can be obtained.37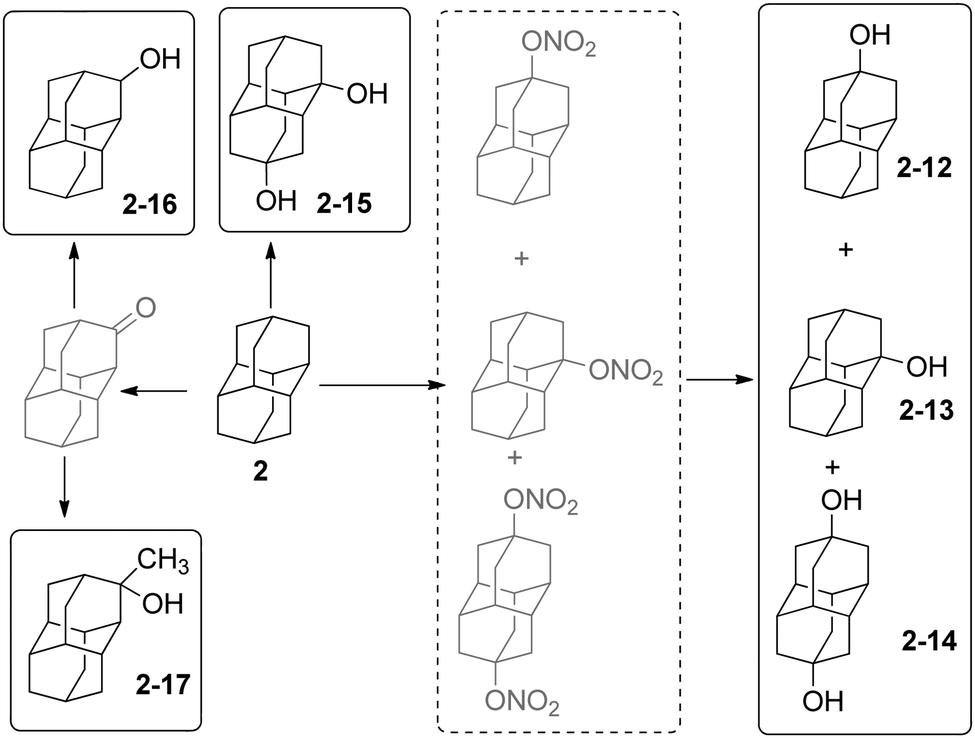 | ||
| Scheme 3 Diamantanols 2-12–2-17. | ||
2.3 Metallation and coupling
A limited number of metallated diamondoids have been prepared, mainly from adamantane (1) (Scheme 4). Grignard reagent 1-adamantylmagnesiumbromide (1-3) has been obtained in 60% yield from the reaction of 1-bromoadamantane 1-1 with an excess of magnesium in the presence of BrCH2CH2Br in Et2O and n-Bu2O; THF was found to be unsuitable.43 1-Adamantyl lithium (1-4) and its congeners 2-adamantyl lithium as well as 1-diamantyl lithium have been directly synthesized from the reactions of their chloride precursors with lithium metal in pentane (82, 85 and 76% respectively).44 In these reactions the control of the metal–solution interface is crucial to avoid diamondoid homologation.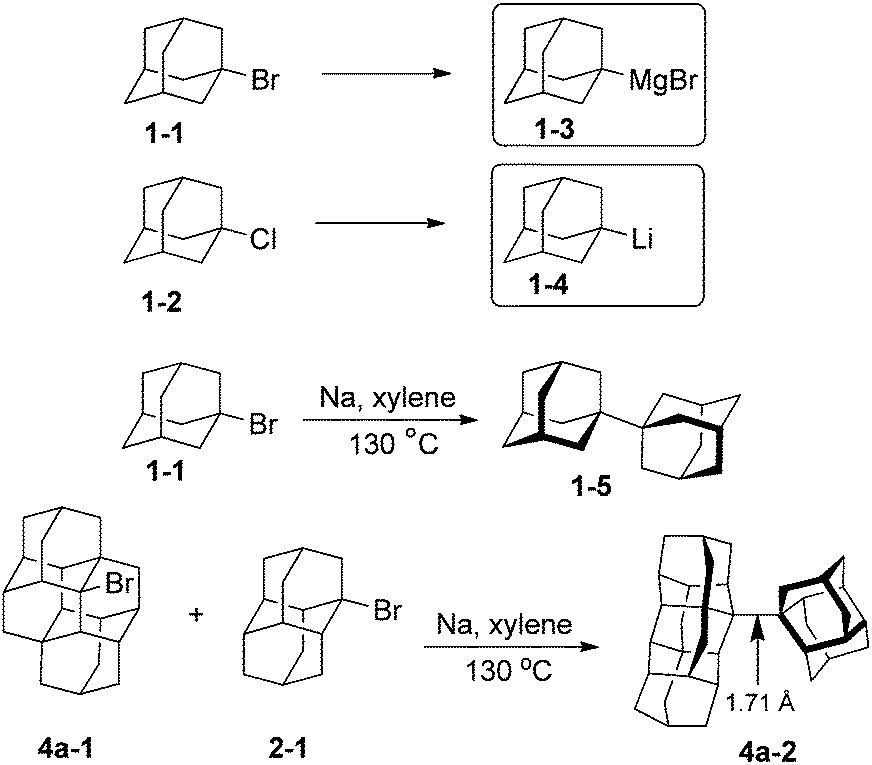 | ||
| Scheme 4 Metallated diamondoids and (homo)coupling reactions. | ||
Metallated derivatives are intermediates in the diamondoid couplings with sodium (Wurtz reaction). Simplest coupled 1,1-diadamantyl (1-5) resembles part of the diamond lattice and may be viewed as a diamondoid of larger size. Recently prepared coupled higher diamondoids represent even larger nanodiamond particles and contain extremely long C–C bonds.45 For instance coupling of the respective bromo derivatives of diamantane 2-1 and tetramantane 4a-1 gives diamondoid 4a-2, which contains the C–C bond of 1.71 Å lengths, the longest so far observed in alkanes.46 Despite pronounced geometrical distortions coupled diamondoids display high thermal stabilities.
2.4 Carboxylation
The preparation of 1-adamantane carboxylic acid (1-6) utilizes the Koch–Haaf reaction of adamantane derivatives with formic acid in sulfuric acid.47 The carboxylation of diamantane derivatives is accompanied by an intermolecular hydride transfer reaction, which results in the formation of a mixture of apical and medial diamantane carboxylic acids. The selective conversion of the 4-bromodiamantane (2-3) to the diamantane-4-carboxylic acid (2-18) is possible under high dilution conditions that prevent the intermolecular hydrogen exchange (Scheme 5).48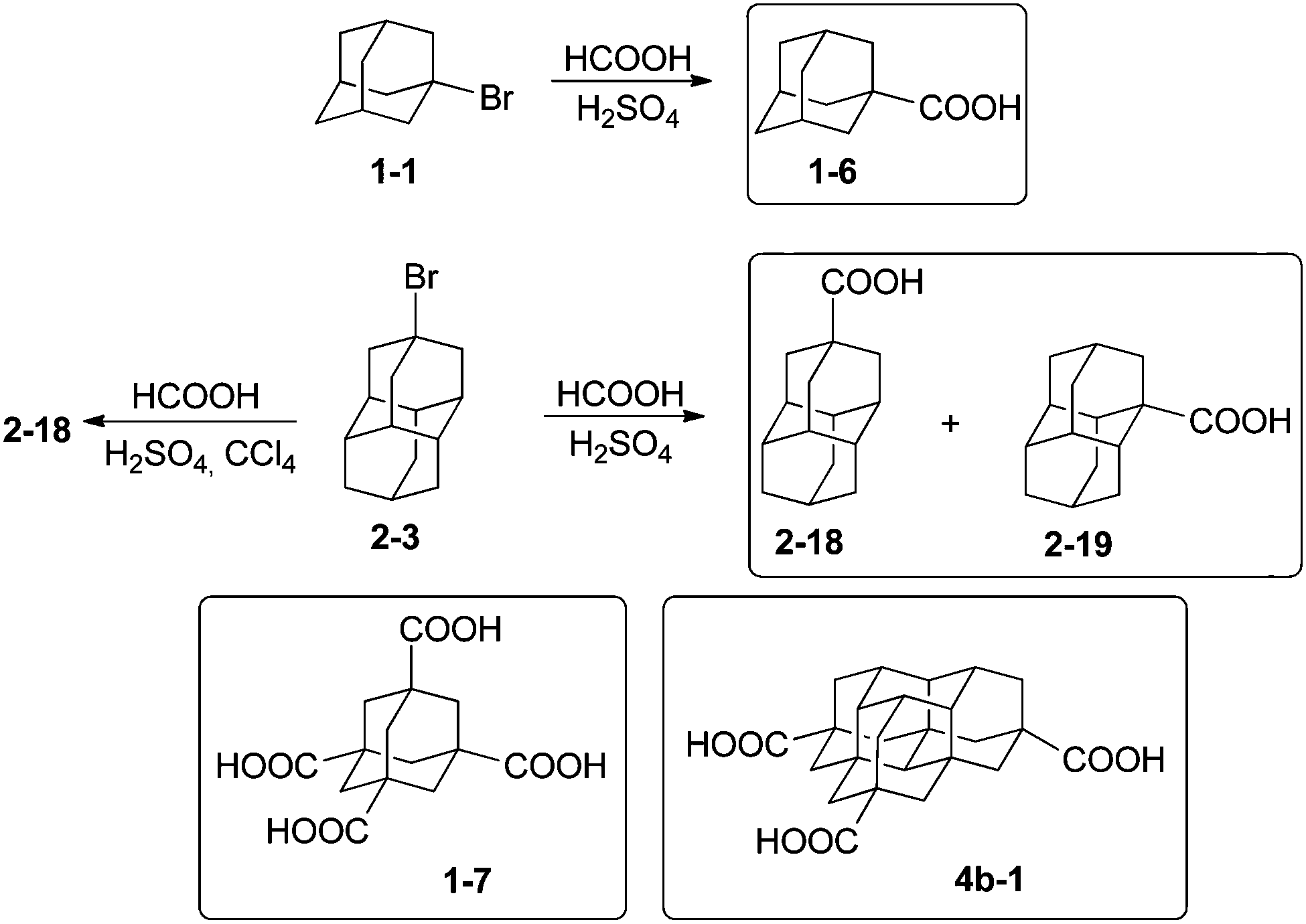 | ||
| Scheme 5 Diamondoid carboxylic acids 1-6–4b-1. | ||
The Koch–Haaf carboxylation was recently applied for the preparation of triamantane, tetramantane, and pentamantane carboxylic acids in good yields.49 The diamondoid polycarboxylic acids are useful as 3D-building blocks, and can be prepared through C–H-photocarbonylation or, better, through hydrolysis of the corresponding nitriles, as was demonstrated in the preparation of adamantane-1,3,5,7-tetracarboxylate (1-7).50 For higher diamondoids direct Koch–Haaf carboxylation allows preparation of [1(2)3]tetramantane-7,11,17-tricarboxylate (4b-1), which can be used as a building block for various tripodal rigid surface anchors.51
2.5 Amination, amidation, and nitration
The development of acetamide and amine derivatives of adamantane has been motivated by their pharmacological applications that have been recently reviewed.52 Monoaminodiamantanes (Scheme 6) and their derivatives have been prepared from diamantane (2), via 1-bromodiamantane (2-1), 4-bromodiamantane (2-3), 1-hydroxydiamantane (2-13), and 4-hydroxydiamantane (2-12). Compounds 2-13 and 2-3 can be converted in one step to the corresponding carboxylic acids, which further form intermediary carbamates that can then be hydrolysed to 1-amino- and 4-aminodiamantane (2-20) and (2-21) with overall yields of about 20% and 40%, respectively.53 1-Bromodiamantane (2-1) gives 1-acetamidodiamantane (2-22) by a Ritter reaction (using CH3CN/H2SO4), 2-22 after hydrolysis forms 2-23 as the hydrochloride salt of 2-20 (overall yield 55%).53 Aminodiamantanes 2-20 and 2-21 form in good yields (63 and 93%, respectively) by acidic exchange of the hydroxyl groups of diamantanols 2-13 and 2-12, respectively: treatment with chloroacetonitrile–H2SO4 in acetic acid gives 2-24 and 2-26, which can be cleaved in the presence of thiourea.53 Aminodiamantanes 2-20 and 2-21 react with m-chloroperbenzoic acid in dichloroethane to give the nitro-functionalized compounds 2-25 and 2-27 in 74% yield.54 4,9-Diamino- and dinitrodiamantane have been also described.53,54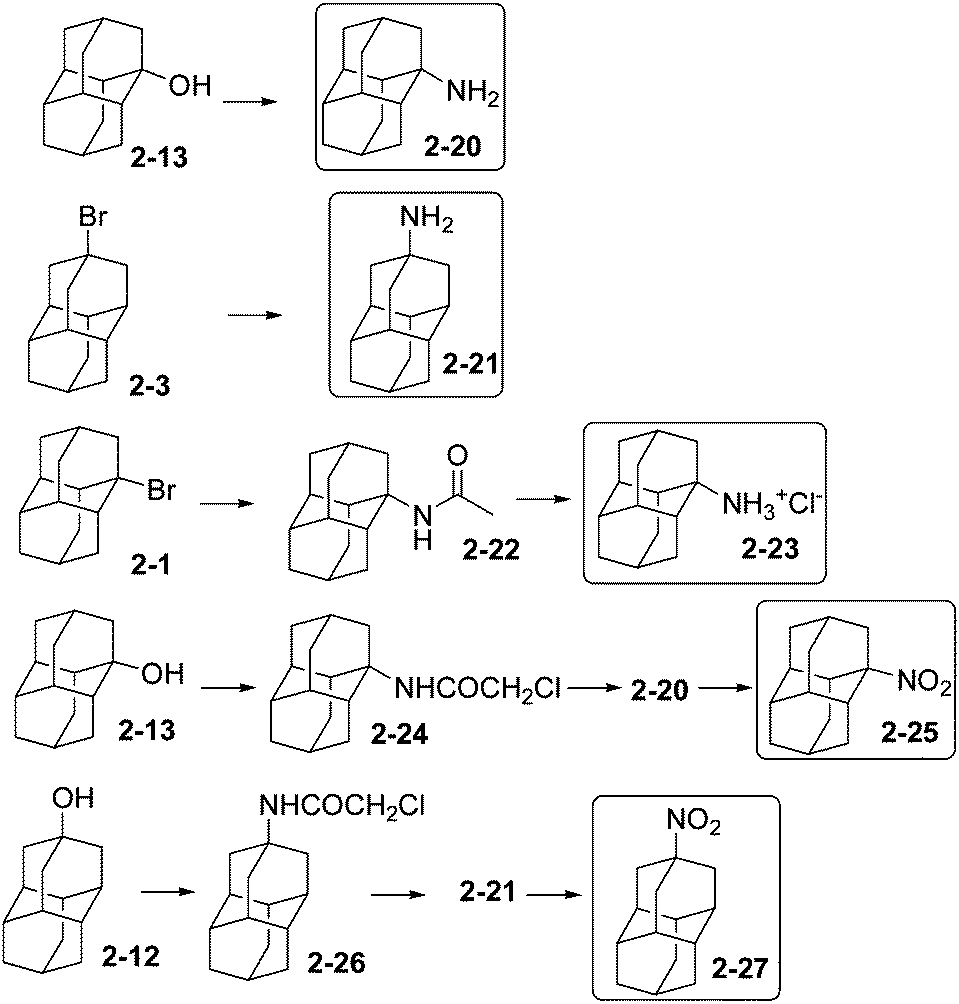 | ||
| Scheme 6 Amino-, amido-, and nitrodiamantanes 2-20–2-27. | ||
2.6 Phosphorylation and phosphination
Diamondoids substituted with alkyl phosphines are ligands for transition metals with exceptional properties. Adamantane (1) can be used for the formation of di(1-adamantyl)phosphinic acid chloride (1-8), which provides access to the organophosphorus compounds di(1-adamantyl)phosphine (1-9) and di(1-adamantyl)chlorophosphine (1-10) (Scheme 7).55 They are obtained in excellent yields of above 85% on a scale of several grams. Compound 1-10 is ideal for generating trialkyl phosphine 1-12.56–58 Another synthetic pathway to adamantylalkylphosphines is the reaction of halophosphines with the precursor adamantyl magnesium bromide 1-3,43 which gives the monoadamantylphosphine 1-11 in excellent 86% yield, but the diadamantyl analogue 1-12 only in moderate 30% yield.59 1-Bromoadamantane (1-1) treated with a large excess of PCl3 in the presence of AlBr3 leads to 1-adamantyl phosphonic acid dichloride (1-13) in 94% yield on a several grams scale.60 1-Adamantyldichlorophosphine (1-15) can be prepared in 68% overall yield from 1-13 through reaction with Lawesson's reagent via 1-adamantyldichlorophosphine sulphide (1-14).61 The primary phosphine 1-16 is an oily product obtained in 75% yield from the reaction of dichlorophosphine 1-15 with LiAlH4.62 4-Diamantylphosphonic acid dichloride (2-28) and di-4-diamantylphosphinic acid chloride (2-29) are prepared from 2 in 22% and 59% yield respectively,63 and 2-29 can be further reduced with HSiCl3 to its corresponding secondary phosphine (84%), which is highly oxygen-sensitive. The triamantylphosphine analogues of the latter have been also synthesized.62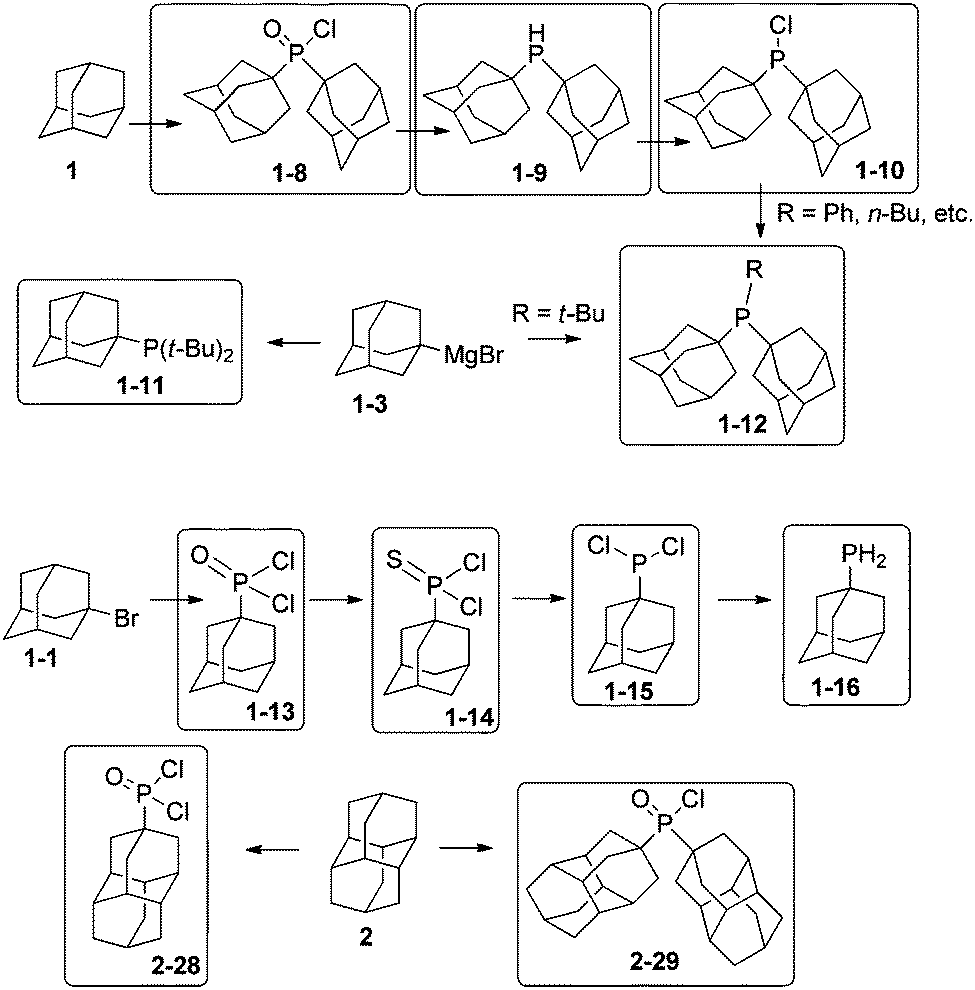 | ||
| Scheme 7 Phosphorus-substituted adamantanes and diamantanes 1-8–1-16 and 2-28, 2-29. | ||
2.7 Olefination and alkylation
Diamondoids substituted with olefinic moieties can be used as monomers in polymerization reactions. Unsaturated compounds such as vinyldiamondoids are also expected to play a role in the immobilization of organic molecules on semiconductors for nanoelectronics. The preparation of 2-adamantylbutadiene-1,3 (1-19) (Scheme 8)64 was conducted from 1via the selective photochemical preparation of 1-acetyladamantane (1-17),65 followed by oxetane 1-18 formation and a dehydrating ring opening catalysed by p-toluenesulfonic acid. Adamantyl-1-carbaldehyde (1-20) undergoes a Wittig reaction to give 1-vinyl adamantane (1-23) in 65% yield.66 Alternatively, the adamantyloxirane 1-21 forms in excellent yield from adamantane-1-carbaldehyde, and its acidic ring opening (HBr–H2SO4) leads to 1-adamantyl-1,2-dibromoethane (1-22), which is debrominated to 1-23 (using Zn in DMF at 150 °C) with an overall yield of 72%.67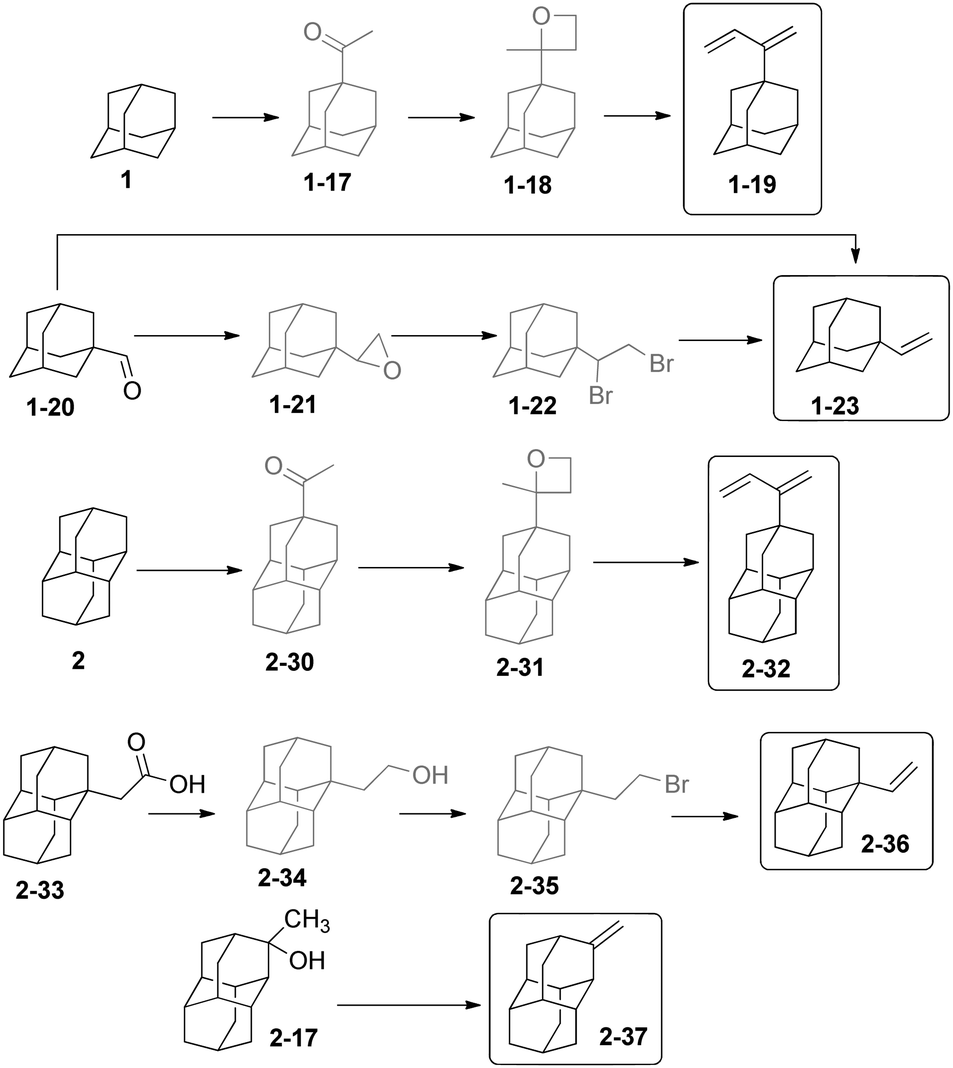 | ||
| Scheme 8 Diene and olefinic adamantane and diamantane 1-19, 1-23, 2-32, 2-36 and 2-37. | ||
Photoacetylation has been also successfully used on the diamondoids, e.g., for 2 to prepare acetyldiamantane 2-30, which allowed the preparation of diamantylbutadiene 2-32 in 94% overall yield via the formation and ring-opening of the corresponding oxetane 2-31.68,69 1-Vinyldiamantane (2-36) has been obtained from diamantane acetic acid 2-33, after reduction to the alcohol 2-34, subsequent bromination to 2-35, and then its dehydrobromination – the overall yield of 2-36 was 62%;67 Its 4,9-divinyl substituted cousin was obtained similarly. Dehydration of 2-17 can be achieved at 135 °C in the presence of H3PO4 to give 2-37 in 86% yield.
Diamondoids substituted with simple alkyl fragments are of practical value for biomedical applications.52,70 For instance, 3,5-dimethyladamantyl-1-amine, also called Memantine®, has been found to give some symptomatic improvement in moderate to severe Alzheimer's disease.70b
Diamondoid halides are useful alkylation reagents. 1-Bromoadamantane (1-1) undergoes reaction with alkyl Grignard reagents R–MgBr (R = Me, Et, CH2Ph) to yield in moderate to good yields compounds 1-24–1-26 (Scheme 9).71 Multistep polyalkylation conditions can also give, for instance, 1-27 and 4,9-dimethyldiamantane. Compound 1-24 can also be obtained in 94% yield by reaction of 1-fluoroadamantane with AlMe3.72 1-Methyldiamantane (2-38) forms in 90% yield from bromide 2-1 by reaction with CH3MgBr.71 Isomeric 2-methyldiamantane (2-39) can be obtained in 85% yield from hydrogenation of olefin 2-37 catalysed by PtO2 under H2 pressure.33b
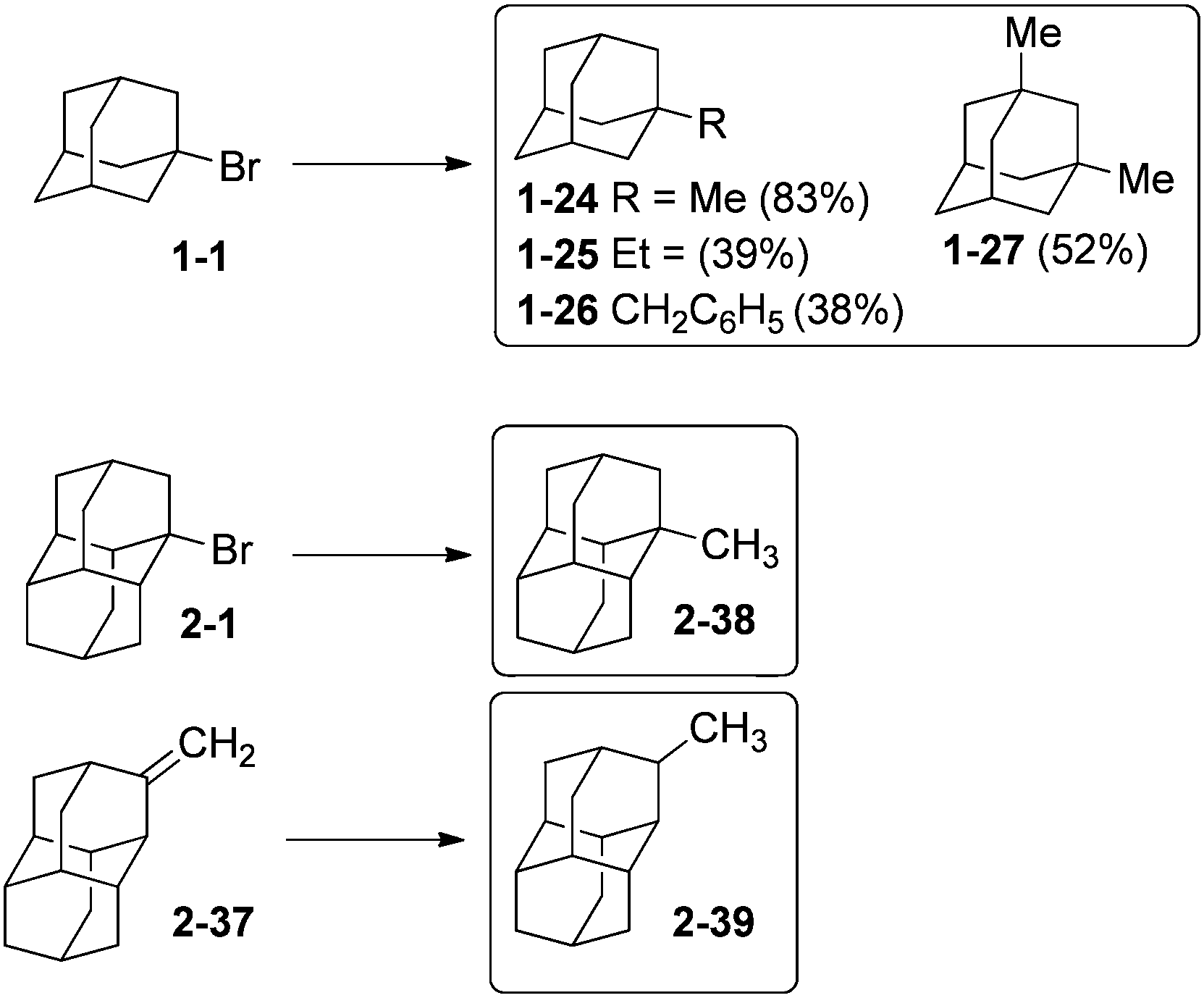 | ||
| Scheme 9 Alkylated adamantanes and diamantanes 1-24–1-27 and 2-38, 2-39. | ||
2.8 Thiolation
Incorporation of thiol groups into diamondoids allows their attachment to noble metal surfaces to form self-assembled monolayers (SAMs) that have applications in molecular electronics. Fokin and Schreiner et al. have shown that treatment of tertiary mono- and dihydroxy hydrocarbon derivatives (diamondoid alcohols) with thiourea in the presence of hydrobromic and acetic acid is a one-step route to prepare the respective tertiary thiols and dithiols.42 This procedure was used for the preparation of diamondoid thiols of adamantane and diamantane in satisfactory yields (Scheme 10): 1-29 (57%), 2-40 (82%), 2-41 (76%) and 2-42 (69%). The thiolation conditions can be extended to higher diamondoids such as triamantane, tetramantanes and pentamantanes. Interestingly, reactions with 1-bromoadamantane (1-1) involving hydrogen sulfide, organolithium reagents with elemental sulfur, potassium thioacetate and ethyl xanthogenate, sodium dimethyldithiocarbamate, hydrosulfide, or thiosulfate resulted at best in unsatisfactory yields of the expected adamantane-1-thiol (1-29);42 conversely, the reaction with thiourea gave satisfactory yield.73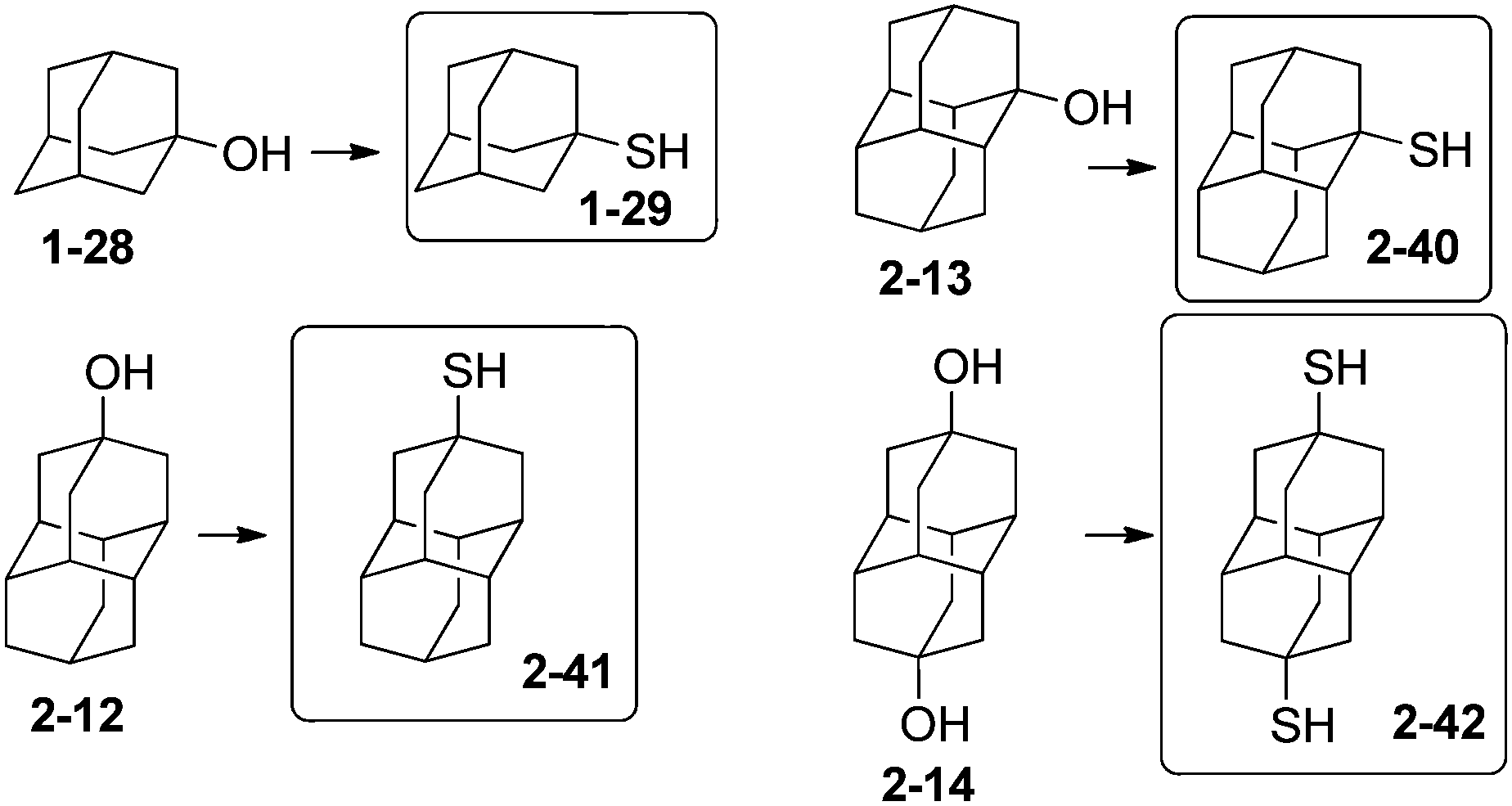 | ||
| Scheme 10 Adamantane- and diamantanethiols 1-29, 2-40–2-42. | ||
3. Unequal difunctionalization of diamondoids
The synthesis of diamondoids having two different reactive functions is a requirement for their use as platforms or linkers. Step by step building of composites can be envisaged based on the differences in reactivity of the functions.3.1 Di- and polyfunctionalized diamondoid halides
Unequally substituted di- and polyhaloadamantanes have been synthesized by successive multistep reactions under PTC conditions.74 Stepwise iodination with iodoform and bromination with CBr4 of 1-chloroadamantane (1-2) (Scheme 11)75 led successively to the formation of 1-chloro-3-iodoadamantane 1-30 (30%) and 1-chloro-3-bromo-5-iodoadamantane 1-31 (35%) as a racemic mixture.74 Chlorination of 1-fluoroadamantane (1-32) with CCl4 gave 1-33 (20%), which was brominated to give 1-34 (21%), which was iodinated with n-C6F13I to give the remarkable tetrahalide 1-bromo-3-chloro-5-fluoro-7-iodoadamantane (1-35) (65%).74 Following this strategy, successive iodination and bromination of 1-fluoroadamantane (1-32) provided first 1-fluoro-3-iodoadamantane (24%) and then 1-fluoro-3-bromo-5-iodoadamantane as a racemic mixture (35%).74 3-Bromoadamantan-1-ol and 3-chloroadamantan-1-ol have also been synthesized and isolated en route to 1,3-diiodoadamantane, which is obtained by addition of hydroiodic acid to any of those compounds.76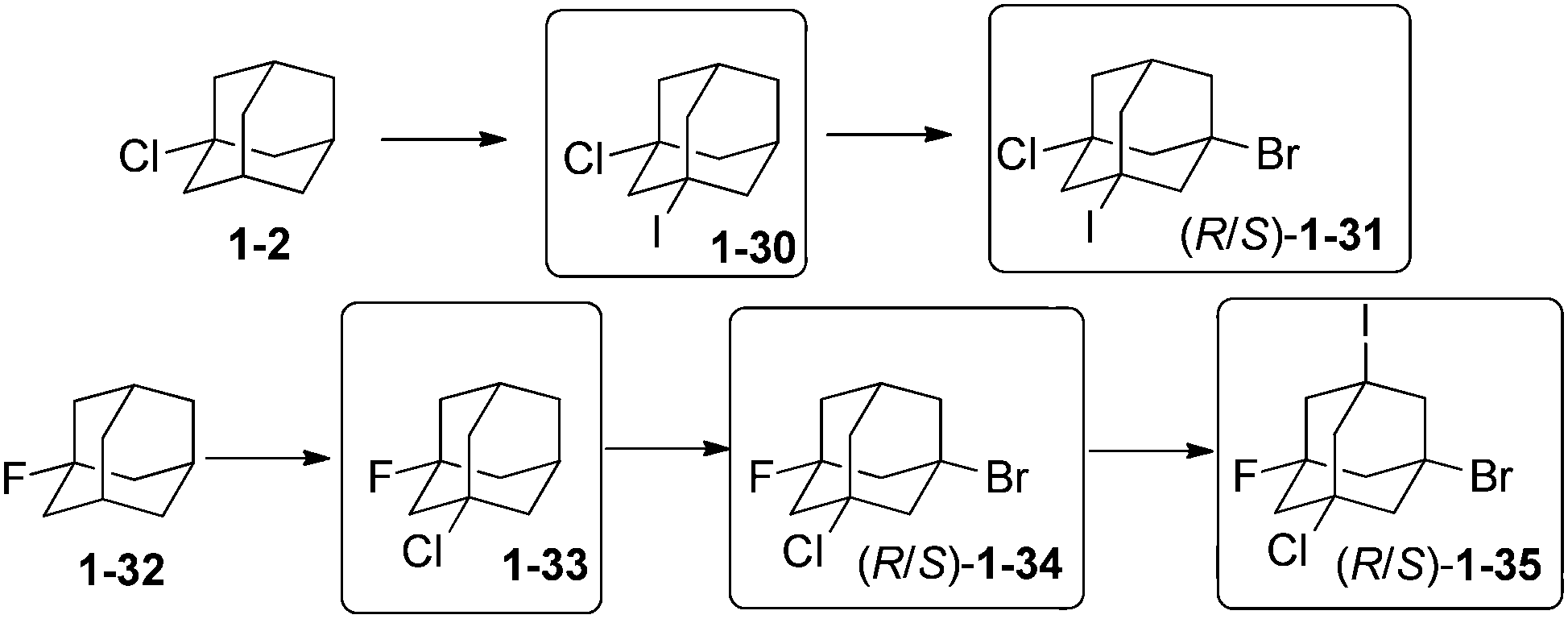 | ||
| Scheme 11 Selection of polyhalogenated adamantanes 1-30–1-35. | ||
The selective protection of one of the hydroxyl functions of 4,9-dihydroxydiamantane (2-14) by a fluoroalkyl ether function is performed with CF3CH2OH in the presence of 20 mol% CF3SO3H to give compound 2-43 (Scheme 12). This selective protection is very important as 2-43 is the precursor for the synthesis of a great number of unequally difunctionalized diamantanes and especially diamantane bromides and fluorides.41
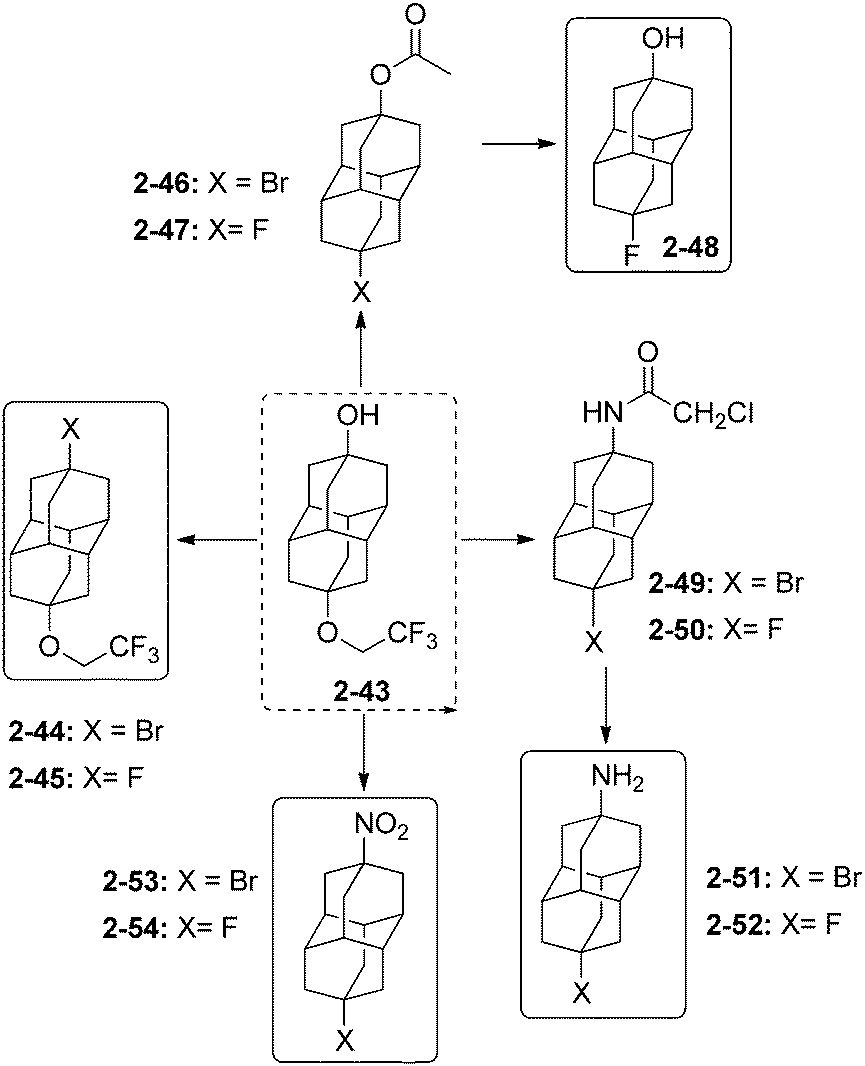 | ||
| Scheme 12 Unequally difunctionalized diamantane halides 2-43–2-54. | ||
Selective bromination of 2-43 at the hydroxyl position with SOBr2 gives 2-44 (97%), which is further converted with AgF in cyclohexane to 2-45 (86%).34 The hydroxyl group of 2-43 can also be protected by an acetoxy group with CH3COCl in pyridine to consecutively give halides 2-46 (63%) in reaction with BBr3 and 2-47 (88%) in reaction with AgF. Fluorodiamantanol 2-48 is obtained in high 92% yield from 2-47 deprotected in excess of NaOH. The amido-protected analogue of 2-43 gives access to the amido-halides 2-49 (97%) and 2-50 (90%) by using consecutively BBr3 and AgF.34 Amino and nitro groups can also be introduced to form the difunctionalized amino-halides 2-51 (58% from its hydroxy analogue reacted with SOBr2), and 2-52 (72% from 2-50 and thiourea), and the nitro-halides 2-53 (83% from hydroxy analogue with SOBr2) and 2-54 (86% from 2-53 and AgF).34
3.2 Unequally substituted diamondoid alcohols and amines
9-Trifluoroethoxydiamantan-4-ol (2-43) (Scheme 13) is very useful to generate the diamantane amino alcohol 2-58 in three synthetic steps (37% overall yield), via acetamide 2-55 (obtained in 45% yield from the action of chloroacetonitrile–H2SO4 in acetic acid on 2-43), followed by conversion of the fluoroalkyl ether function into trifluoroacetoxy ester function (2-57, 97% from reflux in trifluoroacetic acid). 2-58 (86%) is obtained from 2-57 after treatment with thiourea in EtOH–acetic acid, then NaOH.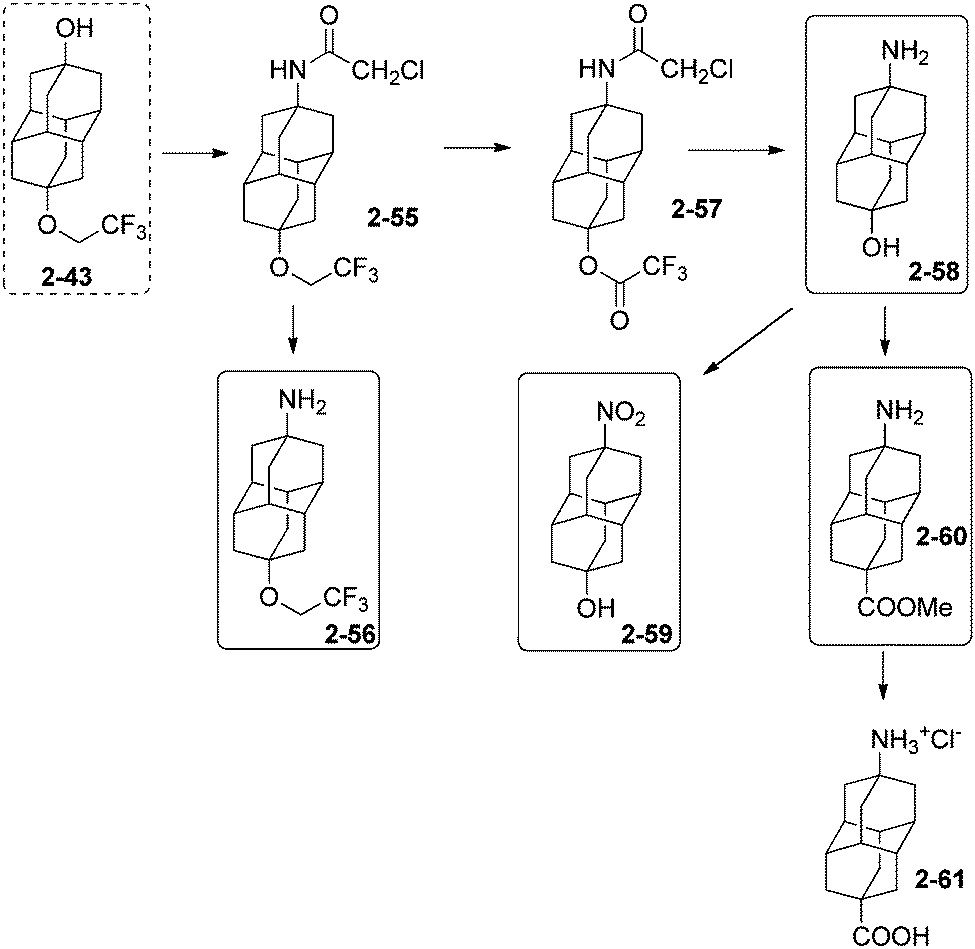 | ||
| Scheme 13 Disubstituted diamantane alcohols and amines and derivatives 2-55–2-61. | ||
Attempts to synthesize the amino alcohol 2-58 in one step from 2-55 by directly using thiourea mixed with acetic or trifluoroacetic acid (trying to convert the ether into a base labile ester) resulted in the predominant formation of amino ether 2-56 (93%). The diamantane amino alcohol 2-58 can be efficiently oxidized to the nitro alcohol 2-59 (89%) by using m-chloroperbenzoic acid in 1,2-dichloroethane. Clean conversion of 2-58 to the corresponding amino acid 2-61 is more difficult (overall yield 34%) but possible via the formation of the methyl ester 2-60 (38%), subsequently hydrolysed in concentrated hydrochloric acid.
4. Applications of functionalized diamondoids
We illustrate herein a selection of salient recent examples of the various fields of applications in which lower diamondoids (adamantane to triamantane derivatives) have been successfully employed.4.1 Homogeneous and heterogeneous catalysis
The applications of diamondoids in homogeneous and heterogeneous catalysis for organic synthesis by transition metals are attractive. Phosphine ligands bearing adamantyl77 or diamantyl78 fragments (Scheme 7) have given spectacular results in terms of catalytic efficiency and scope in various reactions. The bulky electron-rich framework provided by the hydrocarbon cage to phosphine ligands allows for the activation of valuable organic bromide and chloride substrates in metal-catalysed cross-coupling reactions. The following selected examples illustrate this potential well. By using bis(1-adamantyl)(benzyl)phosphine (1-36) as a ligand (Scheme 14) Köllhofer and Plenio have achieved high turnover numbers (TONs) for the palladium-catalysed arylation of terminal alkynes with a variety of aryl bromides (Scheme 14).79 Coupling of various aryl bromides with phenylacetylene was performed at catalyst loadings as low as 0.005 mol%, in excellent yields with Na2PdCl4 as a catalyst and HN(i-Pr)2 as a base and solvent. A TON of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 was obtained for the reaction of the electronically deactivated aryl bromide 2-methoxybromobenzene with phenylacetylene.79
200 was obtained for the reaction of the electronically deactivated aryl bromide 2-methoxybromobenzene with phenylacetylene.79
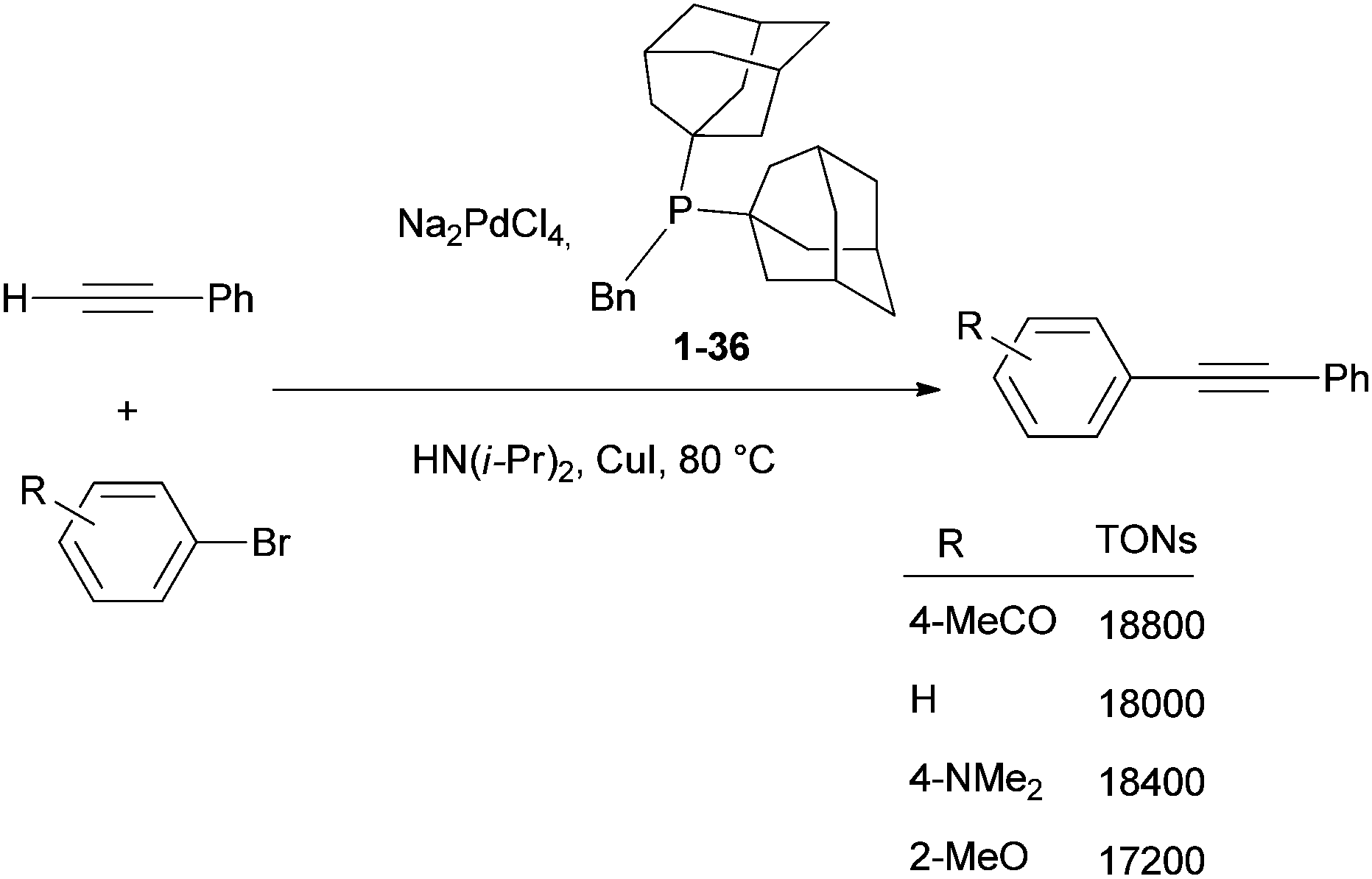 | ||
| Scheme 14 Palladium-catalysed arylation of phenylacetylene using as a catalyst [Pd/(1-Ad)2PBn]. | ||
The same authors have studied heterogenization of bulky electron-rich phosphanes by attaching a bis(1-adamantyl)phosphanyl group to a monomethyl polyethylene glycol ether support. They formed a polymeric phosphane (Ad2P-MeOPEG) soluble in water and polar solvents and insoluble in alkanes. The recoverable catalytic system, combining 1 mol% Na2[PdCl4], 2 mol% Ad2P-MeOPEG, 0.7 mol% CuI, and HN(i-Pr)2 as a base in DMSO at 75 °C, allowed the arylation of phenylacetylene, 1-octyne, and trimethylsilylacetylene with various activated and deactivated aryl bromides in excellent yields.80
4.2 Asymmetric organocatalysis
Organocatalysts incorporating adamantane (1) have also proven to be highly promising. A chiral tetrapeptide platform incorporating an artificial amino acid, γ-aminoadamantane carboxylic acid, is an efficient organocatalyst for enantioselective kinetic resolution of trans-cycloalkane-1,2-diols (Scheme 15).81 The kinetic resolution of chiral trans-cycloalkane-1,2-diols is challenging, and severe limitations exist even with enzymatic approaches (selectivity factors S < 5). The tetramer synthesized by automated solid-phase peptide-coupling methods enantioselectively transfers acyl groups onto trans-cycloalkane-1,2-diols with enantiomeric excess above 99% (S ≫ 50). This kinetic resolution currently provides the highest selectivity for these types of reactions, and this is to a large part due to the rigid and lipophilic adamantyl γ-amino acid.82 The absence of secondary structure in the organocatalyst suggests that the determining factors for stereoselection are established within the complex formed from the charged acylium ion and the peptide catalyst. The stereogenic centers of the artificial peptide determine the final stereochemistry of the diol.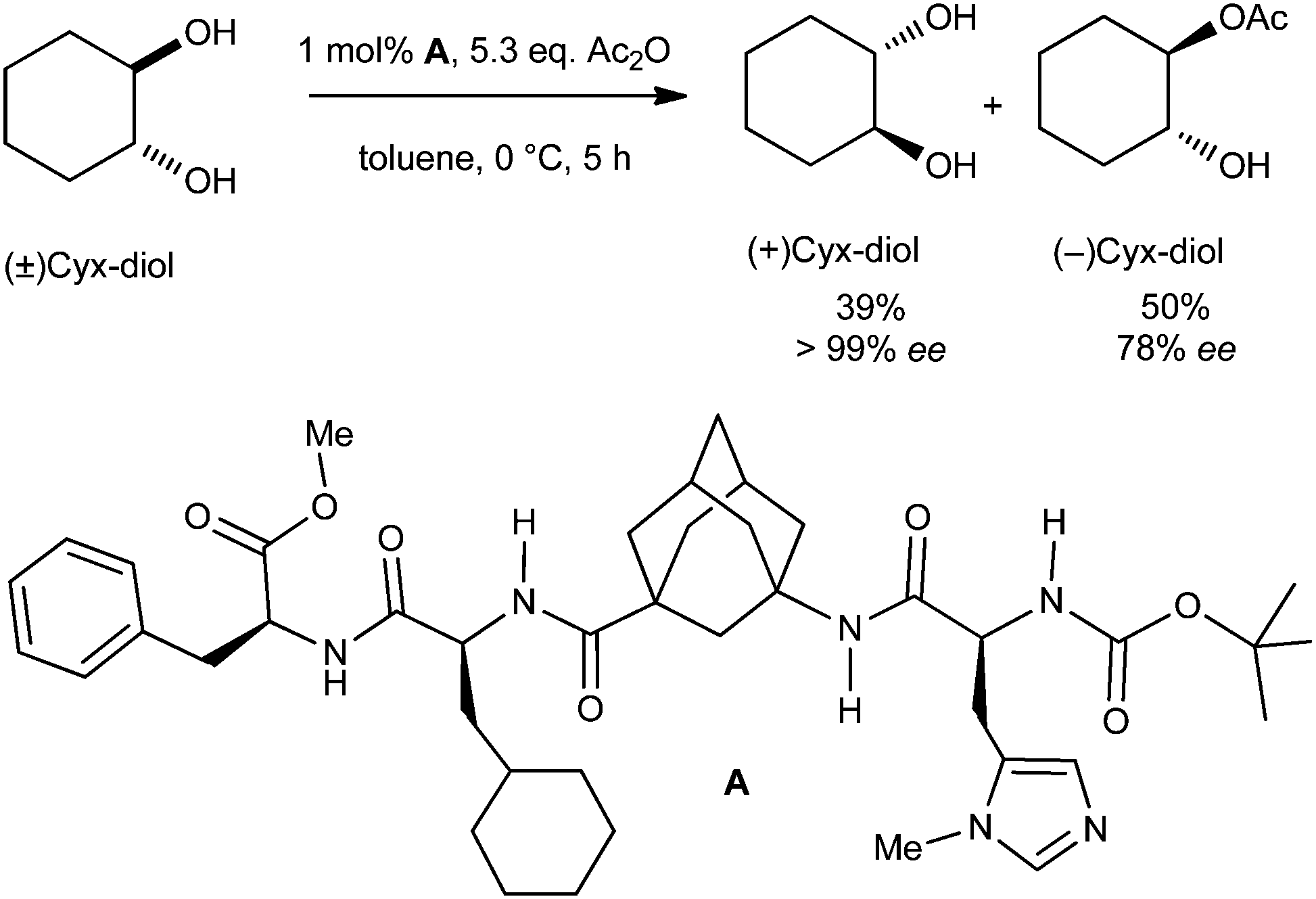 | ||
| Scheme 15 Tetrapeptide chiral platform incorporating γ-aminoadamantane carboxylic acid applied as organocatalyst in kinetic resolution of trans-cycloalkane-1,2-diols (racemic cyclohexane-1,2-diol is shown as an example). | ||
4.3 Biological and medical applications
Simple adamantane derivatives have long been known for their antiviral activity against Influenza A and HIV viruses. Biological properties observed for adamantane derivatives are also associated with the central nervous system (against Parkinson's and Alzheimer's diseases), and some antimicrobial, anti-inflammatory and antifungal activities have been recognized.52,83 Conversely, diamondoid derivatives of higher rank have been less investigated so far for biological and pharmaceutical potential use. This is clearly an oversight by the pharmaceutical industry that is likely due to the unforeseen isolation and chemical functionalization of diamondoids higher than adamantane. The large majority of derivatives where adamantane is replaced by a larger diamondoid moiety still await their preparation and utilization.Adamantane, diamantane, and triamantane motifs can be introduced into DNA by chemical and enzymatic methods.84 This was achieved by the synthesis of diamond modified triphosphates and phosphoramidites from brominated diamondoids. These artificial nucleotides were then incorporated into DNA in two ways, either using DNA polymerase or automated solid phase synthesis. Remarkably, the β-conformation of the DNA double helix tolerates the diamond-modified nucleotides.
4.4 Host–guest molecular recognition
Inclusion complexes of cyclodextrins (CDs) can form host–guest pairs whose stabilities are mainly determined by the shape and hydrophobicity of the guest molecules. Schreiner and Ravoo have reported the synthesis and characterization of inclusion complexes of water-soluble lower diamondoid carboxylic acids and their 5-aminoisophthalic acid derivatives (Scheme 16) into β- and γ-cyclodextrins (β-CD and γ-CD).85 Due to their hydrophobicity and symmetry, it was expected that the cages of these diamondoids would be excellent guests for CDs, with the pendant hydrophilic aromatic moieties (R) not included in the CDs cavity. Indeed, all diamondoids studied interact with β-CD. However, only the bulkiest ones (mainly triamantanes) interact with γ-CD. γ-CD is more flexible than β-CD, and in solution the cavity of γ-CD partially collapses. Therefore, its tendency to replace internal hydration water with a suitable hydrophobic guest is significantly reduced. The stoichiometry of host–guest complexes was found to be 1:1 and their interaction is endothermic in each case, with increasingly negative ΔH for the larger diamondoid acids.
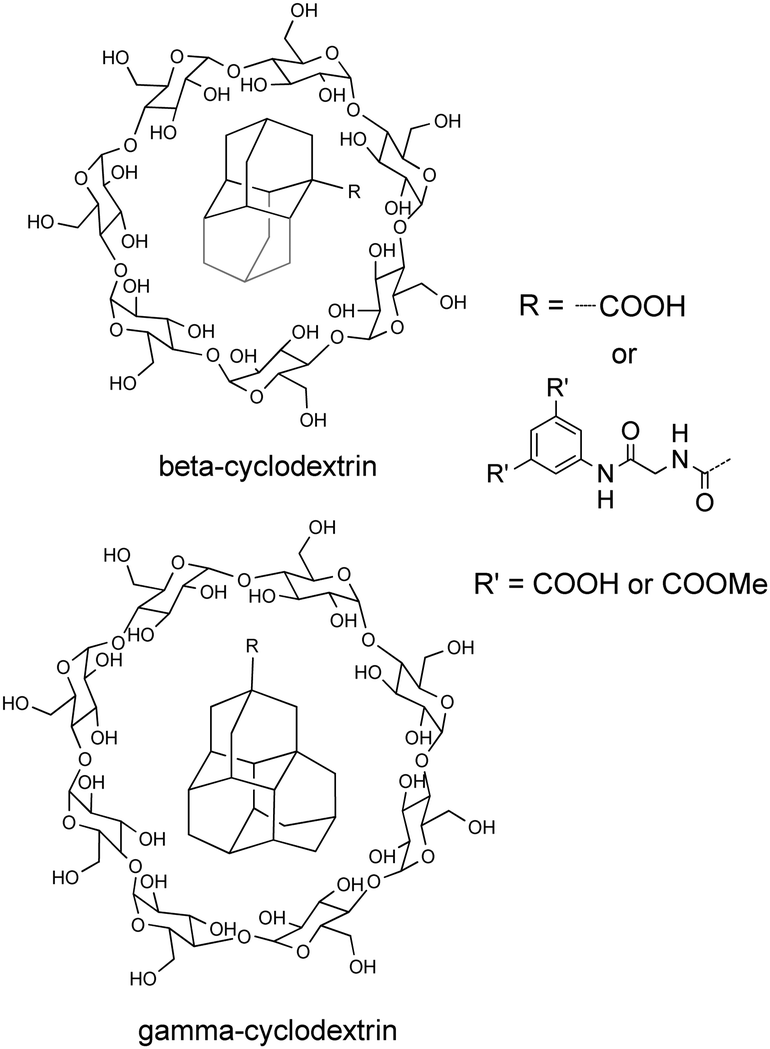 | ||
| Scheme 16 Host–guest inclusion complexes in β-CD (top) and γ-CD (bottom) of derivatives of adamantane (1), diamantane (2), and triamantane (3) bearing a carboxylic acid or peptide side chain. | ||
From another approach of organohybrid design, Stoddart and Zink have built a nanocontainer based on an α-CD blocked by an adamantyl stopper (non-inclusive assembly). This container can encapsulate luminescent rhodamine B and controls its release by removal of its snap-top adamantyl ester motif via enzymatic esterase bond breaking.86 The global system is a biocompatible controlled release system exploiting enzymatic specificity for controlled delivery of small encapsulated molecules (exemplified with rhodamine).
4.5 Polymer materials upgrading
1-Ethynyladamantane, 1,3-diethynyladamantane,87 and 1,4-, 4,9- and 1,6-diethynyldiamantane88 have been synthesized and polymerized or co-polymerized to give thermoset resins thermally stable above 400 °C. Diamondoids have also been incorporated as co-monomers that increase the rigidity and thermal stability of light-emitting polyolefins.89 The incorporation of an adamantane unit in the repeating motif significantly increases the Tg and Td of the polymers. The adamantane unit is an efficient π-conjugation interrupter and prevents interchain interactions. Blue or green light emission and low turn-on voltages were obtained from a single-layer LED fabricated from the polymers.1,3,5,7-Tetrabromoadamantane has been employed to synthesize 1,3,5,7-tetrakis(4-aminophenyl)adamantane that was then used as a cross-linker for the incorporation of adamantane into a commercial epoxy resin network.90 A new epoxy material obtained exhibited superior thermal properties with high glass transition temperatures and low dielectric constants in the GHz frequency range.
1-Adamantylacrylate, 1-vinyladamantane, 1-diamantylacrylate, and 4-diamantylmethacrylate have been employed to form polymers containing side-chain adamantane and diamantane motifs, from either atom-transfer radical-polymerization (ATRP), cationic, or free radical polymerization.91 Soluble colourless polymers of low molar masses exhibited an unusual combination of moderately high refractive index with low optical dispersion, making the polymers potentially applicable as thin film specialty optical plastics.
Diamondoids can be used as additives in polypropylene and polycarbonate polymer formulation, for which they can behave as plasticizers or antiplasticizers.92
4.6 Photoemission from self-assembled monolayers (SAMs)
The functionalized diamondoid [121]tetramantane-6-thiol, which pertains to the family of thiolated diamondoid derivatives (Scheme 10), has been used to form large area SAMs on silver and gold surfaces (Scheme 17).93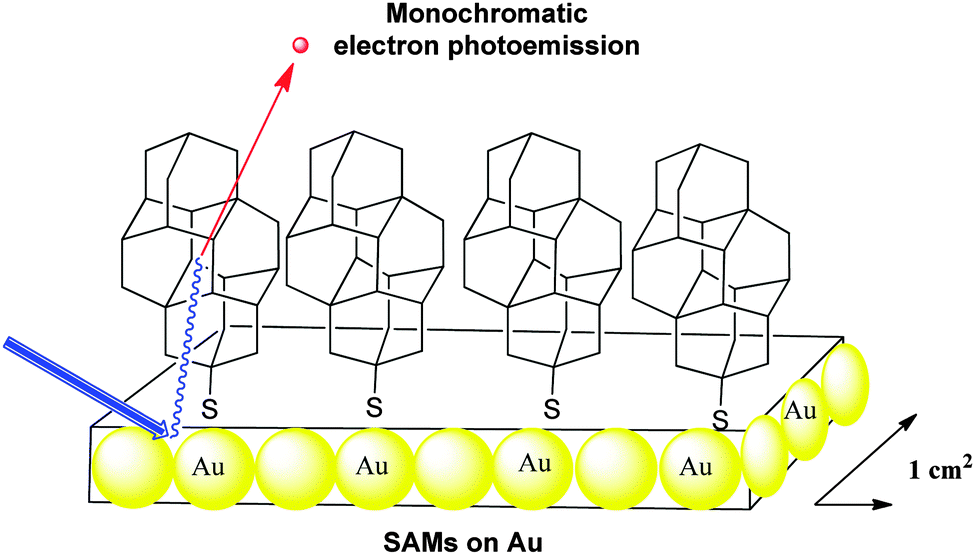 | ||
| Scheme 17 Monochromatic electron photoemission from self-assembled monolayers of [121]tetramantane-6-thiol on gold. | ||
Photoelectron spectra of the layers exhibited a peak at the low-kinetic energy threshold with up to 68% of all emitted electrons emitted within this single energy peak.94 With a narrow energy distribution width of 0.3 electron volts, this source of monochromatic electrons might find application in technologies such as electron microscopy, electron beam lithography, and field-emission flat-panel displays. Comparatively, the spectrum of a [121]tetramantane film shows only a small peak at low kinetic energy.93 The poor electron conductivity within the thicker films versus that through the SAMs, as well as the role that the thiol groups play within the samples could be the origin of this difference. Such strong electron emission does not occur exclusively from the diamondoid surface and the metal substrate is also involved in the process. Measurement of the mean free path for scattering demonstrates that such diamondoid SAMs do have a significantly short interaction length compared to the known related materials.
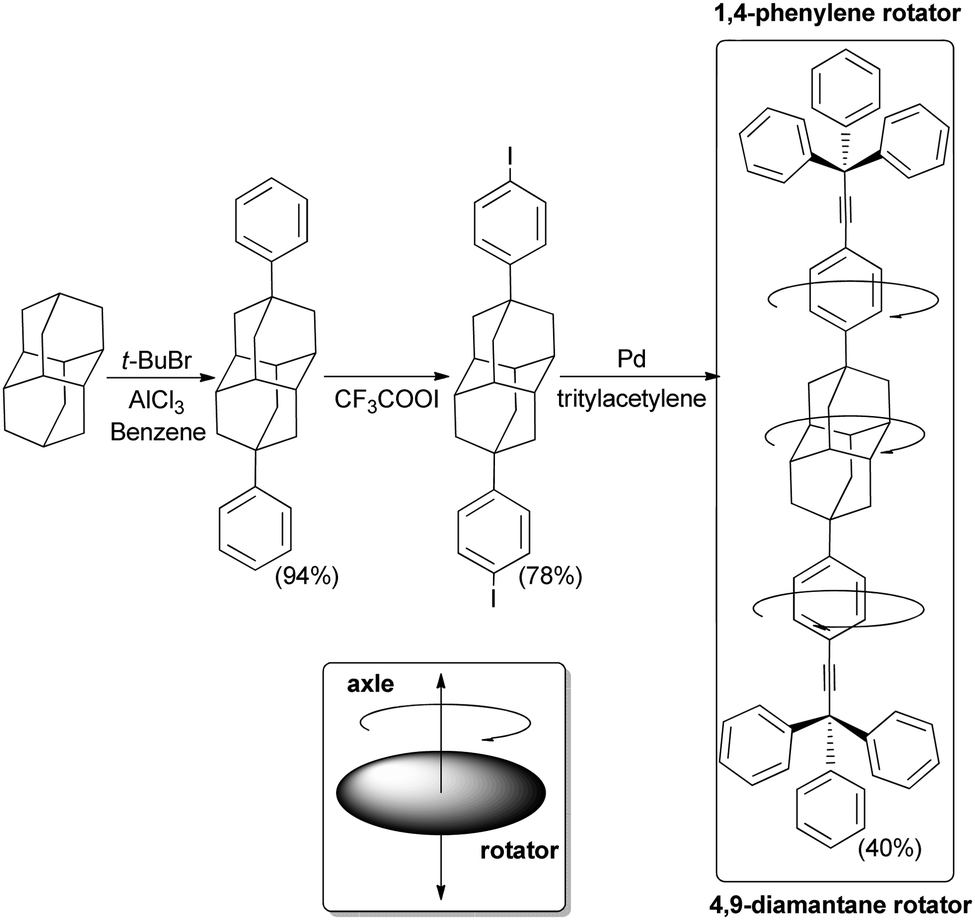
4.7 Molecular machines and rotors
Owing to their unique rigidity and sphericity diamondoid structures have provided decisive “mechanical” functions at the nanometer scale within the framework of organohybrid structures.So-called “nanocars” are supramolecular structures that can translate on surfaces with some controlled directionality.95 These “nanocars” incorporate wheels similar to their real-live models. The synthesis of fluorescent nanocars equipped with adamantane wheels has thus been reported (Scheme 18), and their behaviour was analysed by imaging using a method named single-molecule fluorescence microscopy (SMFM). The “nanocars” were imaged using 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) as the chromophore. A BODIPY moiety was rigidly incorporated into the molecule chassis via acetylenic cross-coupling. The mobility of the nanocars was determined from fluorescence images taken as a function of time. The adamantane four-wheeled nanocar showed a significant improvement in the percentage of moving nanocars over less-spherical carborane-wheeled nanocars having the same “chassis”. The interaction energy between the chosen wheels (diamondoid or carborane) and the glass substrate determines the mobility of the nanocars (weak van der Waals forces or hydrogen bonding, respectively). The final results are consistent with a wheel-like rolling motion associated with the sphericity of adamantane.
 | ||
| Scheme 18 Adamantane incorporated into a molecular machine: a mobile adamantane-wheeled fluorescent “nanocar”. | ||
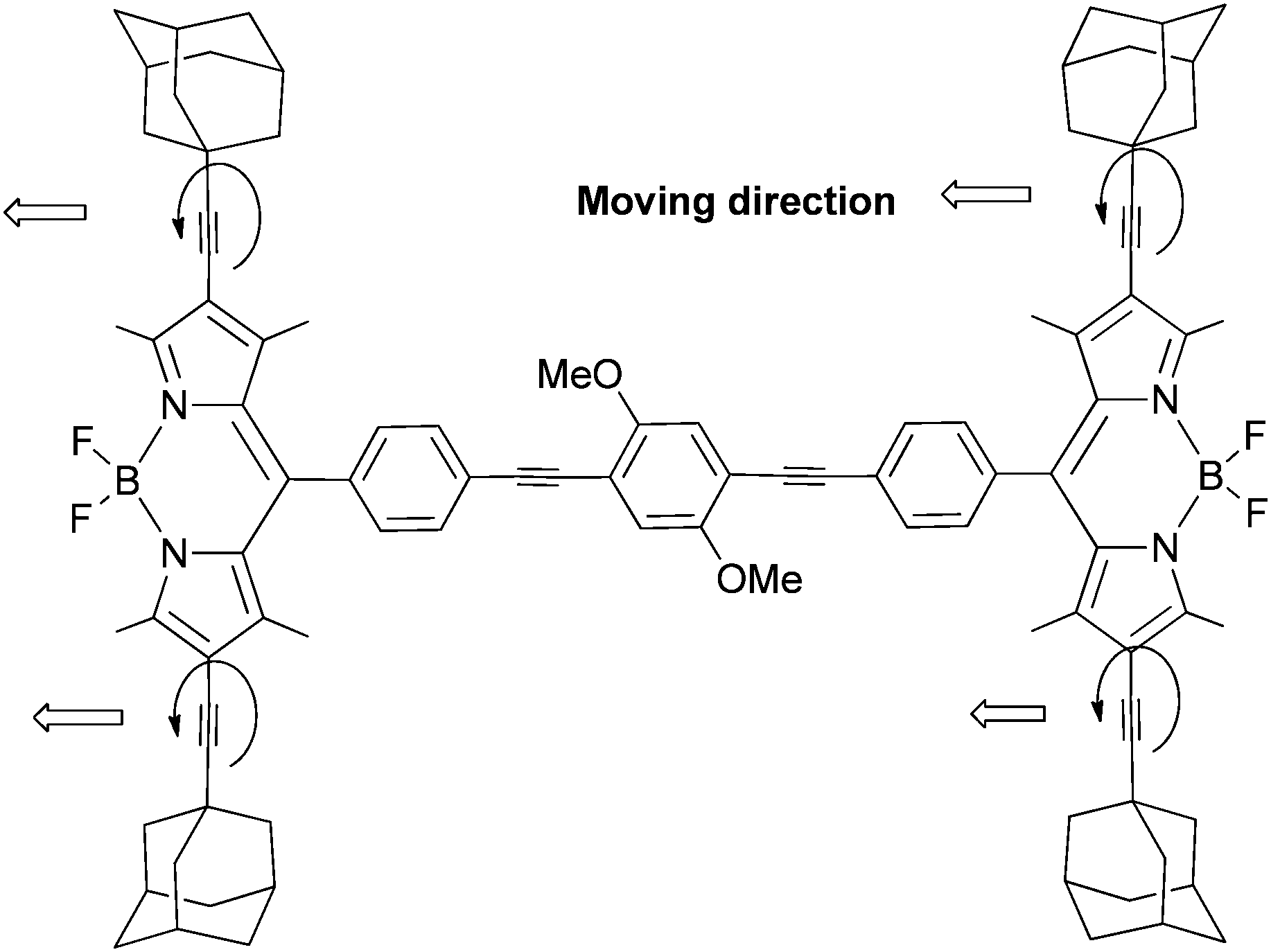
Diamantane has been functionalized to serve as a rotator in a so-called “nanogyroscope” (Scheme 19).96 This was achieved in three synthetic steps with an overall yield of 29%.97
 | ||
| Scheme 19 Diamantane incorporated into a molecular “nanogyroscope” with a fast diamantane rotator within a rigid crystalline environment. | ||
4,9-Diphenyldiamantane was first obtained from one-pot Lewis acidic catalysed bromination and subsequent phenylation. Iodination of the phenyl groups at the para-position of ipso-C followed by a palladium-catalysed arylation of tritylacetylene leads to a “nanogyroscope” in which both phenylene and diamantane rotating units are present (Scheme 19). The interest in controlling rotary dynamics at the molecular level within a rigid crystalline environment led to this construction, with the objective of introducing diamantane as a more cylindrical group with potentially low-energy rotational barriers. Solid state NMR studies have shown that the rotary dynamics of nanogyroscopes depend primarily on steric shielding. In general, while such structures ending with triptycyls are static, analogues with trityl groups can display 180° rotation with rates of ca. 500 Hz at 300 K.97 Remarkably, at 300 K the diamantane group rotates 20000 times faster than the connected phenylene groups, despite their proximity and close environment. These observations confirm that the more cylindrical high order rotators may have much higher rotational frequencies in solids.
4.8 Diamondoids as 3D building blocks for material chemistry
Functionalized diamondoids provide a unique combination of valuable features and properties for building new materials: multivalence, geometrically stiff structure, absence of π–π stacking interactions and great size of the molecular scaffold. In particular, their rigidity makes diamondoids valuable building blocks and platforms for crystal design and crystal engineering. Adamantane-1,3,5,7-tetracarboxylate (1-7, Scheme 5) is an ideal tetrahedral spacer, which was co-polymerized with Cu(NO3)298 or Zn(NO3)299 to form porous metal–organic framework (MOF) materials with a 3D channel system. Utilization of the expanded analogue of 1-7, the adamantane-1,3,5,7-tetrabenzoate (derived from 1-37, Scheme 20), retains the tetrahedral topology of MOF but comprises larger channels.100 The MOFs based on adamantane-1,3-dicarboxylate (derived from 1-38) usually also display the 3D structures.101 The condensations of 1-38 carboxylate derivative with different pyridyl-containing ligands give rise to various coordination architectures.102 A novel one-dimensional (1D) metal–organic nanotube has been synthesized and characterized presenting the first independent 1D metal–organic nanotube with mesohelical structure.103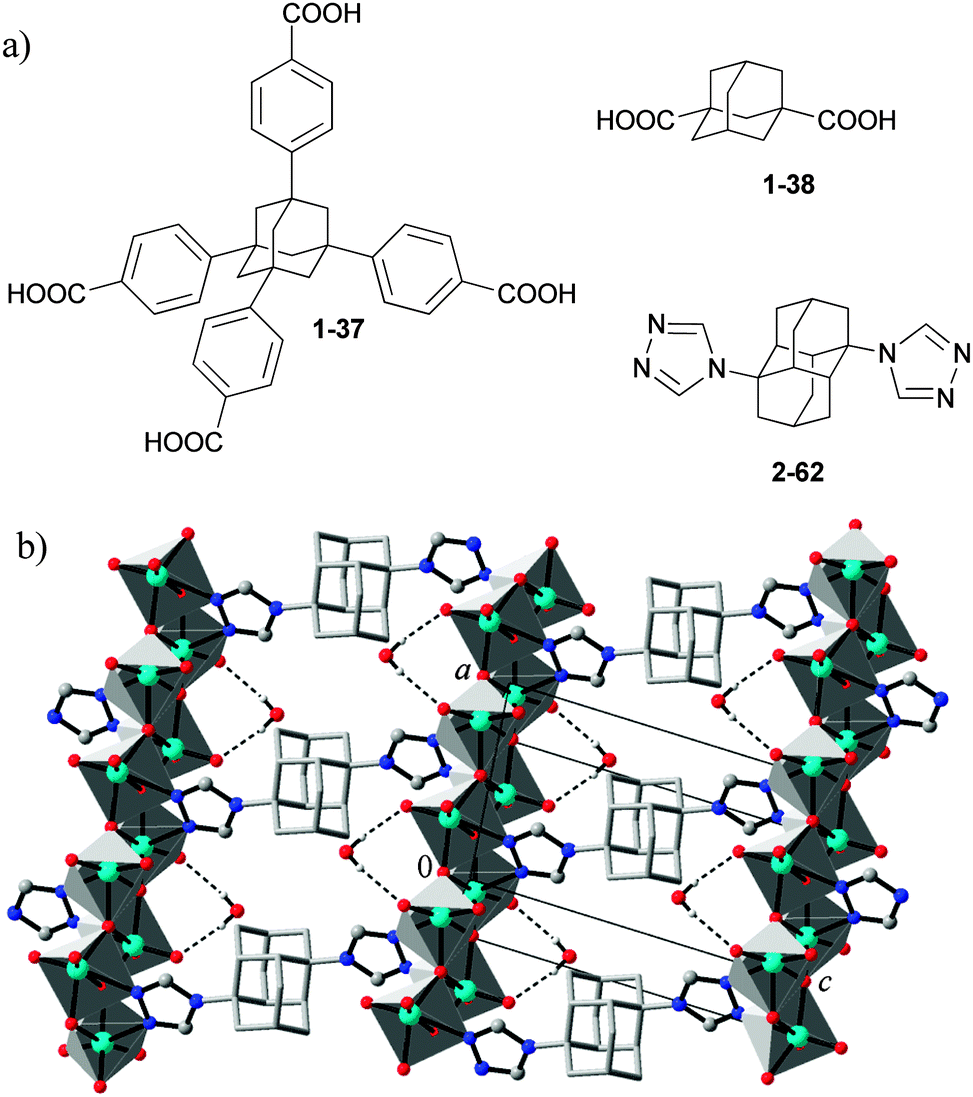 | ||
| Scheme 20 The metal–organic framework building blocks based on adamantane and diamantane (a), and the molybdenum(VI) oxide–organic framework system based on 1,6-bis(1,2,4-triazol-4-yl)diamantane (2-62) (see (b) adapted from ref. 104). | ||
1,6-Bis(1,2,4-triazol-4-yl)diamantane (2-62) and other ditopic adamantane and diamantane based 1,2,4-triazole ligands were used in the preparation of metal oxide–organic frameworks (MOOFs) that complement and extend the general design approaches towards common MOFs (Scheme 20).104
5. Conclusions
Diamondoids (molecular nanodiamonds) represent a unique class of chemical structures with largely untapped potential in chemistry, for the preparation of highly lipophilic pharmacologically active compounds, and as building blocks for novel carbon materials. Here we provide an overview of their origin as well as functionalization with typical chemical functional groups, and provide glimpses of applications that have come forward in the last few years.Practically all bridgehead tertiary derivatives are available or can be synthesized by predictable routes with high chemical yields. Highly valuable halides, alcohols, acids, amines and aryl derivatives are now at the chemist's and materials scientist's disposal. Secondary derivatives are much more difficult to access and the only high-yielding route currently involves the oxidation to the respective ketones followed by subsequent functional group conversion.
Nanodiamonds complement the surging rise of carbon materials that are for the most part based on sp2-materials (e.g., fullerenes, nanotubes, graphenes) with largely different properties. Several fundamental studies on the electronic properties of functionalized nanodiamonds are highly promising in this respect as they show that many of the highly desirable properties of macroscopic diamond (hardness, electron emission, high chemical resistance, low toxicity, fluorescence, biocompatibility, etc.) are readily transferred to the nano scale with the added advantage of chemical functionalizability, purity and reproducibility.
Acknowledgements
The work in Dijon was supported by the “Conseil Régional de Bourgogne” (PhD grant for M. A. G. in project PARI-IME SMT08) and by CNRS from 3MIM P4-program. The diamondoid programme work in Germany was supported over the years by the Deutsche Forschungsgemeinschaft and the National Science Foundation of the USA (DFG-NSF Schr597/12-1), and in part by the Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering, under contract DE-AC02-76SF00515.Notes and references
- H. Schwertfeger, A. Fokin and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 1022 CrossRef CAS PubMed.
- A. Krueger, Adv. Mater., 2008, 20, 2445 CrossRef CAS.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11 CrossRef CAS PubMed.
- A. T. Balaban and P. v. R. Schleyer, Tetrahedron, 1978, 34, 3599 CrossRef CAS . This definition prevents thus the inclusion of cyclohexane and other simple alkanes. In this reference various examples of non-polymantane diamondoids are described.
- G. P. Moss, Pure Appl. Chem., 1999, 71, 513 CrossRef CAS.
- J. Filik, Diamondoid Hydrocarbons, in Carbon-Based Nanomaterials, Materials Science Foundations (monograph series), ed. N. Ali, A. Öchsner and W. Ahmed, Trans Tech, Switzerland, 2010 Search PubMed.
- S. Landa and V. Machacek, Collect. Czech. Chem. Commun., 1933, 5, 1 CAS.
- S. Landa, S. Kriebel and E. Knobloch, Chem. Listy, 1954, 48, 61 CAS.
- P. v. R. Schleyer, J. Am. Chem. Soc., 1957, 79, 3292 CrossRef CAS.
- C. A. Cupas, P. v. R. Schleyer and D. J. Trecker, J. Am. Chem. Soc., 1965, 87, 917 CrossRef CAS.
- T. Courtney, D. E. Johnston, M. A. McKervey and J. J. Rooney, J. Chem. Soc., Perkin Trans. 1, 1972, 2691 RSC.
- S. Hala, S. Landa and V. Hanus, Angew. Chem., Int. Ed. Engl., 1966, 5, 1045 CAS.
- V. Z. Williams, P. v. R. Schleyer, G. J. Gleicher and L. B. Rodewald, J. Am. Chem. Soc., 1966, 88, 3862 CrossRef CAS.
- W. Burns, T. Mitchell, M. McKervey, J. Rooney, G. Ferguson and P. Roberts, J. Chem. Soc., Chem. Commun., 1976, 893 RSC.
- P. v. R. Schleyer, My Thirty Years in Hydrocarbon Cages: From Adamantane to Dodecahedrane, in Cage Hydrocarbons, ed. G. A. Olah, John Wiley, New York, 1990 Search PubMed.
- E. Osawa, A. Furusaki, N. Hashiba, T. Matsumoto, V. Singh, Y. Tahara, E. Wiskott, M. Farcasiu, T. Iizuka, N. Tanaka, T. Kan and P. v. R. Schleyer, J. Org. Chem., 1980, 45, 2985 CrossRef CAS.
- R. Lin and Z. A. Wilk, Fuel, 1995, 74, 1512 CrossRef CAS.
- J. E. Dahl, J. M. Moldowan, K. E. Peters, G. E. Claypool, M. A. Rooney, G. E. Michael, M. R. Mello and M. L. Kohnen, Nature, 1999, 399, 54 CrossRef CAS PubMed.
- J. E. Dahl, S. Liu and R. M. K. Carlson, Science, 2003, 299, 96 CrossRef CAS PubMed.
- J. E. P. Dahl, J. M. Moldovan, Z. Wei, P. A. Lipton, P. Denisevich, R. Gatt, S.-G. Liu, P. R. Schreiner and R. M. K. Carlson, Angew. Chem., Int. Ed., 2010, 49, 9881 CrossRef CAS PubMed.
- J. Reiser, E. McGregor, J. Jones, R. Enick and G. Holder, Fluid Phase Equilib., 1996, 117, 160 CrossRef CAS.
- T. E. Jenkins and J. Lewis, Spectrochim. Acta, Part A, 1980, 36, 259 CrossRef.
- J. E. P. Dahl, J. M. Moldowan, T. M. Peakman, J. C. Clardy, E. Lobkovsky, M. M. Olmstead, P. W. May, T. J. Davis, J. W. Steeds, K. E. Peters, A. Pepper, A. Ekuan and R. M. K. Carlson, Angew. Chem., Int. Ed., 2003, 42, 2040 CrossRef CAS PubMed.
- For chloroadamantane, see: R. Jalal and R. Gallo, Synth. Commun., 1989, 19, 1697 CrossRef CAS.
- For iodoadamantane, see: H. Stone and H. Shechter, J. Org. Chem., 1950, 15, 491 CrossRef CAS.
- For fluoroadamantane, see: S. Hara and M. Aoyama, Synthesis, 2008, 2510 CrossRef CAS PubMed.
- G. L. Baughman, J. Org. Chem., 1964, 29, 238 CrossRef CAS.
- T. M. Gund and P. v. R. Schleyer, Tetrahedron Lett., 1971, 19, 1586 Search PubMed.
- T. M. Gund, P. v. R. Schleyer, G. D. Unruh and G. J. Gleicher, J. Org. Chem., 1974, 39, 2995 CrossRef.
- A. A. Fokin, B. A. Tkachenko, P. A. Gunchenko, D. V. Gusev and P. R. Schreiner, Chem.–Eur. J., 2005, 11, 7091 CrossRef CAS PubMed.
- T. M. Gund, M. Nomura, V. Z. Williams Jr and P. v. R. Schleyer, Tetrahedron Lett., 1970, 56, 4875 CrossRef.
- A. A. Fokin, B. A. Tkachenko, P. A. Gunchenko, D. V. Gusev and P. R. Schreiner, Chem.–Eur. J., 2005, 11, 7091 CrossRef CAS PubMed.
- (a) Methods have been early on developed for the conversion of diamantane into the three possible types of monofunctional derivatives: 1-(medial), 3-(secondary), 4-(apical), see synthetic details in ref. 48; (b) A. A. Fokin, T. S. Zhuk, A. E. Pashenko, P. O. Dral, P. A. Gunchenko, J. E. P. Dahl, R. M. K. Carlson, T. V. Koso, M. Serafin and P. R. Schreiner, Org. Lett., 2009, 11, 3068 CrossRef CAS PubMed.
- H. Schwertfeger, C. Würtele, H. Hausmann, J. E. P. Dahl, R. M. K. Carlson, A. A. Fokin and P. R. Schreiner, Adv. Synth. Catal., 2009, 351, 1041 CrossRef CAS.
- P. R. Schreiner, N. A. Fokina, B. A. Tkachenko, H. Hausmann, M. Serafin, J. E. P. Dahl, S. Liu, R. M. K. Carlson and A. A. Fokin, J. Org. Chem., 2006, 71, 6709 CrossRef CAS PubMed.
- N. A. Fokina, B. A. Tkachenko, A. Merz, M. Serafin, J. E. P. Dahl, R. M. K. Carlson, A. A. Fokin and P. R. Schreiner, Eur. J. Org. Chem., 2007, 4738 CrossRef CAS.
- A. A. Fokin, P. R. Schreiner, N. A. Fokina, B. A. Tkachenko, H. Hausmann, M. Serafin, J. E. P. Dahl, S. Liu and R. M. K. Carlson, J. Org. Chem., 2006, 71, 8532 CrossRef CAS PubMed.
- Z. Cohen, E. Keinan, Y. Mazur and T. H. Varkony, J. Org. Chem., 1975, 40, 2141 CrossRef CAS.
- G. L. Baughman, J. Org. Chem., 1964, 29, 238 CrossRef CAS.
- S. Liu, J. Guo and D. Jia, Pat. CN, 1974515A, 2007 Search PubMed.
- H. Schwertfeger, C. Würtele, M. Serafin, H. Hausmann, R. M. K. Carlson, J. E. P. Dahl and P. R. Schreiner, J. Org. Chem., 2008, 73, 7789 CrossRef CAS PubMed.
- B. A. Tkachenko, N. A. Fokina, L. V. Chernish, J. E. P. Dahl, S. Liu, R. M. K. Carlson, A. A. Fokin and P. R. Schreiner, Org. Lett., 2006, 8, 1767 CrossRef CAS PubMed.
- (a) G. Molle, P. Bauer and J. E. Dubois, J. Org. Chem., 1982, 47, 4120 CrossRef CAS; (b) J. E. Dubois, P. Bauer, G. Molle and J. Daza, C. R. Acad. Sci., Paris Ser. IIc: Chim., 1977, 284, 145 CAS.
- G. Molle, P. Bauer and J. E. Dubois, J. Org. Chem., 1983, 48, 2975 CrossRef CAS.
- P. R. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Yu. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. P. Dahl, R. M. K. Carlson and A. A. Fokin, Nature, 2011, 477, 308 CrossRef CAS PubMed.
- A. A. Fokin, L. V. Chernish, P. A. Gunchenko, E. Yu. Tikhonchuk, H. Hausmann, M. Serafin, J. E. P. Dahl, R. M. K. Carlson and P. R. Schreiner, J. Am. Chem. Soc., 2012, 134, 13641 CrossRef CAS PubMed.
- H. Stetter, M. Schwarz and A. Hirschhorn, Angew. Chem., 1959, 71, 429–430 CrossRef CAS.
- T. M. Gund, M. Nomura and P. v. R. Schleyer, J. Org. Chem., 1974, 39, 2987 CrossRef CAS.
- N. A. Fokina, B. A. Tkachenko, J. E. P. Dahl, R. M. K. Carlson, A. A. Fokin and P. R. Schreiner, Synthesis, 2012, 259 CrossRef CAS PubMed.
- G. S. Lee, J. N. Bashara, G. Sabih, A. Oganesyan, G. Godjoian, H. M. Duong, E. R. Marinez and C. Gutierrez, Org. Lett., 2004, 6, 1705 CrossRef CAS PubMed.
- A. A. Fokin, B. A. Tkachenko, N. A. Fokina, H. Hausmann, M. Serafin, J. E. P. Dahl, R. M. K. Carlson and P. R. Schreiner, Chem.–Eur. J., 2009, 15, 3851 CrossRef CAS PubMed.
- L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516 CrossRef CAS PubMed.
- A. A. Fokin, A. Merz, N. A. Fokina, H. Schwertfeger, S. L. Liu, J. E. P. Dahl, R. K. M. Carlson and P. R. Schreiner, Synthesis, 2009, 909 CrossRef CAS PubMed.
- H. Schwertfeger, C. Würtele and P. R. Schreiner, Synlett, 2010, 493 CAS.
- J. R. Goerlich and R. Schmutzler, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 102, 211 CrossRef CAS.
- A. Köllhofer and H. Plenio, Chem.–Eur. J., 2003, 9, 1416 CrossRef PubMed.
- M. Beller, A. Ehrentraut, C. Fuhrmann and A. Zapf, PCT Int. Appl., WO 2002010178 A1 20020207, 2002 Search PubMed.
- A. Zapf and M. Beller, Chem. Commun., 2005, 431 RSC.
- J. P. Stambuli, S. R. Stauffer, K. H. Shaughnessy and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 2677 CrossRef CAS.
- M. Gouygou, G. Etemad-Moghadam and M. Koenig, Synthesis, 1987, 508 CrossRef CAS.
- G. A. Olah, O. Farooq, Q. Wang and A. Wu, J. Org. Chem., 1990, 55, 1224 CrossRef CAS.
- H. Duddeck, M. Hani, A. Elgamal and A. G. Hanna, Phosphorus, Sulfur Silicon Relat. Elem., 1986, 28, 307 CrossRef CAS.
- H. Schwertfeger, M. M. Machuy, C. Würtele, J. E. P. Dahl, R. M. K. Carlson and P. R. Schreiner, Adv. Synth. Catal., 2010, 352, 609 CrossRef CAS.
- A. A. Fokin, E. D. Butova, L. V. Chernish, N. A. Fokina, J. E. P. Dahl, R. M. K. Carlson and P. R. Schreiner, Org. Lett., 2007, 9, 2541 CrossRef CAS PubMed.
- (a) I. Tabushi and S. Kojo, Tetrahedron Lett., 1973, 26, 2329 CrossRef; (b) I. Tabushi, S. Kojo and K. Fukunishi, J. Org. Chem., 1978, 43, 2370 CrossRef CAS.
- J. A. Wright, M. J. Gaunt and J. B. Spencer, Chem.–Eur. J., 2006, 12, 949 CrossRef CAS PubMed.
- A. A. Fokin, E. D. Butova, A. V. Barabash, N. N. Huu, B. A. Tkachenko, N. A. Fokina and P. R. Schreiner, Synth. Commun., 2013, 43, 1772–1777 CrossRef CAS.
- I. Tabushi, S. Kojo, P. v. R. Schleyer and T. M. Gund, J. Chem. Soc., Chem. Commun., 1974, 591 RSC.
- A. A. Fokin, P. A. Gunchenko, A. A. Novikovsky, T. E. Shubina, B. V. Chernyaev, J. E. P. Dahl, R. M. K. Carlson, A. G. Yurchenko and P. R. Schreiner, Eur. J. Org. Chem., 2009, 5153 CrossRef CAS.
- (a) S. Samnick, S. Ametamey, K. L. Leenders, P. Vontobel, G. Quack, C. G. Parsons, H. Neu and P. A. Schubiger, Nucl. Med. Biol., 1998, 25, 323 CrossRef CAS; (b) B. Reisberg, R. Doody, A. Stöffler, F. Schmitt, S. Ferris and H. J. Möbius, N. Engl. J. Med., 2003, 348, 1333 CrossRef CAS PubMed.
- (a) E. Ōsawa, Z. Majerski and P. v. R. Schleyer, J. Org. Chem., 1971, 36, 205 CrossRef; (b) G. Molle, J. E. Dubois and P. Bauer, Can. J. Chem., 1987, 65, 2428 CrossRef CAS.
- M. Aoyama and S. Hara, Tetrahedron, 2009, 65, 3682 CrossRef CAS PubMed.
- (a) L. Bauer and K. K. Khullar, J. Org. Chem., 1971, 36, 3038 CrossRef CAS; (b) J. M. Kokosa, L. Bauer and R. S. Egan, J. Org. Chem., 1975, 40, 3196 CrossRef CAS; (c) P. A. Cahill, Tetrahedron Lett., 1990, 31, 5417 CrossRef CAS.
- P. R. Schreiner, A. A. Fokin, O. Lauenstein, Y. Okamoto, T. Wakita, C. Rinderspacher, G. H. Robinson, J. K. Vohs and C. F. Campana, J. Am. Chem. Soc., 2002, 124, 13348 CrossRef CAS PubMed.
- A. A. Fokin, O. Lauenstein, P. A. Gunchenko and P. R. Schreiner, J. Am. Chem. Soc., 2001, 123, 1842 CrossRef CAS PubMed.
- A. E. Lukach, A. N. Santiago and R. A. Rossi, J. Org. Chem., 1997, 62, 4260 CrossRef CAS PubMed.
- A. Tewari, M. Hein, A. Zapf and M. Beller, Tetrahedron, 2005, 61, 9705 CrossRef CAS PubMed.
- G. Eastham, N. Tindale, PCT Int. Appl., WO 2005079981 A1 20050901, 2005 Search PubMed.
- A. Köllhofer and H. Plenio, Adv. Synth. Catal., 2005, 347, 1295 CrossRef.
- A. Köllhofer and H. Plenio, Chem.–Eur. J., 2003, 9, 1416 CrossRef PubMed.
- C. E. Müller, L. Wanka, K. Jewell and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 6180 CrossRef PubMed.
- C. B. Shinisha and R. B. Sunoj, Org. Lett., 2009, 11, 3242 CrossRef CAS PubMed.
- W. Danysz, C. G. Parsons, J. Kornhuber, W. J. Schmidt and G. Quack, Neurosci. Biobehav. Rev., 1997, 21, 455 CrossRef CAS.
- Y. Wang, B. A. Tkachenko, P. R. Schreiner and A. Marx, Org. Biomol. Chem., 2011, 9, 7482 CAS.
- J. Voskuhl, M. Waller, S. Bandaru, B. A. Tkachenko, C. Fregonese, B. Wibbeling, P. R. Schreiner and B. J. Ravoo, Org. Biomol. Chem., 2012, 10, 4524 CAS.
- K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y.-W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382 CrossRef CAS PubMed.
- T. G. Archibald, A. A. Malik and K. Baum, Macromolecules, 1991, 24, 5261 CrossRef CAS.
- A. A. Malik, T. G. Archibald and K. Baum, Macromolecules, 1991, 24, 5266 CrossRef CAS.
- S. Zheng, J. Shi and R. Mateu, Chem. Mater., 2000, 12, 1814 CrossRef CAS.
- Q. Wei, A. Lazzeri, F. Di Cuia, M. Scalari and E. Galoppini, Macromol. Chem. Phys., 2004, 205, 2089 CrossRef CAS.
- (a) D. R. Robello, J. Appl. Polym. Sci., 2013, 127, 96 CrossRef CAS; (b) C. Sinkel, S. Agarwal, N. A. Fokina and P. R. Schreiner, J. Appl. Polym. Sci., 2009, 114, 2109 CrossRef CAS.
- A. Ghosh, S. F. Sciamanna, J. E. Dahl, S. Liu, R. M. K. Carlson and D. A. Schiraldi, J. Polym. Sci., Polym. Chem. Ed., 2007, 45, 1077 CAS.
- W. L. Yang, J. D. Fabbri, T. M. Willey, J. R. I. Lee, J. E. Dahl, R. M. K. Carlson, P. R. Schreiner, A. A. Fokin, B. A. Tkachenko, N. A. Fokina, W. Meevasana, N. Mannella, K. Tanaka, X. J. Zhou, T. van Buuren, M. A. Kelly, Z. Hussain, N. A. Melosh and Z.-X. Shen, Science, 2007, 316, 1460 CrossRef CAS PubMed.
- S. Roth, D. Leuenberger, J. Osterwalder, J. E. Dahl, R. M. K. Carlson, B. A. Tkachenko, A. A. Fokin, P. R. Schreiner and M. Hengsberger, Chem. Phys. Lett., 2010, 495, 102 CrossRef CAS PubMed.
- P.-L. E. Chu, L. Y. Wang, S. Khatua, A. B. Kolomeisky, S. Link and J. M. Tour, ACS Nano, 2013, 7, 35 CrossRef CAS PubMed.
- Macroscopic gyroscopes are devices used for measuring or maintaining orientation, based on the principles of angular momentum. Gyroscopes consist of a spinning wheel or disk mounted on an axle, which is free to assume any orientation. Myriad of applications of these devices are found in navigation and stabilizing systems (planes, helicopters, space-crafts, remote control, guiding systems, mobile phones, motion capture systems, etc.).
- S. D. Karlen, R. Ortiz, O. L. Chapman and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2005, 127, 6554 CrossRef CAS PubMed.
- B. Chen, M. Eddaoudi, T. M. Reineke, J. W. Kampf, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 11559 CrossRef CAS.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504 CrossRef CAS PubMed.
- J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239 CrossRef CAS PubMed.
- F. Millange, C. Serre, J. Marrot, N. Gardant, F. Pelle and G. Ferey, J. Mater. Chem., 2004, 14, 642 RSC.
- J. Jin, Y. Wang, W. Zhang, A. S. Lermontov, E. Kh. Lermontova and Q. Shi, Dalton Trans., 2009, 10181 RSC.
- J. Jin, Y. Wang, P. Liu, R. Liu, C. Ren and Q. Shi, Cryst. Growth Des., 2010, 10, 2029 CAS.
- A. B. Lysenko, G. A. Senchyk, J. Lincke, D. Lassig, A. A. Fokin, E. D. Butova, P. R. Schreiner, H. Krautscheid and K. V. Domasevitch, Dalton Trans., 2010, 39, 4223 RSC.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |